

Abstract
Interleukin (IL-)6 is the major pro-inflammatory cytokine within the IL-6 family. IL-6 signals via glycoprotein 130 (gp130) and the membrane-bound or soluble IL-6 receptor (IL-6R), referred to as classic or trans-signaling, respectively. Whereas inflammation triggers IL-6 expression, eventually rising to nanogram/ml serum levels, soluble IL-6R (sIL-6R) and soluble gp130 (sgp130) are constitutively present in the upper nanogram/ml range. Calculations based on intermolecular affinities have suggested that systemic IL-6 is immediately trapped in IL-6·sIL-6R and IL-6·sIL-6R·sgp130 complexes, indicating that sIL-6R and sgp130 constitute a buffer system that increases the serum half-life of IL-6 or restricts systemic IL-6 signaling. However, this scenario has not been experimentally validated. Here, we quantified IL-6·sIL-6R and IL-6·sIL-6R·sgp130 complexes over a wide concentration range. The amounts of IL-6 used in this study reflect concentrations found during active inflammatory events. Our results indicated that most IL-6 is free and not complexed with sIL-6R or sgp130, indicating that the level of endogenous sgp130 in the bloodstream is not sufficient to block IL-6 trans-signaling via sIL-6R. Importantly, addition of the single-domain antibody VHH6, which specifically stabilizes IL-6·sIL-6R complexes but did not bind to IL-6 or sIL-6R alone, drove free IL-6 into IL-6·sIL-6R complexes and boosted trans-signaling but not classic signaling, demonstrating that endogenous sIL-6R has at least the potential to form complexes with IL-6. Our findings indicate that even though high concentrations of sIL-6R and sgp130 are present in human serum, the relative ratio of free IL-6 to IL-6·sIL-6R allows for simultaneous classic and trans-signaling.
Keywords: cytokine, signal transduction, interleukin 6 (IL-6), interleukin 6 receptor (IL6R), glycoprotein, classic signaling, Trans-signaling
Introduction
The pro-inflammatory cytokine interleukin 6 (IL-6)2 controls development of chronic inflammatory diseases, such as rheumatoid arthritis, asthma, or inflammatory bowel diseases. It is also involved in cardiovascular diseases including atherosclerosis and in tumor initiation and progression (1). Inhibition of IL-6 signaling has been successfully translated into the clinic as a powerful anti-inflammatory strategy. IL-6 is secreted in response to infection by leukocytes, but also by fibroblasts, adipocytes, keratinocytes, and endothelial cells, and is induced by other cytokines, including IL-1 or tumor necrosis factor α (TNFα). For signaling, IL-6 binds to the nonsignal transducing IL-6 receptor (IL-6R), followed by complex formation with the signal-transducing co-receptor glycoprotein 130 (gp130). IL-6 signaling activates downstream signaling pathways such as Janus kinases/signal transducers and activators of transcription (Jak/STAT), the phosphatidylinositol 3-kinase cascade and the mitogen-activated protein kinase cascade through gp130 homodimer formation (2). Importantly, free IL-6 has no binding affinity toward gp130, making the IL-6R mandatory for initial IL-6 binding and subsequent activation of gp130 (3). In addition to the membrane-bound IL-6R, a soluble form of IL-6R (sIL-6R) is produced by proteolytic shedding and alternative splicing (4–6). Binding of IL-6 to membrane-bound IL-6R induces classic signaling via gp130, whereas binding of IL-6 to sIL-6R induces trans-signaling, also via gp130. IL-6R is mainly found on hepatocytes and immune cells, thereby restricting the number of cells targeted by IL-6 classic signaling. IL-6 classic signaling induces the acute-phase response and is considered to have homeostatic and anti-inflammatory effects (7). Because gp130 is ubiquitously expressed, trans-signaling via the sIL-6R can activate virtually all cells of the body. IL-6 trans-signaling mainly regulates pro-inflammatory reactions (7). A soluble form of gp130 (sgp130) has been shown to specifically inhibit IL-6 trans-signaling, whereas IL-6 classic signaling is largely unaffected (3, 8). Blocking of trans-signaling was effective in a variety of preclinical chronic and autoimmune disease models (7). Interestingly, under conditions with a high molar excess of sIL-6R over IL-6, sgp130 also interferes with IL-6 classic signaling (8). For selective IL-6 trans-signaling, a designer cytokine called Hyper-IL-6 was generated. It consists of IL-6 connected to the sIL-6R by a flexible peptide linker, and is 100–1000-fold more potent in inducing trans-signaling than natural IL-6·sIL-6R complexes (3). Compared with natural IL-6·sIL-6R complexes, Hyper-IL-6 mimics a situation where all IL-6 molecules are trapped in such complexes. Another designer protein useful both for in vivo therapeutic applications and for in vitro experiments is sgp130Fc, which was constructed by dimerizing two sgp130 extracellular domains with a human IgG1-Fc (3).
Homeostatic production of IL-6 leads to plasma levels of about 1–10 pg/ml (9, 10). During infection, inflammatory diseases, or cancer, IL-6 serum levels are elevated to the lower nanogram/ml range. Fatal sepsis will even result in much higher IL-6 concentrations in the 100–1,000 ng/ml range (11). Importantly, sIL-6R is always present at high concentrations (25–75 ng/ml) in human sera, and these levels increase only moderately during inflammation by a factor of 2–3 (12–14). Finally, sgp130 is found at concentrations between 100 and 400 ng/ml in the serum of healthy humans (12).
Different dissociation constant (Kd) values for IL-6 binding to sIL-6R have been reported, ranging from 0.5 to 34 nm (12, 15, 16). Depending on the Kd, calculations based on intermolecular affinities suggested that some or most systemic IL-6 will be trapped in IL-6·sIL-6R or IL-6·sIL-6R·sgp130 complexes, indicating that sIL-6R and sgp130 constitute a buffer system to increase the serum half-life of IL-6 and/or to restrict systemic IL-6 signaling. The aim of this study was to experimentally determine IL-6·sIL-6R and IL-6·sIL-6R·sgp130 complex formation over a wide concentration range and to compare these data with calculated amounts based on Kd. To directly quantify IL-6·sIL-6R complexes, we used a recently commercialized ELISA and developed an sgp130Fc precipitation assay to assess IL-6·sIL-6R·sgp130Fc complexes.
Results
Specific detection of IL-6·sIL-6R complex formation showed that the amounts of naturally formed IL-6·sIL-6R complexes is far below the calculated theoretical values
In the last 25 years, different Kd values for IL-6·IL-6R complex formation have been described, ranging from 0.5 to 34 nm (12, 15, 16). Based on the Kd and an equation described by Gaillard et al. (12), it is possible to calculate the theoretical proportions of IL-6·sIL-6R complexes and free IL-6 for any given concentration. To enable the comparison of the theoretical data with experimentally quantified IL-6·sIL-6R complexes, we determined the Kd of our recombinant proteins IL-6 and sIL-6R by surface plasmon resonance to be 22 nm for IL-6·sIL-6R complexes (immobilized sIL-6R; Fig. S1A). Albeit our Kd value is in good agreement with the recently reported Kd values by Adams et al. (15) (15.4 nm for immobilized sIL-6R and 34.2 nm for immobilized IL-6), it differs significantly from the Kd values of 0.5 and 0.9 nm initially described by Gaillard et al. (12) and Zohlnhöfer et al. (16). Zohlnhöfer et al. (16) measured the Kd by determination of IL-6 binding toward HepG2 cells, whereas Gaillard et al. (12) also used surface plasmon resonance. gp130 is an affinity converter, leading to formation of a high affinity IL-6·IL-6R·gp130 ternary complex with a Kd in the range of 10 pm (17). To determine whether our recombinant IL-6 has this property, we used Ba/F3–gp130–IL-6R cells, which require IL-6 as a stimulus for proliferation (18). Half-maximal proliferation of Ba/F3–gp130–IL-6R was achieved with 8.4 pm IL-6 (Fig. S1B), which is excellent compared with previous reports using IL-6 and the same cell line (18–21). Despite the fact that our IL-6 appears to have lower binding affinity to IL-6R as compared with some older reports, analysis of its biological activity demonstrates high specificity and activity of our recombinant IL-6, and recent data from Adams et al. (15) match our results very closely. Therefore, we concluded that the Kd determined in this study is appropriate.
Next, we calculated the theoretical amounts of IL-6·sIL-6R complexes based on the physiological concentrations of IL-6 and sIL-6R in health and disease using our Kd of 22 nm (adapted from Ref. 12). For example, systemic inflammation eventually results in IL-6 concentrations up to 10 ng/ml (0.422 nm, molecular mass of IL-6 23.7 kDa), which, in combination with typical endogenous sIL-6R levels around 50 ng/ml (0.97 nm, molecular mass of sIL-6R 51.5 kDa) will result in 95.8% free IL-6 (0.404 nm) and 4.2% IL-6 (0.018 nm) in IL-6·sIL-6R complexes (0.0175 nm) and 98.2% free sIL-6R (0.952 nm) (Table S1). Our previously published experimental data based on cellular proliferation and STAT3 phosphorylation of Ba/F3–gp130 cells also indicated that for these concentrations, the total amount of free IL-6 should have been above 90% with less than 10% in IL-6·sIL-6R complexes (8). To experimentally determine IL-6·sIL-6R complexes, we used the recently commercialized IL-6·sIL-6R Complex DuoSet ELISA from R&D Systems (Minneapolis, MN) (DY8139-05). Before analyzing complex formation using the IL-6·sIL-6R ELISA, we demonstrated that the ELISA specifically detects IL-6·sIL-6R complexes but not IL-6 or sIL-6R alone (Fig. 1A). Next, we combined 25 (0.458 nm), 50 (0.97 nm), or 75 ng/ml (1.45 nm) of sIL-6R, reflecting physiological sIL-6R levels, with 1 to 1,000 ng/ml (0.042 to 42.2 nm) of IL-6. We then quantified the IL-6·sIL-6R complexes by ELISA (Fig. 1B) and calculated the resulting percentages of free and complexed sIL-6R (Fig. 1C). The calculation is based on the molar ratios of IL-6 and sIL-6R in the IL-6·sIL-6R complex (molecular mass of 75.2 kDa) and is described under “Experimental procedures.” Surprisingly, we found that the ELISA is only able to quantify IL-6·sIL-6R complexes for IL-6 concentrations starting with 10 ng/ml, suggesting that this ELISA is unsuitable to detect endogenous IL-6·sIL-6R complexes, because IL-6 is usually below 10 ng/ml, even in acute or chronic inflammatory conditions (22–25). The sensitivity of the ELISA is described to be at 78 pg/ml for the standard protein. We were able to reach this detection limit using the standard protein delivered by R&D, which is a fusion protein of IL-6 and sIL-6R (Fig. 1A), which helps to explain the higher detection limit of our IL-6 and sIL-6R mixtures as compared with the standard protein. Our results showed that even with a 87-fold molar excess of IL-6 (1,000 ng/ml, 42.2 nm) over sIL-6R, only a maximum of 13% of sIL-6R was bound in IL-6·sIL-6R complexes (Fig. 1C, Table S2, 25 ng/ml of recombinant sIL-6R (0.485 nm)). This finding is in stark contrast to the calculated percentage of 65.5% of sIL-6R theoretically bound in IL-6·sIL-6R complexes at concentrations of 1,000 ng/ml of IL-6, using the Kd of 22 nm (Table S1). Furthermore, we observed that for all applied IL-6 concentrations, only about ∼0.16–2% of IL-6 was present in IL-6·sIL-6R complexes, depending on the sIL-6R concentration (Table S3). Next, we reversed the experimental set-up and used three fixed concentrations of IL-6 (1, 10, or 100 ng/ml) and increasing concentrations of sIL-6R (1–1,000 ng/ml) and quantified the IL-6·sIL-6R complexes by ELISA (Fig. 1D). Comparably, only limited amounts of sIL-6R were found in complex with IL-6, with the maximum for 25 ng/ml of sIL-6 and 100 ng/ml of IL-6, about 7% of all sIL-6R molecules were bound in IL-6·sIL-6R complexes (Fig. 1E, Tables S4 and S5). Furthermore, we observed in this setup that even with a 4.5-fold molar excess of sIL-6R (1,000 ng/ml, 19.4 nm) over IL-6 (100 ng/ml, 4.22 nm), only a maximum of ∼4% of IL-6 was bound in IL-6·sIL-6R complexes (Tables S4 and S6). In conclusion, our data using recombinant IL-6 and sIL-6R suggests that the amount of naturally formed IL-6·sIL-6R complexes is far below the calculated theoretical values based on the Kd of 22 nm.
Figure 1.

Quantification of IL-6·sIL-6R complexes. A, validation of the IL-6·sIL-6R ELISA was performed with recombinant IL-6 (0.078–5 ng/ml), sIL-6R (0.078–5 ng/ml), or the IL-6·sIL-6R complex standard (0.078–5 ng/ml). B, recombinant human IL-6 (1–1,000 ng/ml) and sIL-6R (25, 50, or 75 ng/ml) were mixed and IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R ELISA. Combined data from three experiments are shown. Error bars represent the S.D. C, the percentages of sIL-6R present in IL-6·sIL-6R complexes were calculated from B. The calculation is based on the molecular masses of IL-6 (23.7 kDa) and sIL-6R (51.5 kDa), bound in the IL-6·sIL-6R complex (75.2 kDa, ratio = ∼1/3 IL-6 and 2/3 sIL-6R). For example, 5 ng/ml of IL-6/sIL-6R were detected, by combination of 100 ng/ml of IL-6 and 50 ng/ml of sIL-6R. Thus the complex consists of 3.42 ng/ml of sIL-6R and 1.58 ng/ml of IL-6. Consequently, 6.85% of the used sIL-6R and 1.57% of the used IL-6 were bound in the IL-6·sIL-6R complex. Error bars represent the S.D. D, recombinant human sIL-6R (1–1,000 ng/ml) and IL-6 (1, 10, or 100 ng/ml) were mixed and IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R ELISA. Combined data were from three independent experiments. Error bars represent the S.D. E, the percentages of sIL-6R present in IL-6·sIL-6R complexes were calculated from D.
IL-6·sIL-6R complex formation and trans-signaling is boosted by the single domain antibody VHH6 targeting a junctional IL-6·sIL-6R epitope
Subsequently, we analyzed if the amount of IL-6·sIL-6R complexes can be stabilized by the recently described single-domain antibody VHH6. VHH6 specifically binds to an epitope that spans the junction between IL-6 and sIL-6R, but does not interact with IL-6 or sIL-6R alone (15) (Fig. 2A). Importantly, VHH6 did not interfere with the ELISA detection of IL-6·sIL-6R complexes and did not cross-react in the absence of IL-6·sIL-6R (Fig. S1, C and D). First of all, the optimal concentration of VHH6 for maximal stabilization of IL-6·sIL-6R complexes was determined by ELISA. 0.1, 0.25, 0.5, or 1 ng/ml of IL-6 were combined with 50 ng/ml of sIL-6R plus increasing concentrations of VHH6, demonstrating that above a concentration of 0.5 μg/ml of VHH6, no further increase of IL-6·sIL-6R complexes was detected (Fig. 2B, Fig. S1C). Moreover, addition of VHH6 increased sensitivity of IL-6·sIL-6R complex detection about 100-fold, starting already at 0.1 ng/ml of IL-6 (Fig. 2B, Table S7). Our calculation showed that up to 50% of the IL-6 were forced into IL-6·sIL-6R complexes by VHH6.
Figure 2.

VHH6 promotes IL-6 trans-signaling and IL-6·sIL-6R complex formation. A, schematic illustration of VHH6 binding to IL-6·sIL-6R complexes. B, recombinant human IL-6 (0.1–1 ng/ml) and sIL-6R (50 ng/ml) plus increasing amounts of VHH6 (0.1 or 0.5 μg/ml) were mixed and the formed IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R ELISA. Combined data were from three experiments. Error bars represent S.D. C, cellular proliferation of Ba/F3–gp130 cells. VHH6 (0.01–30 μg/ml) was titrated into a setup in which equal numbers of cells were cultured for 3 days without stimulus, with 10 ng/ml of Hyper-IL-6 (HIL-6; control representing 100% trans-signaling complex) or with a combination of 10 ng/ml of IL-6 and 10 ng/ml of sIL-6R. Proliferation was measured using the colorimetric CellTiter-Blue Cell Viability Assay. One representative experiment of three is shown. Error bars represent the S.D. D, cellular proliferation of Ba/F3-gp130 cells. IL-6 (0.01–10 ng/ml) was titrated into a setup in which equal numbers of cells were cultured for 3 days with sIL-6R (10 ng/ml) or with a combination of sIL-6R (10 ng/ml) and VHH6 (10 μg/ml) or HIL-6 alone (10 ng/ml). Proliferation was measured using the colorimetric CellTiter-Blue Cell Viability Assay. One representative experiment of three is shown. Error bars represent the S.D. E, cellular proliferation of Ba/F3-gp130-IL-6R cells. Equal numbers of cells were cultured for 3 days in the presence of HIL-6 (10 ng/ml) or VHH6 (10 μg/ml) and increasing concentrations of IL-6 (0.005–10 ng/ml). Proliferation was measured using the colorimetric CellTiter-Blue Cell Viability Assay. One representative experiment of three is shown. Error bars represent the S.D. F, analysis of STAT3 activation following trans-signaling. Ba/F3–gp130 cells were washed three times, starved, and stimulated with the indicated amounts of HIL-6, IL-6, sIL-6R, and VHH6 for 10 min. Cellular lysates were prepared, and equal amounts of total protein (50 μg/lane) were loaded on SDS gels, followed by immunoblotting using antibodies against phospho-STAT3 or STAT3 (loading control, same samples were used on separated gels). Western blotting data show one representative experiment of three. G, analysis of STAT3 activation following classic signaling. Ba/F3–gp130–IL-6R cells were washed three times, starved, and stimulated with the indicated amounts of IL-6 and VHH6 for 10 min. Cellular lysates were prepared, and equal amounts of total protein (50 μg/lane) were loaded on SDS gels, followed by immunoblotting using antibodies against phospho-STAT3 or STAT3 (loading control, same samples were used on separated gels). Western blotting data show one representative experiment of three.
To analyze the effects of VHH6 in a cell assay, we used IL-6- or IL-6·sIL-6R-induced proliferation of Ba/F3–gp130–IL-6R or Ba/F3–gp130 cells, respectively, to independently evaluate the stabilizing effect of VHH6 on classic and trans-signaling. Proliferation of Ba/F3–gp130 cells depends on IL-6·sIL-6R-mediated trans-signaling, whereas proliferation of Ba/F3–gp130–IL-6R cells depends on IL-6-mediated classic signaling (7). First of all, we demonstrated that the very low proliferation of Ba/F3–gp130 cells induced by a combination of 10 ng/ml of IL-6 and 10 ng/ml of sIL-6R was strongly increased by VHH6 (Fig. 2C). Interestingly, about 300 ng/ml of VHH6 were sufficient to achieve maximal proliferation of Ba/F3–gp130 (Fig. 2C). In another experimental setup with constant concentrations of sIL-6R (10 ng/ml) and VHH6 (10 μg/ml) and increasing concentrations of IL-6 (0.01 to 10 ng/ml), VHH6 reduced the minimal concentration of IL-6 needed to induce cellular proliferation by trans-signaling from 1–3 (42.2–126 pm) to 0.03 ng/ml (1.2 pm) (Fig. 2D). Next, we investigated the effect of VHH6 on classic signaling. IL-6 induced proliferation of Ba/F3–gp130–IL-6R cells in a dose-dependent manner, reaching maximal proliferation at 1 ng/ml of IL-6 (half-maximal proliferation at 0.2 ng/ml or 8.4 pm). VHH6 had, however, a small but significant inhibitory effect on classic signaling (Fig. 2E). Accordingly, VHH6 enhanced trans-signaling-induced STAT3 phosphorylation in Ba/F3–gp130 cells (Fig. 2F), but not the classic signaling-driven STAT3 phosphorylation in Ba/F3–gp130–IL-6R cells (Fig. 2G). Using ImageJ for quantification of Western blots from three independent experiments, our data indicate that the increase in pSTAT3 induced by VHH6 for IL-6 trans-signaling was significant, whereas VHH6 had no such effect on classic signaling (Fig. S2, A and B). Taken together, these data show that VHH6 stabilized IL-6·sIL-6R complexes and enhanced IL-6 trans-signaling but had no boosting effect on classic signaling.
Detection of IL-6·sIL-6R complexes in human serum using recombinant IL-6
After having quantified IL-6·sIL-6R complexes in a standard buffer system, our next goal was to analyze IL-6·sIL-6R complexes in human blood serum. Therefore, we drew blood from 10 healthy volunteers and initially quantified IL-6, sIL-6R, and sgp130 levels. Whereas IL-6 and, as a consequence, also IL-6·sIL-6R complexes were below the detection limit of the ELISAs (<2 pg/ml for IL-6), sIL-6R and sgp130 levels were in the range from 36 to 69 ng/ml (52.9 ± 16.3 ng/ml) and 205 to 330 ng/ml (243 ± 38.9 ng/ml), respectively (Fig. 3A). To quantify the amount of IL-6·sIL-6R complexes in these serum samples, we compared IL-6·sIL-6R standards prepared in R&D Reagent Diluent with standard prepared in human serum. Importantly, the measurement results of the standard curve prepared in serum were on average 8.3 times lower than those obtained with the R&D Reagent Diluent. This strongly suggested that serum interferes with ELISA detection of the IL-6·sIL-6R complex and that endogenous IL-6·sIL-6R complexes in serum would therefore be detected with strongly reduced sensitivity (Fig. 3B). In subsequent experiments, we therefore performed the IL-6·sIL-6R standard curve in R&D Reagent Diluent and included a correction factor (×8.3) for the calculation of IL-6·sIL-6R complexes measured in the serum samples. To determine how much IL-6·sIL-6R complex might be formed in human serum, we added recombinant IL-6 in concentrations ranging from 1 to 1,000 ng/ml to serum samples from 10 healthy volunteers and quantified IL-6·sIL-6R complexes by ELISA. As expected, increasing concentrations of IL-6 resulted in increasing amounts of IL-6·sIL-6R complexes (Fig. 3C). The percentage of sIL-6R present in IL-6·sIL-6R complexes was below 1% for IL-6 concentrations below 25 ng/ml and up to 15.24 ± 3.54% for 1,000 ng/ml IL-6 (Fig. 3D; Table 1, Table S2), which is in good agreement with our values obtained with recombinant proteins in standard buffer. Also the proportions of IL-6 or sIL-6R bound in IL-6·sIL-6R complexes were comparable between serum samples and buffer conditions (Table S2 and S3).
Figure 3.

Quantification of IL-6·sIL-6R complexes in human serum. A, serum level of IL-6, sIL-6R, sgp130, and IL-6·sIL-6R complexes from 10 human healthy volunteers were quantified by ELISA. Bars represent the mean (middle line) and the S.D. (upper and lower line). B, IL-6·IL-6R complex standard from R&D Systems was reconstituted either in the recommended diluent or in human serum. The calculation of the correction factor for IL-6·sIL-6R complex detection in serum (8.3 times) was performed with pooled data from three independent experiments. Error bars represent the S.D. C, recombinant human IL-6 (1–1,000 ng/ml) was titrated into serum samples from 10 human healthy volunteers, and the resulting IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R complex ELISA. Bars represent the mean (middle line) and the S.D. (upper and lower line). D, the percentages of sIL-6R in IL-6·sIL-6R complexes were calculated from C. Error bars represent the S.D. E, recombinant human IL-6 (0.1–1 ng/ml) and VHH6 (0 or 1 μg/ml) were added to serum samples from 3 human healthy volunteers, and the resulting IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R complex ELISA. Combined data were from three experiments. Error bars represent the S.D. F, recombinant human IL-6 (10 or 1,000 ng/ml), VHH6 (1 μg/ml), and endogenous sIL-6R (0.5, 1, 2.5, or 5 ng/ml) in the serum of three human healthy volunteers were mixed and IL-6·sIL-6R complex was quantified by IL-6·sIL-6R complex ELISA. Combined data from three experiments. Error bars represent the S.D.
Table 1.
Overview of IL-6·sIL-6R complex formation based on the Kd calculation (22 nm) or on ELISA measurements from Fig. 1B and 3C

Next, we investigated whether VHH6 also stabilized IL-6·sIL-6R complexes in serum samples. 1 μg/ml of VHH6 and 0.1 to 1 ng/ml of IL-6 were added to serum samples from 3 volunteers, and the resulting IL-6·sIL-6R complexes were quantified by ELISA. Whereas practically no complexes were detected in the absence of VHH6, addition of 1 μg/ml of VHH6 to 1 ng/ml of IL-6 in serum resulted in the formation of about 4.2 ng/ml of IL-6·sIL-6R complexes, demonstrating that virtually all IL-6 molecules were forced into IL-6·sIL-6R complexes by VHH6 (Fig. 3E, Table S7). This showed that VHH6 was also able to stabilize IL-6·sIL-6R complexes in serum samples.
To analyze how much of the endogenous sIL-6R is able to form complexes with recombinant IL-6, we diluted serum samples in PBS and adjusted the sIL-6R concentrations to 0.5, 1, 2.5, or 5 ng/ml. These diluted serum samples were then supplemented with 10 or 1,000 ng/ml of IL-6 in the presence of 1 μg/ml of VHH6. For 1,000 ng/ml of IL-6, ∼6.5 ng/ml of IL-6·sIL-6R complexes were detected in the presence of 5 ng/ml of endogenous sIL-6R, which suggests that at least 89% of the serum sIL-6R was able to bind to IL-6 (Fig. 3F). In Table S7, we compared IL-6·sIL-6R complexes formed with recombinant IL-6 and sIL-6R with those formed with recombinant IL-6 and endogenous sIL-6R in the presence and absence of VHH6.
Endogenous sgp130 might interfere with the ELISA detection of IL-6·sIL-6R complexes in serum samples. To analyze this cross-inhibition, we added saturating amounts of sgp130Fc to recombinant IL-6 and sIL-6R. Sgp130Fc is a dimerized fusion protein of sgp130 and the Fc part of an IgG1 antibody and confers 10-fold higher inhibitory capacity toward trans-signaling compared with natural monomeric sgp130. Importantly, only IL-6·sIL-6R complexes but not IL-6 and sIL-6R alone can bind to sgp130Fc (3). As depicted in Fig. 4A, 1 μg/ml of sgp130Fc completely prevented the detection of recombinant IL-6·sIL-6R complexes by ELISA for 1–1,000 ng/ml of IL-6 and 50 ng/ml of sIL-6R. Next, we used constant concentrations of 100 ng/ml of IL-6 and 50 ng/ml of sIL-6R and increasing amounts of sgp130Fc (5–10,000 ng/ml) to directly compare the inhibitory effect of sgp130Fc on cellular proliferation and on IL-6·sIL-6R detection by ELISA. We chose these IL-6 and sIL-6R concentrations because they were sufficient to provoke sustained proliferation of Ba/F3–gp130 cells (Fig. 4B). Proliferation of Ba/F3–gp130 cells driven by 100 ng/ml of IL-6 and 50 ng/ml of sIL-6R was inhibited by about 50% with 200 ng/ml of sgp130Fc (Fig. 4B). Interestingly, the detection of IL-6·sIL-6R complexes by ELISA was reduced by 50% by a similar concentration of sgp130Fc (100 ng/ml) (Fig. 4C), demonstrating that the cellular proliferation and ELISA data are in good agreement. Having shown that already 100 ng/ml of sgp130Fc was able to reduce IL-6·sIL-6R complex detection by 50%, we attempted to quantify the influence of endogenous sgp130 on IL-6·sIL-6R detection. To achieve this goal, we removed about 98% of endogenous sgp130 from the serum by immunoprecipitation (Fig. 4D). Sgp130-depleted and -containing serum was then supplemented with 50, 100, 200, or 1,000 ng/ml of IL-6, and IL-6·sIL-6R complexes were quantified. Whereas we did not observe a quantitative difference of IL-6·sIL-6R complex detection for 50 and 100 ng/ml of IL-6, higher concentrations of 200 and 1,000 ng/ml of IL-6 resulted in about 17 and 21% more IL-6·sIL-6R complexes detected after sgp130 depletion (Fig. 4E, Table S8). Considering that sgp130Fc is about 10-fold more potent than monomeric sgp130 (3), we predicted that 40 ng/ml of recombinant sgp130Fc would exhibit the same inhibitory capacity as 400 ng/ml of endogenous sgp130 on ELISA detection of IL-6·sIL-6R complexes. As shown in Fig. 4C, 40 ng/ml of sgp130Fc was indeed equivalent to 400 ng/ml of endogenous sgp130 because it inhibited IL-6·sIL-6R complex detection by about 26%. Our data confirms that endogenous sgp130 is able to bind to IL-6·sIL-6R complexes, whereas the amount of endogenous sgp130 in the serum is most likely only able to trap a smaller fraction of IL-6·sIL-6R in IL-6·sIL-6R·sgp130 complexes and therefore does not neutralize most of the endogenous trans-signaling complexes as previously expected.
Figure 4.

Influence of sgp130Fc on the ELISA quantification of IL-6·sIL-6R complexes in human serum. A, recombinant human IL-6 (1–1,000 ng/ml), sIL-6R (50 ng/ml), and sgp130Fc (0 or 1 μg/ml) were mixed and the resulting IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R complex ELISA. One representative experiment of three is shown. Error bars represent the S.D. B, cellular proliferation of Ba/F3-gp130 cells. Equal numbers of cells were cultured for 3 days in the presence of HIL-6 (10 ng/ml) or IL-6 (100 ng/ml) plus sIL-6R (50 ng/ml) and increasing concentrations of sgp130Fc (0.005–10 μg/ml). Proliferation was measured using the colorimetric CellTiter-Blue Cell Viability Assay. One representative experiment of three is shown. Error bars represent the S.D. C, recombinant human IL-6 (100 ng/ml), sIL-6R (50 ng/ml), and sgp130Fc (5–10,000 ng/ml) were mixed and the resulting IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R complex ELISA. Combined data from three experiments. Error bars represent the S.D. D, endogenous sgp130 was removed from the serum of one healthy volunteer by immunoprecipitation with an anti-sgp130 antibody and Protein A-Sepharose. Endogenous sgp130 in the serum before and after precipitation was quantified by sgp130 ELISA. E, recombinant human IL-6 (50–1,000 ng/ml) was added to the endogenous sIL-6R in natural and sgp130-depleted serum of one human healthy volunteer and the resulting IL-6·sIL-6R complexes were quantified by IL-6·sIL-6R complex ELISA. Combined data were from three experiments. Error bars represent the S.D.
In summary, the amount of experimentally determined recombinant and endogenous sIL-6R bound in IL-6·sIL-6R complexes was in general 3–10-fold lower than the calculated theoretical values based on the Kd of 22 nm (Table 1) experimentally determined using the same recombinant cytokines. This was reflected in a ∼3 times higher dissociation Kd value (70.63 ± 11 nm) as obtained from the ELISA data calculated from supporting Table 2 using the IL-6 values within the linear range (10, 25, 50, and 100 ng/ml).
Development of a strategy to determine the amount of IL-6·sIL-6R·sgp130 complexes
Because sgp130 and sgp130Fc inhibited IL-6·sIL-6R detection by ELISA, we developed an alternative strategy to determine IL-6·sIL-6R·sgp130 complexes based on Protein A-Sepharose-mediated precipitation of IL-6·sIL-6R·sgp130Fc complexes followed by quantification of the remaining sIL-6R (present as free sIL-6R and in IL-6·sIL-6R complexes) in the supernatant by ELISA. This procedure allows the calculation of the amounts of sIL-6R trapped in IL-6·sIL-6R·sgp130Fc complexes (Fig. 5A). We used constant amounts of recombinant sIL-6R (50 ng/ml, 0.97 nm) and sgp130Fc (10 μg/ml, 38.5 nm, molecular mass of ∼260 kDa) plus increasing concentrations of IL-6 (1–200 ng/ml, 0.0422–8.43 nm). First, we verified that sIL-6R did not bind to sgp130Fc in the absence of IL-6 by performing ELISA quantification of sIL-6R after sgp130Fc precipitation (Fig. 5B). After addition of increasing amounts of IL-6 with subsequent precipitation of sgp130Fc, reduced concentrations of sIL-6R were detected in the supernatant by ELISA. Precipitation of about 90% of the sIL-6R was achieved with saturating concentrations of IL-6 (starting with 50 ng/ml, 2.1 nm, 2-fold molar excess over sIL-6R) and sgp130Fc. Interestingly, a 1:1 molar ratio of IL-6 to sIL-6R (25 ng/ml of IL-6 = 1.05 nm; 50 ng/ml of sIL-6R = 0.97 nm) resulted in precipitation of ∼60% of the sIL-6R molecules (Fig. 5, B and C, Table 2). Moreover, in the absence of sgp130Fc and when using the same concentrations of IL-6 and sIL-6R (both 50 ng/ml) only 3.24 ± 0.59% sIL-6R proteins were detected in IL-6·sIL-6R complexes (Table 1, Table S2).
Figure 5.

Quantification of IL-6·sIL-6R·sgp130Fc complexes. A, schematic illustration of the precipitation of IL-6·sIL-6R·sgp130Fc by Protein A-Sepharose beads and subsequent analysis of the remaining sIL-6R in the supernatant. B, recombinant human IL-6 (1–200 ng/ml), sIL-6R (50 ng/ml), and sgp130Fc (10 μg/ml) were mixed. The resulting IL-6·sIL-6R·sgp130Fc complexes were precipitated with Protein A-Sepharose and sIL-6R in the supernatant was quantified by ELISA. Combined data were from five experiments. Error bars represent the S.D. C, the percentages of sIL-6R in IL-6·sIL-6R·sgp130Fc complexes were calculated from B. Error bars represent the S.D. D, recombinant human IL-6 (10–1,000 ng/ml), sIL-6R (50 ng/ml), and sgp130Fc (0.04–10 μg/ml) were mixed. The resulting IL-6·sIL-6R·sgp130Fc complexes were precipitated with Protein A-Sepharose and sIL-6R was quantified in the supernatant by ELISA. Mean of combined data were from three experiments. Error bars represent the S.D. E, recombinant human IL-6 (1–1,000 ng/ml), endogenous (serum) sIL-6R, and sgp130Fc (10 μg/ml) were mixed. The resulting IL-6·sIL-6R·sgp130Fc complexes were precipitated with Protein A-Sepharose and sIL-6R was quantified in the supernatant by ELISA. Serum of 10 human healthy volunteers was used. Mean of combined data were from 10 measurements. Error bars represent the S.D. F, the percentages of endogenous (serum) sIL-6R in IL-6·sIL-6R·sgp130Fc complexes were calculated from E. Error bars represent the S.D. G, recombinant human IL-6 (1–1,000 ng/ml), endogenous (serum) sIL-6R, sgp130Fc (10 μg/ml), and VHH6 (0 or 1 μg/ml) were mixed. IL-6·sIL-6R·sgp130Fc complexes were precipitated with Protein A-Sepharose and sIL-6R was quantified in the supernatant by ELISA. Mean of combined data were from three experiments. Error bars represent the S.D. H, recombinant human IL-6 (1 or 2 μg/ml), endogenous (serum) sIL-6R and sgp130Fc (10 or 20 μg/ml) were mixed. The resulting IL-6·sIL-6R·sgp130Fc complexes were precipitated with Protein A-Sepharose and sIL-6R was quantified in the supernatant by ELISA. The precipitation and sIL-6R ELISA quantification was repeated after a second addition of IL-6 (2,000 ng/ml) and sgp130Fc (20 μg/ml). Mean of combined data were from three experiments. Error bars represent the S.D.
Table 2.
Quantification of IL-6·sIL-6R·sgp130 complex formation from Fig. 5B and E
Dashes (-) indicate this concentration has not been assayed.
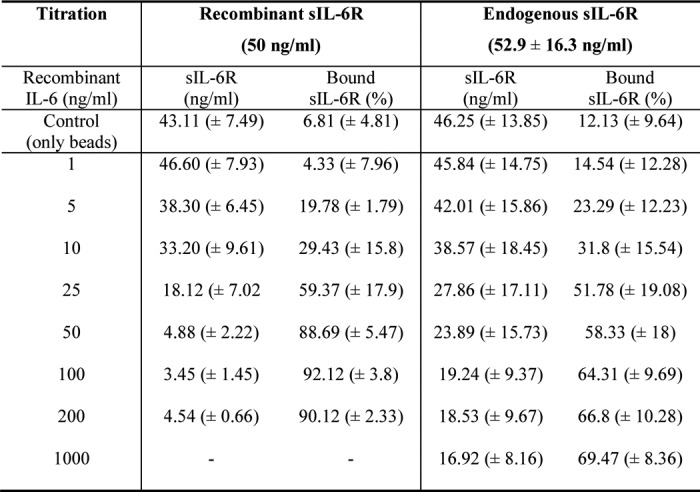
Next, we analyzed how much IL-6·sIL-6R·sgp130 complexes were formed with 40 or 400 ng/ml of sgp130Fc using precipitation of sgp130Fc via Protein A-Sepharose and subsequent ELISA detection of sIL-6R. As depicted in Fig. 5D, 400 ng/ml of sgp130Fc was as efficient as 10 μg/ml of sgp130Fc in precipitating almost all sIL-6R molecules in IL-6·sIL-6R·sgp130 complexes. In line with the data shown in Fig. 4E, 40 ng/ml of sgp130Fc was only able to precipitate about 30% of IL-6·sIL-6R in IL-6·sIL-6R·sgp130 complexes, again suggesting that the level of endogenous sgp130 cannot completely buffer trans-signaling.
Finally, we used serum samples of the same 10 healthy volunteers as described above to analyze IL-6·sIL-6R·sgp130Fc complex formation with the endogenous sIL-6R. We added 10 μg/ml of sgp130Fc and increasing amounts of IL-6 (1–1,000 ng/ml) to the serum samples, precipitated IL-6·sIL-6R· sgp130Fc complexes, and determined the amount of remaining sIL-6R in the supernatant. The highest amounts of IL-6·sIL-6R·sgp130Fc complexes in serum were observed with 200 ng/ml or more of IL-6. Surprisingly, a maximum of 69% of sIL-6R in the serum was forced into IL-6·sIL-6R·sgp130Fc complexes (Fig. 5, E and F, Table 2). Moreover, also the addition of 1 μg/ml of VHH6 was not able to force more than 69% of sIL-6R into IL-6·sIL-6·sgp130Fc complexes (1000 ng/ml of IL-6, 10 μg/ml of sgp130Fc) (Fig. 5G). Our results suggested that at least 30% of the serum sIL-6R was not able to bind to sgp130Fc, even though we have shown that ∼89% of the endogenous sIL-6R was able to form complexes with IL-6 (Fig. 3F). To exclude that IL-6 and/or sgp130Fc were not supplied in sufficient amounts, we increased the amounts of IL-6 (up to a ∼260-fold molar excess of IL-6 over endogenous sIL-6R) and sgp130Fc to precipitate IL-6·sIL-6R·sgp130Fc complexes from serum of three volunteers, which had, however, only a modest effect and increased the precipitation of sIL-6R from 58% (using 1,000 ng/ml IL-6 (65-fold molar excess) and 10 μg/ml of sgp130Fc) to 61% (using 2,000 ng/ml IL-6 (130-fold molar excess) and 20 μg/ml of sgp130Fc). We even used the same serum from which 61% of the sIL-6R was precipitated in the first round and performed a second round of precipitation after another addition of 2,000 ng/ml of IL-6 (∼260 molar excess of IL-6 over endogenous sIL-6R) and 20 μg/ml of sgp130Fc. After the second precipitation, 32% of the initial amount of sIL-6R still remained in the supernatant (Fig. 5H). Our results hence suggest that the endogenous sIL-6R is not completely accessible for binding to sgp130Fc. Taken together, we have developed a robust method to experimentally determine the amount of IL-6·sIL-6R·sgp130 complexes.
Discussion
The rather high endogenous serum levels of sIL-6R and sgp130 are usually considered as a systemic buffer system that can either promote IL-6 trans-signaling via formation of IL-6·sIL-6R complexes or inhibit trans-signaling via IL-6·sIL-6R·sgp130 complexes (7). It is currently unknown how much of these complexes are actually present in vivo. Initial calculations of the proportion of IL-6·sIL-6R complexes were based on the previously published binding affinity (Kd) of 0.5 nm between IL-6 and sIL-6R. They predicted that for low to medium levels of IL-6 (0.001–10 ng/ml), which are characteristic for health (<10 pg/ml) or acute and chronic inflammations, most IL-6 should be trapped in IL-6·sIL-6R complexes. In combination with published normal levels of 50 ng/ml of sIL-6R in serum, levels of 0.1 ng/ml of IL-6 should result in 66% of total IL-6 bound in IL-6·sIL-6R complexes, 1 ng/ml of IL-6 in 65% complexed IL-6 and 10 ng/ml of IL-6 in 59% complexed IL-6 (12). Controversially, our and another recent study were not able to reproduce these low affinities and documented Kd values were in the 14 to 34 nm range (15). Importantly, the excellent biological activity of our recombinant IL-6 resulted in strong stimulation of Ba/F3–gp130–IL-6R cells at very low doses with a half-maximal activation at 8.4 pm IL-6, which is in good agreement with the Kd of about 10 pm for affinity of the IL-6·IL-6R complexes toward gp130 (17). Moreover, if the high ratios of IL-6·sIL-6R complexes for low IL-6 concentrations based on the Kd of 0.5–0.8 nm (12, 16) were true, there would be no plausible explanation why the combination of free IL-6 and sIL-6R is 100–1000-fold less potent to stimulate Ba/F3–gp130 cell proliferation and STAT3 phosphorylation than the fusion protein Hyper-IL-6 (18). Our data presented here challenges the predictions based on the initially described Kd values for IL-6 and sIL-6R and shows that at concentrations of 10 ng/ml of IL-6 and 50 ng/ml of sIL-6R, only ∼1% IL-6 is in complex with sIL-6R. Our data would further explain the low biological activity of IL-6·sIL-6R mixtures on receiver cells, as previously determined (18). Importantly, the low IL-6·sIL-6R complex formation was not only found for recombinant sIL-6R but also for natural sIL-6R in human serum. This shows that the differences between calculated and experimentally determined IL-6·sIL-6R complexes cannot be simply explained by, e.g. a major fraction of inactive sIL-6R in our recombinant protein preparations or by other structural differences between recombinant and natural sIL-6R, such as glycosylation patterns or the exact composition of the C terminus (usually tagged in recombinant sIL-6R, but untagged and heterogeneous due to generation by shedding or alternative splicing in natural sIL-6R). Moreover, the low amounts of IL-6·sIL-6R complexes detected by ELISA were not caused by interfering binding of endogenous sgp130, because we showed experimentally that endogenous sgp130 had only little effect on IL-6·sIL-6R complex detection.
By using VHH6, a single domain antibody that has been shown to promote formation of the IL-6·sIL-6R complex (15), we showed that most IL-6 and sIL-6R proteins were indeed able to form heterodimeric complexes. VHH6 stabilizes the IL-6·sIL-6R in a way so that the dissociation of the complex is slowed (15) and shifts the complex formation from its normal equilibrium toward IL-6·sIL-6R complexes. Biological activity of these VHH6-stabilized IL-6·sIL-6R complexes was shown by ELISA and by a major enhancement of trans-signaling in terms of proliferation and STAT3 phosphorylation of Ba/F3-gp130 cells. Interestingly, VHH6 did not enhance classic signaling.
A molar excess of recombinant sgp130Fc efficiently inhibits IL-6 trans-signaling and also classic signaling, albeit only if the molar sIL-6R level exceeds that of IL-6 (3, 8). The mechanism of action of sgp130, including its influence on classic signaling, might have direct implications for therapeutic application of sgp130Fc in IL-6-dependent diseases with documented molar excesses of sIL-6R over IL-6 (12, 26). Using sgp130Fc to force IL-6·sIL-6R complexes in ternary IL-6·sIL-6R·sgp130 complexes, followed by precipitation and quantification of the remaining sIL-6R in the supernatant, we demonstrated efficient IL-6·sIL-6R·sgp130Fc complex formation by recombinant proteins and precipitation of up to 90% of the sIL-6R. For unknown reasons, only about 70% of the endogenous sIL-6R in human serum could be forced into IL-6·sIL-6R·sgp130Fc complexes, even though the binding of the endogenous sIL-6R to IL-6 was not disturbed.
As mentioned above, sIL-6R and sgp130 are traditionally considered as a systemic buffer system either promoting IL-6 trans-signaling via IL-6·sIL-6R complexes or inhibiting trans-signaling via IL-6·sIL-6R·sgp130 complexes (7). Alternatively, these proteins could also form transient complexes that associate and dissociate rapidly such as to increase the half-life of IL-6 and enabling both classic and trans-signaling. Our data suggest that endogenous sIL-6R and sgp130 only form transient complexes with IL-6. Consequently, systemic trans-signaling is considered of minor importance due to the low amounts of circulating IL-6·sIL-6R complexes. The same is true for the consideration of endogenous sgp130 as a natural inhibitor of trans-signaling. Our data clearly indicate that the systemic levels of endogenous sgp130 are by far not sufficient to inhibit trans-signaling. However, it is important to keep in mind that all of our calculations and experiments were based on sIL-6R and sgp130 levels present in systemic circulation. It is currently unknown if these concentrations are higher at local sites of inflammation, where massive production of sIL-6R by protease-mediated shedding takes places (27) and where also sgp130 may be present in much higher concentrations due to shedding (28). Especially for paracrine and autocrine trans-signaling, even small amounts of IL-6·sIL-6R complexes might be sufficient to stimulate gp130 on adjacent cells.
In summary, IL-6·sIL-6R and IL-6·sIL-6R·sgp130 complexes were formed with much lower frequency than previously anticipated, which leaves more space for classic rather than for trans-signaling even under conditions where sIL-6R is in a high molar excess over IL-6. Moreover, our results strongly support the therapeutic use of efficacy-enhanced trans-signaling inhibitors like the sgp130Fc derivative olamkicept and mutants thereof (29) in chronic inflammatory diseases, as only such compounds with much higher binding affinity and inhibitory capacity will be able to actually block pathological trans-signaling. However, even olamkicept leaves physiological trans-signaling intact (7).
Experimental procedures
Cells and reagents
Ba/F3 cells (ACC-300) were purchased from the Leibniz Institute DSMZ/German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and were grown in Dulbecco's modified Eagle's medium high glucose culture medium (GIBCO®, Life Technologies, Darmstadt, Germany) supplemented with 10% fetal bovine serum (GIBCO®, Life Technologies), 60 mg/liter of penicillin and 100 mg/liter of streptomycin (Genaxxon Bioscience GmbH, Ulm, Germany) at 37 °C with 5% CO2. Generation of Ba/F3–gp130 and Ba/F3–gp130–IL-6R cells was described previously (30). Ba/F3–gp130 cells were maintained in the presence of 10 ng/ml of human Hyper-IL-6, a fusion protein of IL-6 and soluble IL-6R, which mimics IL-6 trans-signaling (18). Ba/F3–gp130–IL-6R cells were cultured with 10 ng/ml of human IL-6. Hyper-IL-6, human IL-6, sIL-6R, and sgp130Fc were expressed and purified as described previously (3, 18, 31). Quality control assays for these recombinant proteins included silver-stained native and SDS-polyacrylamide gels, binding ELISAs with immobilized sgp130Fc and Ba/F3–gp130 cell proliferation assays according to the manufacturer's standard procedures (CONARIS Research Institute AG, Kiel, Germany). VHH6 was expressed and purified as described previously (15). Phospho-STAT3 (Tyr-705) (D3A7) and STAT3 (124H6) antibodies were obtained from Cell Signaling Technology (Frankfurt, Germany). The α-human-gp130 (BR-3) antibody was obtained from Abcam (Cambridge, UK). The anti-hIL-6R mAb 4–11 was produced as described previously (30). Biotinylated human IL-6R detection antibody (BAF227) was obtained from R&D Systems. Peroxidase-conjugated secondary mAbs (numbers 31432 and 31462) were obtained from Pierce (Thermo Scientific, St. Leon-Rot, Germany). Human IL-6·sIL-6R Complex DuoSet ELISA (DY8139-05) and human sgp130 DuoSet ELISA (DY228) were obtained from R&D Systems. Human IL-6 ELISA Kit (KHC0061) was obtained from Invitrogen.
Calculation of IL-6·sIL-6R and IL-6·sIL-6R·sgp130(Fc) complexes
To calculate the amount of IL-6·sIL-6R in blood serum, the IL-6·sIL-6R complex standard (R&D Systems) was diluted in R&D Reagent Diluent and human serum. The measurements showed that the standard curve in serum is lower by a factor of ∼8.3 compared with the curve in Reagent Diluent. Therefore, we decided to multiply the serum measurement values of IL-6·sIL-6R complexes by 8.3. The amount of bound sIL-6R in the IL-6·sIL-6R complex was calculated based on the molecular masses of IL-6 and sIL-6R (IL-6, 23.7 kDa; sIL-6R, 51.5 kDa; IL-6·sIL-6R complex, 75.2 kDa). For example, if a concentration of 5 ng/ml of IL-6·sIL-6R complex resulted from the combination of 100 ng/ml of IL-6 and 50 ng/ml of sIL-6R (molecular ratio: ∼1/3 IL-6 (31.5%) and ∼2/3 sIL-6R (68.5%) of the complex), 5 ng/ml × 0.685 = 3.425 ng/ml of sIL-6R were contained in the complex, corresponding to 3.425 ng/ml per 50 ng/ml × 100 = 6.85% of the total sIL-6R. The percentages of bound IL-6 were calculated in an analogous fashion.
Ethical approval for IL-6·sIL-6R analyses using serum from healthy human volunteers
Ethical approval for this study was obtained from the institutional review board of the Heinrich-Heine-University (study number 5829R). All participants gave prior written informed consent. Peripheral blood from healthy volunteers was collected by venipuncture. Serum was prepared using Serum/CAT Blood Collection Tubes (BD Biosciences, number 367819) according to the manufacturer's instructions. All human study procedures were performed in accordance with the principles outlined by the Declaration of Helsinki.
Enzyme-linked immunosorbent assay
The ELISA for human IL-6R was described previously (30). Accordingly, microtiter plates were coated with mouse anti-human IL-6R mAb 4–11 (diluted to 1 μg/ml in PBS) overnight at room temperature. After blocking with 5% sucrose and 1% BSA in PBS, samples were added. The biotinylated goat anti-human IL-6R antibody BAF227 (R&D Systems) was used as detection antibody, followed by incubation with streptavidin-horseradish peroxidase (R&D Systems). The enzymatic reaction was performed using the peroxidase substrate BM blue POD (Roche Applied Science, Mannheim, Germany) and the absorbance was read at 450 nm on a Tecan infinite M200 PRO reader (Tecan, Maennedorf, Switzerland). ELISAs for human IL-6·sIL-6R complex and sgp130 as well as IL-6 (Invitrogen) were performed according to the manufacturer's instructions.
Proliferation assays
Ba/F3–gp130 and Ba/F3–gp130–IL-6R cells were washed and 5,000 cells of each cell line were cultured for 3 days in a final volume of 100 μl with or without cytokines and/or inhibitors. Proliferation of the Ba/F3 cell lines was determined 72 h after cytokine stimulation using the CellTiter-Blue® Cell Viability Assay (Promega, Karlsruhe, Germany). The CellTiter-Blue® Reagent was used to estimate the number of viable cells by recording the fluorescence (excitation 560 nm, emission 590 nm) using the Infinite M200 PRO plate reader (Tecan) immediately after adding 20 μl of reagent per well (time point 0) and 60 min after incubation. The fluorescent signal from the CellTiter-Blue® reagent is proportional to the number of viable cells. All of the values were measured in triplicates per experiment. Fluorescence values were normalized by subtraction of the values obtained at time point 0. All experiments were performed at least three times, and one representative experiment was selected.
SPR measurements
To measure the kinetics of interaction between IL-6 and IL-6R, we applied the same conditions as recently reported (15) by immobilizing IL-6 on a CM5 chip of a BIAcore ×100 Plus instrument (GE Healthcare, Freiburg, Germany). The experiment was carried out at 25 °C in HBS-P+ buffer, containing 10 mm HEPES, pH 7.4, 0.15 m NaCl, 0.05% (v/v) surfactant P20 (GE Healthcare). A single cycle model was used to determine the kinetic parameters for the IL6–IL6R interaction. After immobilization of IL-6 on the CM5 chip, IL-6R was injected at a flow rate of 30 μl/min at increasing concentrations (0–120 nm). Association of IL-6 in each defined concentration was monitored in periods of 180 s and the global dissociation was measured at the end of the final injection in periods of 360 s. The data were fitted using 1:1 binding model.
Western blotting
Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were blocked and probed with the indicated primary antibodies (α-STAT3). After washing, membranes were incubated with streptavidin-HRP or secondary peroxidase-conjugated antibodies, respectively. The ImmobilonTM Western Reagents (Millipore Corporation, Billerica, MA) and the ChemoCam Imager (INTAS Science Imaging Instruments GmbH, Göttingen, Germany) were used for signal detection. Control STAT3 blots (loading control) were produced with the same samples on separate gels.
Immunoprecipitation of sgp130Fc
The sIL-6R (recombinant or endogenous) was incubated for 1 h at room temperature with IL-6, then 10 μg/ml of sgp130Fc was added under constant agitation. 50 μl of Protein A-Sepharose beads (Roche Holding GmbH, Mannheim, Germany) per milliliter was added to the samples, followed by 1 h incubation under constant agitation at room temperature. Afterward, the samples were centrifuged (5 min, 300 × g, room temperature) and the supernatant was harvested. Subsequently, the amount of free sIL-6R remaining in the supernatant was determined by sIL-6R ELISA.
Immunoprecipitation of endogenous sgp130 from blood serum
For the isolation of endogenous sgp130 from human blood serum, 50 μl of Protein A-Sepharose beads were incubated with 0.5 μg of α-human-gp130 (BR-3) antibody (Abcam, Cambridge, UK) overnight at 4 °C in 1 ml of PBS. Unbound antibody was removed by centrifugation (5 min, 300 × g, 4 °C). 50 μl of α-human–CD130-coupled Protein A-Sepharose beads were used per milliliter of serum. The samples were incubated for 4 h at 4 °C under gentle agitation. Afterward the samples were centrifuged (5 min, 300 × g, 4 °C) and the serum was isolated from the Protein A-Sepharose beads. Subsequently, the remaining amount of sgp130 and sIL-6R was determined by ELISA.
Programs and statistical analysis
Western blots were analyzed by ImageJ (rsb.info.nih.gov/ij/). Data are expressed as mean ± S.D. calculated from at least three independent experiments unless otherwise stated. Statistical analysis was performed using a two-way analysis of variance test followed by a Bonferroni test, using GraphPad Prism software (GraphPad, La Jolla, CA). p values are indicated as within the figures: p ≤ 0.05 = *; p ≤ 0.01 = **; p ≤ 0.001 = ***; and p ≤ 0.0001 = ****.
Author contributions
P. B., S. H., G. H. W., M. A., L. L., M. R. A., and J. M. M. investigation; P. B. and S. H. methodology; H. J. H. and J. S. formal analysis; J. S. conceptualization; J. S. resources; J. S. data curation; J. S. software; J. S. supervision; J. S. funding acquisition; J. S. validation; J. S. visualization; J. S. writing-original draft; J. S. project administration; J. S. writing-review and editing.
Supplementary Material
This work was supported by Deutsche Forschungsgemeinschaft Project Grant AH 92/8-1 (to A. K. and M. R. A.), and the Bundesministerium für Bildung und Forschung (BMBF) project InTraSig (to J. S.). G. H. W. is employed by CONARIS Research Institute AG (Kiel, Germany), which is commercially developing sgp130Fc proteins as therapeutics for inflammatory diseases.
This article contains Figs. S1 and S2 and Tables S1–S8.
- IL
- interleukin
- HIL-6
- Hyper-IL-6
- gp130
- glycoprotein 130
- IL-6R
- interleukin 6 receptor
- STAT
- signal transducers and activators of transcription
- sgp130
- soluble gp130.
References
- 1. Scheller J., Chalaris A., Schmidt-Arras D., and Rose-John S. (2011) The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 1813, 878–888 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- 2. Garbers C., Hermanns H. M., Schaper F., Müller-Newen G., Grötzinger J., Rose-John S., and Scheller J. (2012) Plasticity and cross-talk of Interleukin 6-type cytokines. Cytokine Growth Factor Rev. 23, 85–97 10.1016/j.cytogfr.2012.04.001 [DOI] [PubMed] [Google Scholar]
- 3. Jostock T., Mullberg J., Ozbek S., Atreya R., Blinn G., Voltz N., Fischer M., Neurath M. F., and Rose-John S. (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167 10.1046/j.1432-1327.2001.01867.x [DOI] [PubMed] [Google Scholar]
- 4. Horiuchi S., Koyanagi Y., Zhou Y., Miyamoto H., Tanaka Y., Waki M., Matsumoto A., Yamamoto M., and Yamamoto N. (1994) Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur. J. Immunol. 24, 1945–1948 10.1002/eji.1830240837 [DOI] [PubMed] [Google Scholar]
- 5. Matthews V., Schuster B., Schütze S., Bussmeyer I., Ludwig A., Hundhausen C., Sadowski T., Saftig P., Hartmann D., Kallen K.-J., and Rose-John S. (2003) Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J. Biol. Chem. 278, 38829–38839 10.1074/jbc.M210584200 [DOI] [PubMed] [Google Scholar]
- 6. Müllberg J., Schooltink H., Stoyan T., Günther M., Graeve L., Buse G., Mackiewicz A., Heinrich P. C., and Rose-John S. (1993) The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 23, 473–480 10.1002/eji.1830230226 [DOI] [PubMed] [Google Scholar]
- 7. Rose-John S. (2017) The soluble Interleukin 6 receptor: Advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 102, 591–598 10.1002/cpt.782 [DOI] [PubMed] [Google Scholar]
- 8. Garbers C., Thaiss W., Jones G. W., Waetzig G. H., Lorenzen I., Guilhot F., Lissilaa R., Ferlin W. G., Grötzinger J., Jones S. A., Rose-John S., and Scheller J. (2011) Inhibition of classic signaling is a novel function of soluble GP130 which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 286, 42959–42970 10.1074/jbc.M111.295758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akira S., Taga T., and Kishimoto T. (1993) Interleukin-6 in biology and medicine. Adv. Immunol. 54, 1–78 10.1016/S0065-2776(08)60532-5 [DOI] [PubMed] [Google Scholar]
- 10. Kishimoto T., Akira S., Narazaki M., and Taga T. (1995) Interleukin-6 family of cytokines and gp130. Blood 86, 1243–1254 [PubMed] [Google Scholar]
- 11. Jones S. A. (2005) Directing transition from innate to acquired immunity: defining a role for IL-6. J. Immunol. 175, 3464–3468 [DOI] [PubMed] [Google Scholar]
- 12. Gaillard J., Pugnière M., Tresca J., Mani J., Klein B., and Brochier J. (1999) Interleukin-6 receptor signaling: II. bio-availability of interleukin-6 in serum. Eur. Cytokine Netw. 10, 337–344 [PubMed] [Google Scholar]
- 13. Mitsuyama K., Toyonaga A., Sasaki E., Ishida O., Ikeda H., Tsuruta O., Harada K., Tateishi H., Nishiyama T., and Tanikawa K. (1995) Soluble interleukin-6 receptors in inflammatory bowel disease: relation to circulating interleukin-6. Gut 36, 45–49 10.1136/gut.36.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montero-Julian F. A. (2001) The soluble IL-6 receptors: serum levels and biological function. Cell. Mol. Biol. 47, 583–597 [PubMed] [Google Scholar]
- 15. Adams R., Burnley R., Valenzano C., Qureshi O., Doyle C., Lumb S., Del Carmen Lopez M., Griffin R., McMillan D., Taylor R., Meier C., Mori P., Griffin L., Wernery U., et al. (2017) Discovery of a junctional epitope antibody that stabilizes IL-6 and gp80 protein:protein interaction and modulates its downstream signaling. Sci. Rep. 7, 37716 10.1038/srep37716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zohlnhöfer D., Graeve L., Rose-John S., Schooltink H., Dittrich E., and Heinrich P. (1992) The hepatic interleukin-6 receptor: down-regulation of the interleukin-6 binding subunit (gp80) by its ligand. FEBS Lett. 306, 219–222 10.1016/0014-5793(92)81004-6 [DOI] [PubMed] [Google Scholar]
- 17. Hibi M., Murakami M., Saito M., Hirano T., Taga T., and Kishimoto T. (1990) Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63, 1149–1157 10.1016/0092-8674(90)90411-7 [DOI] [PubMed] [Google Scholar]
- 18. Fischer M., Goldschmitt J., Peschel C., Kallen K. J., Brakenhoff J. P. J., Wollmer A., Groetzinger J., and Rose-John S. (1997) A designer cytokine with high activity on human hematopoietic progenitor cells. Nature Biotech. 15, 142–145 10.1038/nbt0297-142 [DOI] [PubMed] [Google Scholar]
- 19. Crabé S., Guay-Giroux A., Tormo A. J., Duluc D., Lissilaa R., Guilhot F., Mavoungou-Bigouagou U., Lefouili F., Cognet I., Ferlin W., Elson G., Jeannin P., and Gauchat J. F. (2009) The IL-27 p28 subunit binds cytokine-like factor 1 to form a cytokine regulating NK and T cell activities requiring IL-6R for signaling. J. Immunol. 183, 7692–7702 10.4049/jimmunol.0901464 [DOI] [PubMed] [Google Scholar]
- 20. Kallen K. J., Grötzinger J., Lelièvre E., Vollmer P., Aasland D., Renné C., Müllberg J., Myer zum Büschenfelde K. H., Gascan H., and Rose-John S. (1999) Receptor recognition sites of cytokines are organized as exchangeable modules. Transfer of the leukemia inhibitory factor receptor-binding site from ciliary neurotrophic factor to interleukin-6. J. Biol. Chem. 274, 11859–11867 10.1074/jbc.274.17.11859 [DOI] [PubMed] [Google Scholar]
- 21. Friederichs K., Schmitz J., Weissenbach M., Heinrich P. C., and Schaper F. (2001) Interleukin-6-induced proliferation of pre-B cells mediated by receptor complexes lacking the SHP2/SOCS3 recruitment sites revisited. Eur. J. Biochem. 268, 6401–6407 10.1046/j.0014-2956.2001.02586.x [DOI] [PubMed] [Google Scholar]
- 22. Bataille R., Jourdan M., Zhang X. G., and Klein B. (1989) Serum levels of interleukin 6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J. Clin. Invest. 84, 2008–2011 10.1172/JCI114392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frieling J. T., van Deuren M., Wijdenes J., van der Meer J. W., Clement C., van der Linden C. J., and Sauerwein R. W. (1995) Circulating interleukin-6 receptor in patients with sepsis syndrome. J. Infect. Dis. 171, 469–472 10.1093/infdis/171.2.469 [DOI] [PubMed] [Google Scholar]
- 24. Nechemia-Arbely Y., Barkan D., Pizov G., Shriki A., Rose-John S., Galun E., and Axelrod J. (2008) IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 19, 1106–1115 10.1681/ASN.2007070744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowell M. A., Richards P. J., Horiuchi S., Yamamoto N., Rose-John S., Topley N., Williams A. S., and Jones S. A. (2003) Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J. Immunol. 171, 3202–3209 10.4049/jimmunol.171.6.3202 [DOI] [PubMed] [Google Scholar]
- 26. Rabe B., Chalaris A., May U., Waetzig G. H., Seegert D., Williams A. S., Jones S. A., Rose-John S., and Scheller J. (2008) Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 111, 1021–1028 [DOI] [PubMed] [Google Scholar]
- 27. Scheller J., Chalaris A., Garbers C., and Rose-John S. (2011) ADAM17: a molecular switch controlling inflammatory and regenerative responses. Trends Immunol. 32, 380–387 10.1016/j.it.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 28. Wolf J., Waetzig G., Chalaris A., Reinheimer T., Wege H., Rose-John S., and Garbers C. (2016) Different soluble forms of the interleukin-6 family signal transducer gp130 fine-tune the blockade of interleukin-6 trans-signaling. J. Biol. Chem. 291, 16186–16196 10.1074/jbc.M116.718551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tenhumberg S., Waetzig G. H., Chalaris A., Rabe B., Seegert D., Scheller J., Rose-John S., and Grötzinger J. (2008) Structure guided optimization of the interleukin-6 transsignaling antagonist sgp130. J. Biol. Chem. 283, 27200–27207 10.1074/jbc.M803694200 [DOI] [PubMed] [Google Scholar]
- 30. Chalaris A., Rabe B., Paliga K., Lange H., Laskay T., Fielding C. A., Jones S. A., Rose-John S., and Scheller J. (2007) Apoptosis is a natural stimulus of IL6R shedding and contributes to the pro-inflammatory trans-signaling function of neutrophils. Blood 110, 1748–1755 10.1182/blood-2007-01-067918 [DOI] [PubMed] [Google Scholar]
- 31. Mackiewicz A., Rose-John S., Schooltink H., Laciak M., Górny A., and Heinrich P. C. (1992) Soluble human interleukin-6-receptor modulates interleukin-6-dependent N-glycosylation of α1-protease inhibitor secreted by HepG2 cells. FEBS Lett. 306, 257–261 10.1016/0014-5793(92)81012-B [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.