

Abstract
Several oxime-containing small molecules have useful properties, including antimicrobial, insecticidal, anticancer, and immunosuppressive activities. Phosphonocystoximate and its hydroxylated congener, hydroxyphosphonocystoximate, are recently discovered oxime-containing natural products produced by Streptomyces sp. NRRL S-481 and Streptomyces regensis NRRL WC-3744, respectively. The biosynthetic pathways for these two compounds are proposed to diverge at an early step in which 2-aminoethylphosphonate (2AEPn) is converted to (S)-1-hydroxy-2-aminoethylphosphonate ((S)-1H2AEPn) in S. regensis but not in Streptomyces sp. NRRL S-481). Subsequent installation of the oxime moiety into either 2AEPn or (S)-1H2AEPn is predicted to be catalyzed by PcxL or HpxL from Streptomyces sp. NRRL S-481 and S. regensis NRRL WC-3744, respectively, whose sequence and predicted structural characteristics suggest they are unusual N-oxidases. Here, we show that recombinant PcxL and HpxL catalyze the FAD- and NADPH-dependent oxidation of 2AEPn and 1H2AEPn, producing a mixture of the respective aldoximes and nitrosylated phosphonic acid products. Measurements of catalytic efficiency indicated that PcxL has almost an equal preference for 2AEPn and (R)-1H2AEPn. 2AEPn was turned over at a 10-fold higher rate than (R)-1H2AEPn under saturating conditions, resulting in a similar but slightly lower kcat/Km. We observed that (S)-1H2AEPn is a relatively poor substrate for PcxL but is clearly the preferred substrate for HpxL, consistent with the proposed biosynthetic pathway in S. regensis. HpxL also used both 2AEPn and (R)-1H2AEPn, with the latter inhibiting HpxL at high concentrations. Bioinformatic analysis indicated that PcxL and HpxL are members of a new class of oxime-forming N-oxidases that are broadly dispersed among bacteria.
Keywords: enzyme kinetics, flavin adenine dinucleotide (FAD), natural product biosynthesis, nicotinamide, oxidase, substrate specificity, oxime, phosphonates, phosphonic acid
Introduction
Oxime-containing small molecules often possess useful biological properties, including antibacterial (1), antifungal (2), insecticidal (3, 4), antitumor (5), immunosuppressive (6), and acetylcholinesterase reactivation activities (7, 8). Some of these bioactive oximes are natural products, with the most commonly identified being the glucosinolates (Fig. 1). These amino acid–derived natural products, produced by plants of the order Capparales, contain a sulfonated oxime responsible for the distinctive flavor and odor of many cruciferous vegetables (9). Other natural product oximes include the vibralactoximes, which are biosynthetic derivatives of the lipase inhibitor vibralactone, produced by the fungus Boreostereum vibrans (10); the DNA methyltransferase inhibitor psammaplin A, produced by the sea sponge Psammaplinaplysilla (11); and the β-lactam antibiotic nocardicin A, produced by the actinobacterium Nocardia uniformis subsp. tsuyamanesis (1) (Fig. 1).
Figure 1.
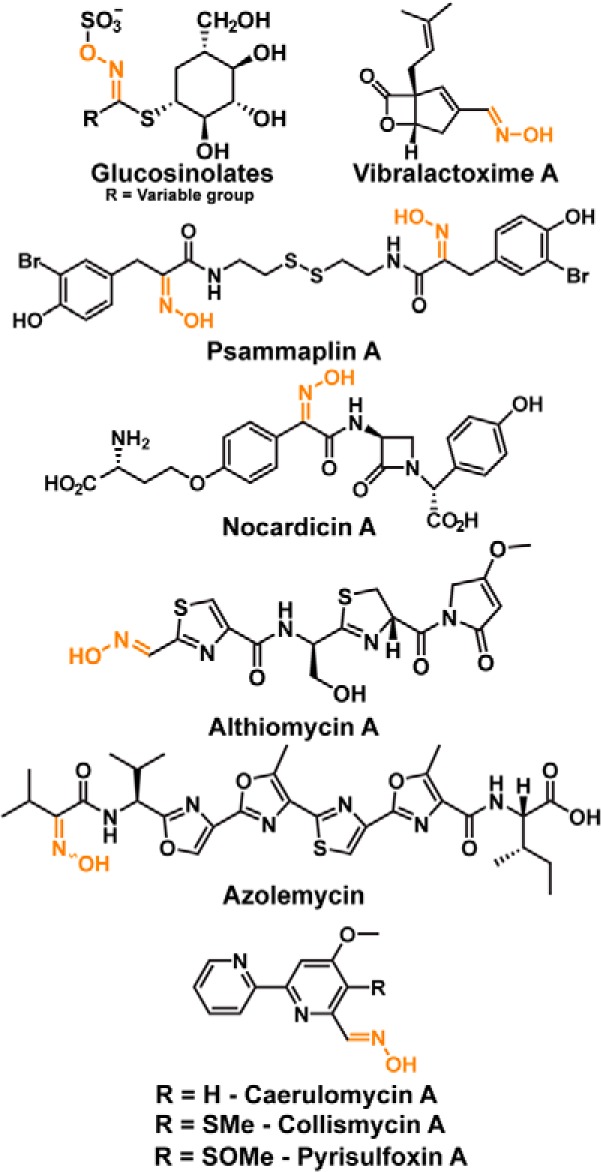
Oxime-containing natural products.
Three different enzyme families are known to produce oxime moieties: (1) cytochrome P450–dependent monooxygenases, (2) ferritin-like enzymes, and (3) flavin-dependent N-monooxygenases. Glucosinolates are usually made via a pathway involving a cytochrome P450–dependent monooxygenase that catalyzes successive N-oxidations to yield an oxime derivative of an amino acid (9, 12); however, these molecules are probably made via an unknown FAD-NAD(P)H-dependent oxygenase in Brassica napus and Brassica camestris (13, 14). In bacteria, the oxime in nocardicin A is installed by a cytochrome P450–dependent monooxygenase known as NocL (1, 15), whereas a metal-dependent N-oxygenase, AlmD, is responsible for oxime formation in althiomycin (16). Finally, the oxime in the ribosomally synthesized peptide azolemycin is post-translationally installed by the flavin-dependent monooxygenase AzmF (17), whereas the oxime in caerulomycin A is introduced using a two-component flavin-dependent monooxygenase, CrmH (18).
Our laboratory recently identified two new phosphonate natural products with an unusual thiohydroximate moiety (19). These include phosphonocystoximic acid, produced by Streptomyces sp. NRRL S-481, and its hydroxylated congener, hydroxyphosphonocystoximic acid, made by a number of Streptomyces species, including Streptomyces regensis NRRL WC-3744, which also makes hydroxynitrilaphos (20). Significantly, many hydroxyphosphonocystoximate producers make small amounts of phosphonocystoximic acid, as well as a suite of biosynthetic intermediates that differ solely by hydroxylation at the carbon α to the phosphorus atom, suggesting that similar molecular machinery is used to produce both natural products (19). This idea is strongly supported by analysis of the biosynthetic genes clusters associated with production of these oxime-containing natural products.
Eighteen genes are shared between the clusters responsible for the synthesis of phosphonocystoximate (the pcx gene cluster) and hydroxyphosphonocystoximate (the hpx gene cluster) based on significant amino acid identity between the encoded proteins (Fig. S1 and Table S1). The first nine genes (hpxA–I and pcxA–I) in both clusters are highly homologous and found in the same order. Nine additional genes are also shared between the two clusters, with their organization conserved for the most part (pcxK–pcxU and hpxK–hpxU). Thus, it seems likely that most of the biosynthetic steps required for synthesis of the two congeners are similar. However, the gene clusters also show significant differences. Key among these differences is a gene encoding a putative 2-oxoglutarate–dependent dioxygenase designated HpxV, found solely in S. regensis. Based on the presence of this gene and the observed metabolic intermediates produced by the two strains, a scheme for biosynthesis of phosphonocystoximate and hydroxyphosphonocystoximate biosynthesis was proposed (Fig. 2 and Ref. 19). The proposed pathways diverge at an early step, with hydroxylation of 2AEPn3 by HpxV yielding 1H2AEPn solely in S. regensis. Consistent with this model, we recently found that purified HpxV catalyzes this reaction, generating the S-enantiomer of 1H2AEPn.4 The mechanism of oxime formation in the two pathways, however, remains a mystery, because homologs of known oxime-forming enzymes are not found in either biosynthetic gene cluster. Nevertheless, putative NAD(P)H-dependent and flavin-dependent oxygenases produced by the pcxL and hpxL genes in Streptomyces sp. NRRL S-481 and S. regensis, respectively, have been suggested as possible candidates for this enzymatic activity (19).
Figure 2.
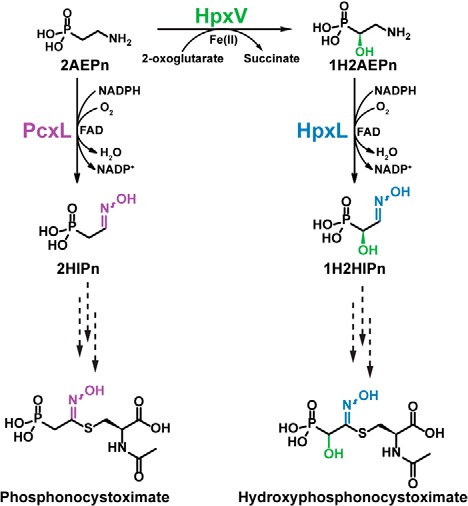
The proposed biosynthetic route of oxime formation in phosphonocystoximates. HpxV is a 2-oxoglutarate- and ferrous iron-dependent 2AEPn dioxygenase that converts 2AEPn to (S)-1H2AEPn. PcxL and HpxL are FAD- and NADPH-dependent amine oxidases that catalyze 2AEPn or 1H2AEPn oxidation to yield 2-hydroxyiminoethylphosphonic acid (1) and 1-hydroxy-2-hydroxyiminoethylphosphonic acid (3), respectively. Multiple steps are required to convert these oxime-containing intermediates to the final products (dashed-line arrows).
Here we present in vitro biochemical evidence that supports this proposal. The data reveal PcxL and HpxL as the founding members of a novel family of oxime-forming enzymes required for the production of the hydroxylated and nonhydroxylated forms of thiohydroximate phosphonic acid natural products.
Results
Bioinformatic analysis suggests that HpxL and PcxL are novel oxime-forming amine oxygenases
Several lines of bioinformatics evidence suggest that the PcxL and HpxL proteins are responsible for the installation of the oxime moiety in the phosphonocystoximic acids produced by Streptomyces sp. S-481 and S. regensis. First, conserved FAD- and NAD(P)-binding domains are observed in both proteins, consistent with a role in redox chemistry. Second, both have a structural similarity to the N-terminal domain of l-ornithine N-hydroxylase from Pseudomonas (Protein Data Bank entry 3S5W) and Kutzneria (Protein Data Bank entry 4TLX) based on the protein-fold recognition algorithm, Phyre2 (21). Taken together, these data suggest that PcxL and HpxL may catalyze FAD- and NAD(P)-dependent amine oxidation. Additional support for the proposed amine oxidase function is provided by their weak similarity (∼35% identity) to the FAD-NAD(P)H-dependent enzymes involved in the biosynthetic pathways of cremeomycin (22) and fosfazinomycin (23) (Table 1). These enzymes oxidize the primary amine of aspartic acid to form nitrosuccinic acid, which is believed to serve as the substrate for downstream N–N bond formation (22). Finally, the existence of the characterized oxime-forming FAD- and NAD(P)H-dependent amine monooxygenases in the biosynthetic pathways of azolemycin (AzmF) (17) and caerulomycin A (CrmH) (18) is also consistent with the proposed amine oxidase function for PcxL and HpxL. However, it should be stressed that AzmF and CrmH are not related to PcxL and HpxL, based on a comparison of their primary amino acid sequences (Table 1). Moreover, these proteins are significantly smaller than PcxL and HpxL (389 and 407 amino acids versus 742 and 750 amino acids). Similarly, the human liver flavin-containing monooxygenases FMO1 (NCBI accession number NP_001269621.1) and FMO3 (NCBI accession number NP_008825.4), which oxidize phenethylamine to the oxime (24), show no similarity to HpxL and PcxL. Thus, if PcxL and HpxL are indeed FAD-NAD(P)H-dependent enzymes that catalyze oxime formation, they belong to a novel protein family.
Table 1.
Known characterized N-oxidases and their homology to HpxL, which is encoded by the hydroxyphosphonocystoximic gene cluster
PcxL is found in the homologous phosphonocystoximic biosynthetic gene cluster. FzmM and CreE catalyze the N-oxidation of aspartic acid to nitrosuccinic acid during fosfazinomycin and cremeomycin biosynthesis, respectively (22, 23). AzmF and CrmH are characterized as flavin-dependent N-oxidases that catalyze oxime formation in azolemycin and caerulomycin biosynthesis (17, 18). ClmM is the predicted FAD- and NAD(P)H-dependent N-oxidase of collismycin biosynthesis (40), and AlmD is the metal-dependent N-oxidase involved in oxime formation of alithomycin biosynthesis (16). Note that only FzmM and CreE can be considered bona fide homologs of PcxL and HpxL.
| Enzyme | NCBI accession no. | Organism | Natural product | % Identity to HpxL | E-Value | Lengtha |
|---|---|---|---|---|---|---|
| HpxL | WP_030990682.1 | S. regensis WC-3744 | Hydroxyphosphonocystoximate | 100 | 0 | 750 |
| PcxL | WP_051704824.1 | Streptomyces sp. S-481 | Phosphonocystoximate | 61 | 0 | 742 |
| AzmF | AMQ23503.1 | Streptomyces sp. FXJ1.264 | Azolemycin | 10 | 2.9 | 389 |
| ClmM | CCC55915.1 | Streptomyces sp. CS40 | Collismycin A | 9 | 1.4 | 402 |
| CrmH | AFD30961.1 | Actinoalloteichus sp. WH1-2216-6 | Caerulomycin A | 8 | 1.1 | 407 |
| AlmD | CCA29200.1 | Myxococcus xanthus DK897 | Althiomycin | 8 | 3.4 | 331 |
| FzmM | WP_053787792.1 | Streptomyces sp. XY332 | Fosfazinomycin | 30 | 6E-14 | 644 |
| CreE | ALA99202.1 | Streptomyces cremeus | Cremeomycin | 35 | 3E-29 | 666 |
a Number of amino acids.
HpxL and PcxL catalyze NADPH oxidation in the presence of FAD and 2AEPn
To examine the biochemical properties of PcxL and HpxL, His6 affinity-tagged alleles were overexpressed in Escherichia coli. Recombinant proteins were then purified and assayed for their ability to oxidize nicotinamide cofactors in the presence of 2AEPn, FAD, or FMN (Table 2). Both enzymes catalyze the oxidation of NADPH and NADH at a significant rate in the absence of additional substrates. Oxidation of reduced nicotinamides is stimulated by the addition of FAD but not by the addition of FMN, suggesting that the former is the preferred cofactor. It should be noted that the as-purified enzymes contain bound FAD, as indicated by the bright yellow coloration of the preparations (Fig. S2A). Additionally, mass spectrometric analysis corroborates the presence of FAD in these protein (Fig. S2, B–E), whereas UV-visible spectra clearly show the presence of a flavin that is reduced upon the addition of NADPH (Fig. S2, F–H). We believe the presence of FAD bound to the proteins upon purification accounts for the basal activity seen in the absence of added FAD. Significantly, oxidation of NADPH, but not NADH, is substantially increased upon the addition of 2AEPn, with the highest rates observed for the combination of NADPH, FAD, and 2AEPn. Thus, activity in the absence of 2AEPn is likely due to an uncoupled, oxygen-dependent reaction, which is common for flavin-dependent enzymes (25). Additionally, when this reaction is run in the absence of oxygen, there is no oxidation of NADPH by PcxL in either the presence or absence of 2AEPn (data not shown).
Table 2.
Oxidation of nicotinamide cofactors by HpxL and PcxL
The apparent rate in the presence/absence of different flavin cofactors and 2AEPn is shown. 2AEPn* = rate in the presence of 2AEPn after subtraction of the uncoupled rate (i.e. before the addition of 2AEPn). 2AEPn was used at a concentration of 5 mm, whereas FAD and FMN were used at 50 μm and NADPH and NADH at 300 μm. Each experiment was completed in triplicate.
| Cofactor(s) | Substrate |
kobs |
|
|---|---|---|---|
| HpxL | PcxL | ||
| s−1 × 10−3 | |||
| NADPH | None | 2.7 ± 0.4 | 14.4 ± 0.7 |
| 2AEPn | 7.2 ± 0.6 | 232 ± 8 | |
| NADPH/FAD | None | 12.5 ± 0.1 | 21 ± 1 |
| 2AEPn | 23.2 ± 0.7 | 310 ± 20 | |
| 2AEPn* | 10.7 ± 0.6 | 290 ± 20 | |
| NADPH/FMN | None | 5.2 ± 0.5 | 14.3 ± 0.7 |
| 2AEPn | 10.0 ± 0.2 | 210 ± 20 | |
| 2AEPn* | 4.8 ± 0.7 | 200 ± 20 | |
| NADH | None | 5.5 ± 0.1 | 24 ± 1 |
| 2AEPn | 1.7 ± 0.2 | 29 ± 2 | |
| NADH/FAD | None | 14 ± 2 | 85 ± 5 |
| 2AEPn | 15.0 ± 0.7 | 83 ± 5 | |
| 2AEPn* | 0.1 ± 0.1 | 0 ± 3 | |
| NADH/FMN | None | 5.9 ± 0.3 | 26.6 ± 0.2 |
| 2AEPn | 7.0 ± 0.4 | 27 ± 4 | |
| 2AEPn* | 1.0 ± 0.2 | 1 ± 4 | |
HpxL and PcxL oxidize 2AEPn to 2-hydroxyiminoethylphosphonic acid and 2-nitroethylphosphonic acid
A series of NMR experiments were conducted to establish the products of PcxL- and HpxL-catalyzed 2AEPn oxidation. Based on 31P NMR spectroscopy, both enzymes form three phosphorus-containing products after prolonged incubation with NADPH, FAD, and 2AEPn (Fig. 3, A and B). Analysis of these molecules using proton-phosphorus–coupled HMBC NMR spectroscopy allowed assignment of the major peak as a mixture of the (E)- and (Z)-isomers of 2-hydroxyiminoethylphosphonic acid (1), with smaller amounts of 2-nitroethylphosphonic acid (2) (Figs. S3 and S4). Accordingly, aldoxime protons display a characteristic proton NMR spectroscopy chemical shift that will be between 6.5 and 8.0 ppm (26). A single proton associated with each of the products, which we assigned as (E)-1 and (Z)-1 at 6.82 and 7.37 ppm, respectively. This assignment is supported by a recent report of the total synthesis of the phosphonocystoximic acids, which involves the synthesis of ethyl-protected aldoximes as intermediates (27). The aldoxime protons for the protected (E)-isomer have a chemical shift of 6.82 ppm, whereas the aldoxime proton associated with the protected (Z)-isomer has a chemical shift of 7.41 ppm (27). Our assignment of the third product as 2-nitroethylphosphonic acid (2) is based on the observation of phosphorus-correlated protons at 2.03 and 4.48 ppm, which is consistent with literature values of synthetically produced 2-nitroethylphosphonic acid (28). Although both enzymes form a mixture of (E)- and (Z)-isomers when using 2AEPn as substrate, HpxL preferentially forms (E)-1 (Fig. S3), whereas PcxL preferentially forms (Z)-1 (Fig. S4).
Figure 3.

Phosphorus NMR spectroscopy analysis of N-oxidase activity in the presence of 2AEPn or (S)-1H2AEPn. A, HpxL and PcxL form three major products in vitro when incubated for 16 h with NADPH, FAD, and 2AEPn as the substrate, whereas no product formation is observed in the absence of enzyme. B, these products have been assigned as the (E)- and (Z)-isomers of 2-iminoethylphosphonic acid (1) and 2-nitroethylphosphonic acid (2) based on the 1H-31P HMBC analysis (Figs. S2 and S3). Note that the (E)- and (Z)-isomers of 1 have identical 31P chemical shifts and thus display only a single peak in the one-dimensional 31P spectrum shown in A. C, HpxL and PcxL both generate two products in vitro using (S)-1H2AEPn as the substrate. D, these products were assigned as (Z)-1-hydroxy-2-hydroxyiminoethylphosphonic acid (3) and 1-hydroxy-2-nitroethylphosphonic acid (4) based on the 1H-31P HMBC analysis (Fig. S4 and S5). Chemical-mixing experiments with the control reaction were performed to confirm that substrate had been completely consumed. Substrates and cofactors were added at the following concentrations: 100 μm FAD, 500 μm NADPH, 3 mm 2AEPn or HpxV-generated (S)-1H2AEPn, 25 μm PcxL or HpxL, 25 mm phosphite, and 10 μm PTDH17x.
We performed an identical experiment using 1H2AEPn as substrate to determine the products that would be formed by the enzymes. PcxL and HpxL catalyze oxidation of the amine moiety in (S)-1H2AEPn as a substrate (Fig. 3, C and D). In contrast to the products obtained with 2AEPn, the enzymes form only the (Z)-isomer of 1-hydroxy-2-hydroxyiminoethylphosphonic acid (3) when using (S)-1H2AEPn as the substrate. This is evident by the presence of only one set of protons correlating with the phosphorus atom with chemical shifts of 4.15 and 7.44 ppm (Figs. S5 and S6). The enzymes also form smaller amounts of 1-hydroxy-2-nitroethylphosphonic acid (4) (Figs. S5 and S6).
To determine the molar ratio of substrates consumed to cofactor used, we determined the stoichiometry of the reaction by monitoring substrate (2AEPn and oxygen) and cofactor (NADPH) consumption during short incubations. We observed that the molar ratio of oxygen to NADPH was ∼1:1 for both enzymes; however, the molar ratio of NADPH to 2AEPn consumption was much higher: 2.5:1 (NADPH/2AEPn) for PcxL and 4.0:1 for HpxL (Table 3). This yields an overall reaction stoichiometry of 2AEPn to NADPH to oxygen consumption of 1/2.5/2.7 for PcxL and 1/4.0/3.8 for HpxL. Significantly, 2 was not observed during the short incubations used in these experiments (data not shown). Thus, the presence of this product in longer incubations is probably due to overoxidation of the substrate.
Table 3.
Stoichiometry of PcxL- and HpxL-catalyzed reactions
Oxygen and NADPH consumption was measured in 5-min reactions using a Clark-type electrode and UV/visible spectrophotometry, respectively, to determine the O2/NADPH ratio. A separate 3-h reaction was used to obtain the NADPH/2AEPn ratio with 31P NMR used to monitor both substrates. Each experiment was performed in triplicate.
| Substrate | Enzyme | O2/NADPH |
NADPH/2AEPn |
Combined ratio |
||||
|---|---|---|---|---|---|---|---|---|
| nmol NADPH | nmol O2 | Ratio | nmol NADPH | nmol 2AEPn | Ratio | 2AEPn/ O2/NADPH | ||
| 2AEPn | PcxL | 966 ± 0.4 | 1040 ± 2 | 1:1.1 | 1970 ± 3 | 810 ± 30 | 2.5:1 | 1:2.5:2.7 |
| HpxL | 163 ± 1 | 156 ± 2 | 1.1:1 | 1420 ± 30 | 350 ± 50 | 4.0:1 | 1:4.0:3.8 | |
HpxL and PcxL both preferentially oxidize 1H2AEPn over 2AEPn but with different stereochemical preferences
To determine the substrate preference of the N-oxidases in vitro, we performed initial rate kinetics on PcxL and HpxL using 2AEPn and the (R)- and (S)-enantiomers of 1H2AEPn as substrates (Table 4 and Fig. 4). Although both enzymes are capable of using all of these substrates, they manifest significantly different substrate preferences. PcxL shows the highest rate with 2AEPn as substrate, but it shows a greater affinity for (R)-1H2AEPn. Thus, the catalytic efficiency of PcxL is similar for these two substrates. In contrast, (S)-1H2AEPn is a relatively poor substrate for PcxL, whereas HpxL turns over (S)-1H2AEPn and 2AEPn at similar rates but has a 10-fold higher affinity for the former. Thus, (S)-1H2AEPn, which is predicted to be the in vivo substrate, is the preferred substrate for this enzyme. (R)-1H2AEPn is a relatively poor HpxL substrate and actually inhibits the enzyme at higher concentrations (Fig. 4F).
Table 4.
Initial rate kinetics for HpxL and PcxL
Rates were determined by monitoring NADPH consumption via UV-visible spectrophotometer. Vmax, Km, and KI were determined by fitting the data with either the Michaelis-Menten (MM) or substrate inhibition (SI) model. If the data were fitted to the MM model, a KI was not applicable (NA). Only the best fit is shown. Reaction components included: 50 mm sodium phosphate buffer, pH 7.8, 300 μm NADPH, 50 μm FAD, substrate ranging from 62.5 μm to 150 mm, and enzyme concentrations of 1 μm or 5 μm for PcxL and 5 μm or 10 μm for HpxL. The substrates used were 2AEPn (Sigma-Aldrich), synthetic (R)-1H2AEPn, and synthetic (S)-1H2AEPn. This experiment was performed in triplicate for each curve.
| Enzyme | Substrate | Kinetic model | Vmax | Apparent Km | kcat/Km | KI |
|---|---|---|---|---|---|---|
| s−1 | mm | m−1 s−1 | mm | |||
| PcxL | 2AEPn | MM | 1.22 ± 0.02 | 3.3 ± 0.5 | 370 | NA |
| (R)-1H2AEPn | MM | 0.39 ± 0.01 | 0.48 ± 0.02 | 810 | NA | |
| (S)-1H2AEPn | MM | 0.32 ± 0.03 | 6 ± 1 | 53 | NA | |
| HpxL | 2AEPn | MM | 0.032 ± 0.002 | 4.3 ± 0.8 | 7.4 | NA |
| (R)-1H2AEPn | SI | 0.06 ± 0.03 | 4 ± 2 | 15 | 2 ± 1 | |
| (S)-1H2AEPn | MM | 0.039 ± 0.001 | 0.39 ± 0.03 | 100 | NA |
Figure 4.

Kinetic curves for PcxL and HpxL in the presence of different substrates. PcxL (A) and HpxL (B) with 2AEPn as the substrate were fit to a standard Michaelis-Menten model. PcxL (C) and HpxL (D) with (S)-1H2AEPn were fit to a standard Michaelis-Menten model. PcxL (E) with (R)-1H2AEPn as a substrate was fit to a standard Michaelis-Menten model, and HpxL (F) with (R)-1H2AEPn as substrate was fit to the substrate inhibition model. Extracted kinetic constants are shown in Table 4.
HpxL and PcxL are members of a large family of putative N-oxidases that is common in phosphonate biosynthetic pathways
The low similarity of HpxL and PcxL to other known N-oxidases suggests that they are members of larger protein family (Table 1). Indeed, BLAST searches of GenBankTM retrieved thousands of hits using HpxL and PcxL as query sequences (data not shown). Phylogenetic analysis of the top 500 HpxL homologs reveals numerous discrete clades within the family. To provide clues as to the function of the proteins within this family, we examined the genomic context surrounding these homologs using the bioinformatics tool RODEO, which retrieves adjacent genes and assigns putative functions based on PFAM membership (29). Significantly, almost all genes encoding proteins within the HpxL and PcxL clade fall within gene clusters devoted to the synthesis of phosphonic acids based on co-localization with genes encoding phosphoenolpyruvate mutase (PepM), which encodes the first step in most phosphonate biosynthetic pathways (Fig. 5A) (19, 30, 31). As described above, the known N-oxidases FzmM and CreE are bona fide members of this protein family; however, they are only distantly related to HpxL and PcxL and do not fall within the top 500 homologs shown in Fig. 5A. Indeed, the BLAST search results using HpxL as the query must be extended to include the top 5000 hits before FzmM and CreE are included.
Figure 5.
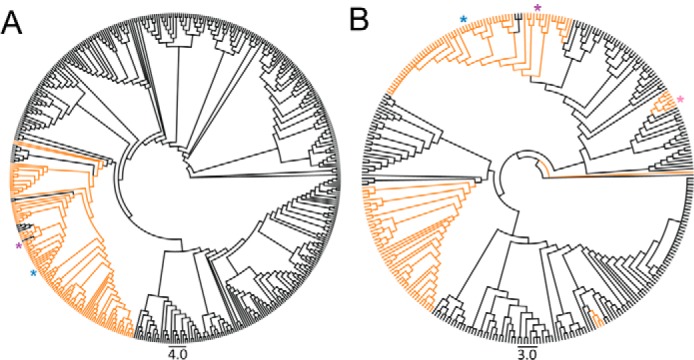
Cladograms showing the phylogenetic distribution of putative N-oxidases related to HpxL and PcxL. A, phylogeny of the 500 closest HpxL homologs. hpxL-containing gene clusters that also contain pepM are highlighted in orange. B, phylogeny of actinobacterial PepM sequences. pepM-containing gene clusters that contain an HpxL homolog are highlighted in orange. HpxL is denoted with a blue asterisk, PcxL is denoted with a purple asterisk, and FzmM is denoted with a pink asterisk. Gene clusters were arbitrarily defined to include the 15 genes upstream or downstream of the N-oxidase.
We performed a similar analysis using previously identified phosphonate biosynthetic gene clusters as the starting point (19). 23% of the phosphonate gene cluster families contain a HpxL homolog, with only three families having characterized products: phosphonocystoximic acids, hydroxyphosphonocystoximic acids, and fosfazinomycins. Thus, at least 15 novel uncharacterized phosphonic acid natural-product biosynthetic pathways encode an HpxL homolog. Based on the similarity of these enzymes within the N-oxidase tree, we can predict that these 15 novel phosphonic acids will likely involve the oxidation of 2AEPn or 1H2AEPn to form an oxime-containing natural product.
Discussion
PcxL and HpxL are 2AEPn and 1H2AEPn amine oxygenases
The data presented here show that PcxL and HpxL are novel FAD- and NADPH-dependent oxime-forming amine oxygenases that utilize amine-containing phosphonate substrates. Although these enzymes were shown to oxidize several small amino phosphonates, the substrate preferences of the two enzymes support the proposed biosynthetic pathways shown in Fig. 2. Accordingly, we expected that the substrate preference of the N-oxidases would be influenced by the presence of the proposed 2AEPn dioxygenase, HpxV, in the gene cluster of S. regensis NRRL WC-3744 (Fig. 2, Fig. S1, and Table S1), with HpxL having a preference for (S)-1H2AEPn and PcxL having a preference for 2AEPn (19). The finding that the catalytic efficiency of HpxL is 10-fold higher for (S)-1H2AEPn than the other substrates tested was fully consistent with this model. Our conclusions regarding PxcL are less certain, because the enzyme displays a similar catalytic efficiency with both 2AEPn and (R)-1H2AEPn. Nevertheless, it is very likely that 2AEPn is the natural substrate given the absence of known genes to produce hydroxylated phosphonate substrates in Streptomyces sp. NRRL S-481.
The stoichiometry of the reaction suggests overoxidation of substrate during formation of the aldoxime
Based on the finding that NADPH and O2 are always consumed in a 1:1 ratio by PcxL and HpxL (Table 3), oxime formation should require two separate hydroxylation steps, each consuming 1 molar equivalent of NADPH and 1 molar equivalent of oxygen. Each step would also produce 1 molar equivalent of H2O by the reduction of O2 with NADPH. We predict that this reaction will proceed through hydroxyamino and dihydroxyamino intermediates, with subsequent loss of water producing a nitroso intermediate, which would tautomerize to afford the final aldoxime product observed in our reactions (Fig. 6). An infrequent, third NADPH- and O2-dependent hydroxylation of the dihydroxyamino/nitroso intermediates, or the oxime products, would then afford small amounts of the nitro derivatives seen in our data (Fig. 3 and Figs. S3–S6). The fact that we see substantially higher levels of O2 consumption relative to aldoxime formation is consistent with this idea, as is the observation of NADPH oxidation in the absence of phosphonate substrates (i.e. the uncoupled reaction). We expect that the latter would produce hydrogen peroxide, although this was not tested directly.
Figure 6.

Proposed stepwise oxidation of 2AEPn to yield 1 or 2. This may explain the 1:2.5 substrate:NADPH ratio observed for oxime formation in vitro. We propose that each N-oxidation would require 1 molar equivalent of NADPH and molecular oxygen. This would require two N-oxidations and a dehydration to yield 2-nitrosoethylphosphonic acid, which could tautomerize to form 2-hydroxyiminoethylphosphonic acid. An enzyme-catalyzed overoxidation of either 2-nitrosoethylphosphonic acid or 2-hydroxyiminoethylphosphonic acid could potentially yield 2-nitroethylphosphonic acid (shown by gray arrows). Compounds shown in purple are all products observed in the reactions catalyzed by HpxL and PcxL using 2AEPn as the substrate (Fig. 3).
PcxL and HpxL are new members of a class of flavin-dependent amine oxidases common in nature
Sequence analysis shows that HpxL and PcxL are members of a very large protein family that is widely distributed in bacteria. Only four members of this family have been characterized biochemically, including HpxL, PcxL, and the distantly related enzymes FzmM and CreE. Although the former, HpxL and PcxL, oxidize amino phosphonates to aldoximes (and small amounts of nitro derivatives), the latter oxidize aspartate to nitrosuccinic acid (22, 23). Given the phylogenetic distance between these distant homologs, it seemed reasonable to assume that most members of the family are amine oxidases. Interestingly, members of this presumptive amine oxidase family are common in actinobacterial phosphonate biosynthetic gene clusters, with nearly one-fourth of those examined containing a homolog. These enzymes are widely distributed on the PepM maximum-likelihood tree, suggesting that HpxL and PcxL homologs were recruited into phosphonic acid biosynthetic gene clusters multiple times; however, all of these homologs are closely related, falling into the same clade within the amine oxidase tree (Fig. 5A). Moreover, each of the gene clusters containing these homologs also contains the biosynthetic machinery needed to make 2AEPn or 1H2AEPn. Taken together, these data suggest that numerous phosphonate biosynthetic pathways include 1 or 3 as early intermediates (see Fig. 3). Thus, although the phosphonocystoximates are the only known oxime-containing phosphonate natural products, it seems certain that numerous additional examples await discovery.
Experimental procedures
General procedures
Nuclear magnetic resonance (NMR) spectra were collected using an Agilent Technologies 600-MHz NMR spectrometer with D2O as the lock solvent (minimum 20% v/v). All chemicals used in this study were purchased from Sigma-Aldrich unless otherwise noted.
N-Oxidase cloning, overexpression, and purification
The genes encoding the N-oxidases were amplified via PCR using primer pairs MG140 (5′-CTGGTGCCGCGCGGCAGCCATATGacgaccgacaattccccagcca-3′) and MG141 (5′-AGTGGTGGTGGTGGTGGTGCTCGAGtcatgcgggggtcctttcgatgtcc-3′) for hpxL and primer pairs MG142 (5′-CTGGTGCCGCGCGGCAGCCATATGcgttccaaccgactcgccatcgtcg-3′) and MG143 (5′-AGTGGTGGTGGTGGTGGTGCTCGAGtcacactcccgtggtgatgcgcgcg-3′) for pcxL from fosmids containing the phosphonocystoximic acid gene clusters (19). The plasmid pET28B was amplified using primers pET28-NdeI-R (5′-CATATGGCTGCCGCGCGGCACCAGGCCGCTG-3′) and pET28B-XhoI-F (5′-CTCGAGCACCACCACCACCACCACTGAGATCCGG-3′). The uppercase letters represent sequence homology to the plasmid pET28B, the underlined sequences represent inserted restriction enzyme sites, and the lowercase letters represent sequence homology to the gene to be amplified. The primer pair MG140 and MG141 was designed to mutate the GTG start codon of hpxL to an ATG start codon for expression in E. coli. The gene fragments were subsequently cloned into pET28B using Gibson Assembly® (New England Biolabs, Ipswich, MA) to yield pMNG016 for hpxL and pMNG017 for pcxL. The sequences were verified at the University of Illinois Urbana-Champaign Core Sequencing Facility.
The PcxL and HpxL proteins were affinity-purified after expression in E. coli. PcxL and HpxL were overexpressed in transformed E. coli Rosetta DE3 cells containing either plasmid pMNG016 or pMNG017. Proteins were purified using one of two methods. Method 1 involved cells grown in Studier's ZYM5052 autoinduction medium (32) with the following modification. The trace elements solution was replaced with 1 mg/liter of each iron(II) sulfate heptahydrate, zinc(II) sulfate heptahydrate, and manganese(II) chloride dihydrate. The cultures were shifted to 18 °C when absorbance (A600) reached 0.6–0.8 and the protein was overexpressed for 16 h. Cells were collected via centrifugation and resuspended in lysis buffer (300 mm sodium chloride, 50 mm sodium phosphate (dibasic), 10% glycerol, and 10 mm imidazole, pH 8.0). The cell suspension was mixed with 1 mg/ml lysozyme, 100 units of DNase I, and 100 μg of RNase A for 30 min at 4 °C. The cell suspension was passed twice through a French press cell disruptor (Thermo Electron Corp., Waltham, MA) at 1000 psi. The suspension was centrifuged at 14,000 × g for a minimum of 30 min, and the pellet was discarded. The liquid portion was passed over a 5-ml HisTrap FPLC protein column (GE Healthcare Life Sciences) and washed with lysis buffer until the protein was no longer eluting from the column as determined by absorbance at A260 nm. Protein was eluted from the column on an ÄTKApurifier FPLC (GE Healthcare Life Sciences) using a gradient from 10 to 250 mm imidazole with 60 column volumes over 1 h at a flow rate of 5 ml/min. Fractions containing purified protein, as determined by running the samples on a SDS-PAGE, were pooled and concentrated using a 30-kDa molecular mass Amicon® Ultra spin filter (EMD Millipore, Burlington, MA). Method 2 involved cells grown in Luria broth until the absorbance (A600) reached 0.6–0.8. The cultures were chilled on ice for 30 min, and protein expression was induced upon the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 1 mm. Cells were shaken at 18 °C for 16 h after induction. Pelleted cells were resuspended in lysis buffer (see above) containing 1 mg/ml lysozyme, 100 units of DNase I, and 100 μg of RNase A and mixed at 4 °C for 30 min. The cell suspension was passed twice through a French press cell disruptor (Thermo Electron Corp.) at 1000 psi. The suspension was centrifuged at 14,000 × g for 30 min, and the pellet was discarded. The soluble fraction containing soluble protein was mixed with 5 ml of loose nickel–nitrilotriacetic acid resin for 30 min at 4 °C. The resin was washed with wash buffer (300 mm sodium chloride, 50 mm sodium phosphate (dibasic), 10% glycerol, and 20 mm imidazole, pH 8.0) until protein was no longer eluting from the resin as determined by absorbance at A260 nm. Protein was eluted from the column by incubating the resin with elution buffer (300 mm sodium chloride, 250 mm sodium phosphate (dibasic), 10% glycerol, and 20 mm imidazole, pH 8.0) for 15 min and collecting the flow-through. This flow-through was concentrated using a 30-kDa molecular mass Amicon® Ultra spin filter (EMD Millipore). All samples, regardless of which purification method was used, were exchanged into storage buffer (300 mm sodium chloride, 50 mm sodium phosphate (dibasic), and 10% glycerol) using a PD-10 desalting column (GE Healthcare Life Sciences).
Bioinformatics characterization of HpxL and PcxL
The following amino acid sequences were downloaded from NCBI: HpxL (accession number WP_030990682.1), PcxL (accession number WP_051704824.1), AzmF (accession number AMQ23503.1), ClmM (accession number CCC55915.1), CrmH (accession number AFD30961.1), AlmD (accession number CCA29200.1), FzmM (accession number WP_053787792.1), and CreE (accession number ALA99202.1). These sequences were aligned using the standard settings for a MUSCLE alignment using Geneious (Geneious version 8.1.2 (33)), which provided the percent identity between the proteins shown in Table 1.
Cofactor preference of the PcxL and HpxL
Cofactor preference experiments were monitored using a Cary 4000 UV-visible spectrophotometer (Agilent Technologies, Santa Clara, CA). NADH and NADPH consumption was monitored by observing the absorption at 340 nm. Reaction components included: 50 mm sodium phosphate buffer, pH 7.8, 300 μm nicotinamide cofactor (either NADPH or NADH), 50 μm flavin cofactor (FAD or FMN), 10 mm 2AEPn, and 10 μm N-oxidase (PcxL or HpxL). Reaction components except for substrate and enzyme were mixed in a 200-μl quartz cuvette. Enzyme was added, and the background rate of the enzyme was collected for approximately 1 min. Reactions were initiated upon addition of 2AEPn, and three replicates were collected for each condition.
Flavin identification
LC/MS detection of flavin
HpxL (13.8 mg) and PcxL (5.4 mg) stocks were brought to room temperature and concentrated to 50 μl using a 30-kDa molecular mass Amicon® Ultra spin filter (EMD Millipore), and the flow-through was discarded. The protein was washed six times with 500 μl of ddH2O. After washing, the PcxL was colorless, whereas HpxL retained some bound flavin as noted by the yellow color of the protein (Fig. S2). The collected flavin was flash-frozen and placed on a lyophilizer until dry. The flavin was resuspended in ddH2O. We determined the concentration of the flavins based on absorbance at 450 nm using the extinction coefficient of ϵ = 12,000 m−1cm−1 for FAD (HpxL-extracted flavin = 37.1 μm and PcxL-extracted flavin = 30.9 μm). The extracted flavin (10 μl) was injected onto a 150 × 4.6 mm Synergi 4-μm Fusion-RP column connected to an Agilent 1200 series LC/MSD SL mass spectrometer (Agilent) monitoring for the masses corresponding to FAD (m/z = 786.2) and FMN (m/z = 457.1) in positive mode. The following solvents were used: A = 95% ddH2O, 5% acetonitrile with 0.1% formic acid and B = 5% ddH2O, 95% acetonitrile with 0.1% formic acid. The sample was run with a linear gradient from times 0–12 min: time 0 min = 5% B, 12 min = 95% B, 15 min = 95% B, 18 min = 5% B, and 25 min = 5% B. The flow rate was 1 ml/min.
Absorption spectra of HpxL and PcxL
HpxL (3.6 mg) and PcxL (6.1 mg) stocks were used to determine the absorption spectra of the proteins on a Cary 4000 UV-visible spectrophotometer (Agilent) scanning from 200 to 800 nm. NADPH was added to the protein to a final concentration of 500 μm, and the absorption spectrum of the protein was measured after 1 min of incubation. Flavin that was washed away from the proteins was also measured.
Stoichiometry determination
Oxygen–NADPH ratio
Oxygen consumption was monitored on a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, England). NADPH consumption was monitored (A340) on a Cary 4000 UV-visible spectrophotometer (Agilent). Reaction components included 50 mm sodium phosphate buffer (air-saturated), pH 7.8, 300 μm NADPH, 50 μm FAD, 50 mm 2AEPn, and 1 μm PcxL or 5 μm HpxL. Reactions were initiated by the addition of enzyme and monitored for 5 min. Three replicates were performed on each instrument. The number of moles of both oxygen and NADPH consumed by the enzyme in 5 min was determined and compared.
NADPH–substrate ratio
NADPH consumption was determined using a Cary 4000 UV-visible spectrophotometer (Agilent) and via 31P NMR. 2AEPn consumption was determined using 31P NMR. The reaction components included 50 mm sodium phosphate buffer (air-saturated), pH 7.8, 3 mm NADPH, 500 μm FAD, 5 mm 2AEPn, and 25 μm PcxL or 25 μm HpxL. Three replicates were performed. The reactions were incubated for 30 min at room temperature and were quenched using 1 reaction volume of 100% methanol. Dimethylphosphinic acid was added to the reaction as an internal quantification standard to yield a final concentration of 10 mm. The number of moles of substrate and NADPH consumed was determined and directly compared.
Generation of (S)-1H2AEPn by HpxV
To yield an N-terminal His6-tagged HpxV, hpxV was amplified using the primers Orf10_LIC_F (5′-TACTTCCAATCCAATGCAcctactcacccacgcactc-3′) and Orf10_LIC_R (5′-TTATCCACTTCCAATGTTATTActacttgctcgaccacatg-3′) and cloned into the ligation-independent cloning vector pET His6 TEV LIC cloning vector (2B-T). The N-terminal His6-tagged HpxV was overexpressed in transformed E. coli BL21 DE3 containing the pET His6 TEV LIC cloning vector (2B-T) with hpxV. HpxV was overexpressed and purified using method 2 as described above. (S)-1H2AEPn was generated using HpxV, a 2AEPn-dependent α-ketoglutarate–dependent dioxygenase from S. regensis NRRL WC-3744. The reactions contained 50 mm PBS, pH 7.4, 40 mm α-ketoglutarate, 1 mm iron(II) ammonium sulfate, 7.5 mm 2AEPn, 4 mm l-ascorbic acid, and 200 μm HpxV. Reactions were incubated for 13 h at 30 °C. The reaction was quenched by precipitating the protein by the addition of methanol to yield a 90% methanol precipitation. The precipitated protein was removed via centrifugation, and the reaction mixture was concentrated on a rotary evaporator. The concentration of 1H2AEPn generated was determined by 31P NMR spectroscopy using phosphite as an internal reference standard for quantification at a final concentration of 10 mm. In some experiments synthetic (S)-1H2AEPn was used (see below).
Product formation experiments
HpxL or PcxL were incubated with 2AEPn or HpxV-generated (S)-1H2AEPn to monitor which products were formed by the N-oxidases. The reactions were set up with the following components: 25 mm sodium phosphate buffer, pH 7.8, 500 μm NADPH, 100 μm FAD, 25 μm PcxL or HpxL, 10 μm phosphite dehydrogenase, 25 mm phosphite, and 3 mm of substrate, pH 7.8. Reactions were incubated for 16 h at room temperature. Product formation was monitored using 31P NMR spectroscopy, 1H NMR spectroscopy, and 31P-1H HMBC spectroscopy. A mutated phosphite dehydrogenase (PTDH17x) that can use either NADH or NADPH as the nicotinamide coenzyme (34) was used to regenerate NADPH in the substrate specificity experiments. This enzyme was overexpressed in E. coli BL21 DE3 cells containing a plasmid with PTDH17x cloned into pET15b. Cells were grown in modified Studier's ZYM5052 medium (as described above) at 37 °C until A600 reached 0.6–0.8, the culture was shifted to 18 °C, and the protein was overexpressed for 16 h. The cell suspension was passed twice through a French press cell disruptor (Thermo Electron Corp.) at 1000 psi. The suspension was centrifuged at 14,000 × g for a minimum of 30 min, and the pellet was discarded. Soluble cell lysate was passed over free nickel–nitrilotriacetic acid immobilized metal affinity chromatography (IMAC) resin and washed with wash buffer (20 mm Tris-HCl, 500 mm NaCl, and 10% glycerol (% v/v), pH 7.6). The bound protein was washed stepwise with the wash buffer containing increasing levels of imidazole at concentrations of 10, 25, and 50 mm, respectively, until there was no protein detected by monitoring A260 nm. Protein was eluted from the column using wash buffer containing 250 mm imidazole and concentrated using a 10-kDa molecular mass Amicon® Ultra spin filter (EMD Millipore). The protein was exchanged into protein storage buffer (50 mm HEPES, 200 mm KCl, and 10% glycerol (% v/v), pH 7.5) using a PD-10 desalting column (GE Healthcare Life Sciences).
Stereospecific synthesis of (R)- and (S)-1H2AEPn
Enantiopure (enantiomeric excess (ee) > 99%) (R)- and (S)-1H2AEPn was generated following previously published methods (35).
Kinetic assays
Enzyme assays were monitored using a Cary 4000 UV-visible spectrophotometer (Agilent). NADPH consumption was monitored by observation of the absorption at 340 nm. Reaction components included: 50 mm sodium phosphate buffer, pH 7.8, 300 μm NADPH, 50 μm FAD, substrate concentrations ranging from 62.5 μm to 150 mm, and enzyme concentrations of 1 or 5 μm for PcxL and 5 or 10 μm for HpxL. Briefly, the reaction components except for substrate and enzyme were mixed in a 200-μl quartz cuvette. Enzyme was added, and the background rate of the enzyme was collected for approximately 1 min. Reactions were initiated upon the addition of substrate. Three replicates were obtained for each concentration of substrate. Because of the time it takes to complete 1 NADPH consumption curve for HpxL (between 15 and 30 min), all of the initial rates are approximate initial rates. The rate over ∼30 s once the enzyme reaches steady state was read, and this was used as the “initial rate.”
Construction of phylogenetic trees and assignment of genomic neighborhoods
To construct the N-oxidase phylogenetic tree, the top 500 BLAST (36) hits obtained using HpxL (WP_030990672.1) as a query sequence were downloaded on February 18th, 2017. These sequences were aligned using MAFFT with default settings (37), and the maximum likelihood tree was constructed with FastTree using default settings (38). The accession numbers of the 500 amino acid sequences used in the tree were run through RODEO (29) to identify the putative N-oxidase proteins involved in a phosphonate gene cluster. A positive hit constitutes a phosphoenolpyruvate mutase enzyme being within 15 genes upstream or downstream of the N-oxidase.
To construct the PepM phylogenetic tree, five Actinobacteria PepM amino acid sequences were used as a BLAST query (19): Streptomyces luridus PepM (ACZ13456), Streptomyces viridochromogenes (AAU00071), Streptomyces fradiae (ACG70831),Strepomyces rubellomurinus (ABB90393), and S. regensis HpxU (WP_030646403.1). The search was limited to Actinobacteria, with Mycobacteria excluded from the search due the presence of large numbers of isocitrate lyase family proteins that lack the diagnostic PepM motif. All hits for each search were sorted to remove duplicates, and the characteristic EDKxxxxxNS motif was identified manually to distinguish actual PepM sequences from other members of the isocitrate lyase superfamily (39). There were 199 amino acid sequences identified as putative PepM sequences using this approach. These amino acid sequences were aligned using MAFFT (37), and the maximum likelihood tree was constructed using FastTree (38). RODEO (29) was used to identify phosphonate gene clusters that contain an N-oxidase homolog. A positive hit constitutes an N-oxidase being within 15 genes upstream or downstream of the PepM.
Author contributions
M. N. G., J. P. C., K. S. J., K. P., and W. W. M. conceptualization; M. N. G., J. P. C., and W. W. M. data curation; M. N. G., J. P. C., and W. W. M. formal analysis; M. N. G. and W. W. M. supervision; K. P. and W. W. M. funding acquisition; M. N. G., J. P. C., K. S. J., K. P., and W. W. M. investigation; M. N. G. and W. W. M. visualization; M. N. G., J. P. C., K. S. J., K. P., and W. W. M. methodology; M. N. G., J. P. C., and W. W. M. writing-original draft; M. N. G. and W. W. M. project administration; M. N. G., J. P. C., and W. W. M. writing-review and editing; K. P. and W. W. M. resources.
Supplementary Material
Acknowledgments
We thank N. Petronikolou for constructing the HpxV expression vector. NMR spectroscopy data collected in this study was done at the Carl R. Woese Institute for Genomic Biology Core Facilities on a 600-MHz NMR funded by National Institutes of Health Grant S10-RR028833.
This work was supported by National Institutes of Health Grant P01 GM077596 B from NIGMS and Grant P27987-N28 from the Austrian Science Fund (FWF). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
M. N. Goettge and W.W. Metcalf, unpublished data.
- 2AEPn
- 2-aminoethylphosphonate
- 1HAEPn
- 1-hydroxy-2-aminoethylphosphonate
- PepM
- phosphoenolpyruvate mutase
- TEV
- tobacco etch virus.
References
- 1. Kelly W. L., and Townsend C. A. (2002) Role of the cytochrome P450 NocL in nocardicin A biosynthesis. J. Am. Chem. Soc. 124, 8186–8187 10.1021/ja025926g [DOI] [PubMed] [Google Scholar]
- 2. Jang J. H., van Soest R. W., Fusetani N., and Matsunaga S. (2007) Pseudoceratins A and B, antifungal bicyclic bromotyrosine-derived metabolites from the marine sponge Pseudoceratina purpurea. J. Org. Chem. 72, 1211–1217 10.1021/jo062010+ [DOI] [PubMed] [Google Scholar]
- 3. Bacher M., Brader G., Hofer O., and Greger H. (1999) Oximes from seeds of Atalantia ceylanica. Phytochemistry 50, 991–994 10.1016/S0031-9422(98)00638-4 [DOI] [Google Scholar]
- 4. Moya P., Castillo M., Primo-Yúfera E., Couillaud F., Martínez-Máñez R., Garcerá M. D., Miranda M. A., Primo J., and Martínez-Pardo R. (1997) Brevioxime: A new juvenile hormone biosynthesis inhibitor isolated from Penicillium brevicompactum. J. Org. Chem. 62, 8544–8545 10.1021/jo970397y [DOI] [PubMed] [Google Scholar]
- 5. Hong S., Shin Y., Jung M., Ha M. W., Park Y., Lee Y. J., Shin J., Oh K. B., Lee S. K., and Park H. G. (2015) Efficient synthesis and biological activity of psammaplin A and its analogues as antitumor agents. Eur. J. Med. Chem. 96, 218–230 10.1016/j.ejmech.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 6. Kim Y. S., Jeong H. Y., Kim A. R., Kim W. H., Cho H., Um J., Seo Y., Kang W. S., Jin S. W., Kim M. C., Kim Y. C., Jung D. W., Williams D. R., and Ahn Y. (2016) Natural product derivative BIO promotes recovery after myocardial infarction via unique modulation of the cardiac microenvironment. Sci. Rep. 6, 30726 10.1038/srep30726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eyer P. (2003) The role of oximes in the management of organophosphorus pesticide poisoning. Toxicol. Rev. 22, 165–190 10.2165/00139709-200322030-00004 [DOI] [PubMed] [Google Scholar]
- 8. Wong L., Radic Z., Brüggemann R. J., Hosea N., Berman H. A., and Taylor P. (2000) Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry 39, 5750–5757 10.1021/bi992906r [DOI] [PubMed] [Google Scholar]
- 9. Halkier B. A., and Gershenzon J. (2006) Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 57, 303–333 10.1146/annurev.arplant.57.032905.105228 [DOI] [PubMed] [Google Scholar]
- 10. Chen H. P., Zhao Z. Z., Li Z. H., Dong Z. J., Wei K., Bai X., Zhang L., Wen C. N., Feng T., and Liu J. K. (2016) Novel natural oximes and oxime esters with a vibralactone backbone from the basidiomycete Boreostereum vibrans. ChemistryOpen 5, 142–149 10.1002/open.201500198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arabshahi L., and Schmitz F. J. (1987) Brominated tyrosine metabolites from an unidentified sponge. J. Org. Chem. 52, 3584–3586 10.1021/jo00392a016 [DOI] [Google Scholar]
- 12. Du L., Lykkesfeldt J., Olsen C. E., and Halkier B. A. (1995) Involvement of cytochrome P450 in oxime production in glucosinolate biosynthesis as demonstrated by an in vitro microsomal enzyme system isolated from jasmonic acid-induced seedlings of Sinapis alba L. Proc. Natl. Acad. Sci. U.S.A. 92, 12505–12509 10.1073/pnas.92.26.12505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bennett R., Donald A., Dawson G., Hick A., and Wallsgrove R. (1993) Aldoxime-forming microsomal enzyme systems involved in the biosynthesis of glucosinolates in oilseed rape (Brassica napus) leaves. Plant Physiol. 102, 1307–1312 10.1104/pp.102.4.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bennett R. N., Kiddle G., Hick A. J., Dawson G. W., and Wallsgrove R. M. (1996) Distribution and activity of microsomal NADPH-dependent monooxygenases and amino acid decarboxylases in cruciferous and non-cruciferous plants, and their relationship to foliar glucosinolate content. Plant Cell Environ. 19, 801–812 10.1111/j.1365-3040.1996.tb00417.x [DOI] [Google Scholar]
- 15. Kelly W. L., and Townsend C. A. (2005) Mutational Analysis of nocK and nocL in the nocardicin A producer Nocardia uniformis. J. Bacteriol. 187, 739–746 10.1128/JB.187.2.739-746.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cortina N. S., Revermann O., Krug D., and Müller R. (2011) Identification and characterization of the althiomycin biosynthetic gene cluster in Myxococcus xanthus DK897. ChemBioChem 12, 1411–1416 10.1002/cbic.201100154 [DOI] [PubMed] [Google Scholar]
- 17. Liu N., Song L., Liu M., Shang F., Anderson Z., Fox D. J., Challis G. L., and Huang Y. (2016) Unique post-translational oxime formation in the biosynthesis of the azolemycin complex of novel ribosomal peptides from Streptomyces sp. FXJ1.264. Chem. Sci. 7, 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Y., Zhang Q., Li S., Lin Q., Fu P., Zhang G., Zhang H., Shi R., Zhu W., and Zhang C. (2013) Insights into caerulomycin A biosynthesis: A two-component monooxygenase CrmH-catalyzed oxime formation. J. Am. Chem. Soc. 135, 18750–18753 10.1021/ja410513g [DOI] [PubMed] [Google Scholar]
- 19. Ju K. S., Gao J., Doroghazi J. R., Wang K. K., Thibodeaux C. J., Li S., Metzger E., Fudala J., Su J., Zhang J. K., Lee J., Cioni J. P., Evans B. S., Hirota R., Labeda D. P., van der Donk W. A., van der Metcalf W. W. (2015) Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc. Natl. Acad. Sci. U.S.A. 112, 12175–12180 10.1073/pnas.1500873112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cioni J. P., Doroghazi J. R., Ju K. S., Yu X., Evans B. S., Lee J., and Metcalf W. W. (2014) Cyanohydrin phosphonate natural product from Streptomyces regensis. J. Nat. Prod. 77, 243–249 10.1021/np400722m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sugai Y., Katsuyama Y., and Ohnishi Y. (2016) A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat. Chem. Biol. 12, 73–75 10.1038/nchembio.1991 [DOI] [PubMed] [Google Scholar]
- 23. Huang Z., Wang K. A., and van der Donk W. A. (2016) New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 7, 5219–5223 10.1039/C6SC01389A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin J., and Cashman J. R. N-Oxygenation of Phenethylamine to the trans-oxime by adult human liver flavin-containing monooxygenase and retroreduction of phenethylamine hydroxylamine by human liver microsomes. J. Pharmacol. Exp. Ther. 282, 1269–1279 [PubMed] [Google Scholar]
- 25. Walsh C. T., and Wencewicz T. A. (2013) Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 30, 175–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pretsch E., Buhlmann P., and Badertscher M. (2009) Structure Determination of Organic Compounds, 4th Ed., p. 209, Springer-Verlag, Berlin [Google Scholar]
- 27. Pallitsch K., Happl B., and Stieger C. (2017) Determination of the absolute configuration of (-)-hydroxynitrilaphos and related biosynthetic questions. Chem. Eur. J. 23, 15655–15665 10.1002/chem.201702904 [DOI] [PubMed] [Google Scholar]
- 28. Anderson V. E., Weiss P. M., and Cleland W. W. (1984) Reaction intermediate analogs for enolase. Biochemistry. 23, 2779–2786 10.1021/bi00307a038 [DOI] [PubMed] [Google Scholar]
- 29. Tietz J. I., Schwalen C. J., Patel P. S., Maxson T., Blair P. M., Tai H. C., Zakai U. I., and Mitchell D. A. (2017) A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol. 13, 470–478 10.1038/nchembio.2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Metcalf W. W., and van der Donk W. A. (2009) Biosynthesis of phosphonic and phosphinic acid natural products. Annu. Rev. Biochem. 78, 65–94 10.1146/annurev.biochem.78.091707.100215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu X., Doroghazi J. R., Janga S. C., Zhang J. K., Circello B., Griffin B. M., Labeda D. P., and Metcalf W. W. (2013) Diversity and abundance of phosphonate biosynthetic genes in nature. Proc. Natl. Acad. Sci. U.S.A. 110, 20759–20764 10.1073/pnas.1315107110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 33. Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., and Drummond A. (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woodyer R. D., Shao Z., Thomas P. M., Kelleher N. L., Blodgett J. A., Metcalf W. W., van der Donk W. A., and Zhao H. (2006) Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem. Biol. 13, 1171–1182 10.1016/j.chembiol.2006.09.007 [DOI] [PubMed] [Google Scholar]
- 35. van Staalduinen L. M., McSorley F. R., Schiessl K., Séguin J., Wyatt P. B., Hammerschmidt F., Zechel D. L., and Jia Z. (2014) Crystal structure of PhnZ in complex with substrate reveals a di-iron oxygenase mechanism for catabolism of organophosphonates. Proc. Natl. Acad. Sci. U.S.A. 111, 5171–5176 10.1073/pnas.1320039111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 37. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Price M. N., Dehal P. S., and Arkin A. P. (2010) FastTree 2: Approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen C. M., Ye Q. Z., Zhu Z. M., Wanner B. L., and Walsh C. T. (1990) Molecular biology of carbon-phosphorus bond cleavage. Cloning and sequencing of the phn (psiD) genes involved in alkylphosphonate uptake and C-P lyase activity in Escherichia coli B. J. Biol. Chem. 265, 4461–4471 [PubMed] [Google Scholar]
- 40. Garcia I., Vior N. M., González-Sabín J., Braña A. F., Rohr J., Moris F., Méndez C., and Salas J. A. (2013) Engineering the biosynthesis of the polyketide-nonribosomal peptide collismycin A for generation of analogs with neuroprotective activity. Chem. Biol. 20, 1022–1032 10.1016/j.chembiol.2013.06.014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.