

Abstract
Retinol dehydrogenase 11 (RDH11) is a microsomal short-chain dehydrogenase/reductase that recognizes all-trans– and cis–retinoids as substrates and prefers NADPH as a cofactor. Previous work has suggested that RDH11 contributes to the oxidation of 11-cis–retinol to 11-cis–retinaldehyde during the visual cycle in the eye's retinal pigment epithelium. However, the role of RDH11 in metabolism of all-trans–retinoids remains obscure. Here, we report that microsomes isolated from the testes and livers of Rdh11−/− mice fed a regular diet exhibited a 3- and 1.7-fold lower rate of all-trans–retinaldehyde conversion to all-trans–retinol, respectively, than the microsomes of WT littermates. Testes and livers of Rdh11−/− mice fed a vitamin A–deficient diet had ∼35% lower levels of all-trans–retinol than those of WT mice. Furthermore, the conversion of β-carotene to retinol via retinaldehyde as an intermediate appeared to be impaired in the testes of Rdh11−/−/retinol-binding protein 4−/− (Rbp4−/−) mice, which lack circulating holo RBP4 and rely on dietary supplementation with β-carotene for maintenance of their retinoid stores. Together, these results indicate that in mouse testis and liver, RDH11 functions as an all-trans–retinaldehyde reductase essential for the maintenance of physiological levels of all-trans–retinol under reduced vitamin A availability.
Keywords: vitamin A, carotenoid, retinoic acid, retinol, dehydrogenase, reductase, retinaldehyde
Introduction
Retinol dehydrogenase 11 (RDH11)2 is a member of the short-chain dehydrogenase/reductase (SDR) superfamily of proteins that was originally identified at the level of transcript in human prostate (1). The first in vitro biochemical characterization of the recombinant human RDH11 (originally named prostate SDR1 (PSDR1) and also known as retinaldehyde reductase 1 (RalR1)) revealed that this enzyme recognizes as substrates the aldehydes and alcohols derived from vitamin A (2). Specifically, it was found that RDH11 exhibits the highest catalytic efficiency for the reduction of all-trans–retinaldehyde to all-trans–retinol with NADPH as the preferred cofactor. The mouse ortholog of human RDH11 (named SCALD for short-chain aldehyde reductase) was identified as one of the genes regulated by the transcription factor sterol regulatory element-binding protein (SREBP) (3). Although mouse RDH11 shares 85% protein sequence identity with its human ortholog, the two proteins exhibit somewhat different substrate specificity and tissue distribution. Both human and mouse enzymes are highly active as retinaldehyde reductases, but the mouse enzyme can also reduce certain medium chain (>C6) lipid-derived aldehydes such as trans-2-nonenal, nonanal, and cis-6-nonenal (3), whereas human RDH11 exhibits little or no activity toward C9 aldehydes (4). Human RDH11 protein has a widespread tissue distribution, with the highest levels detected in the kidney, followed by testis, liver, jejunum, prostate, lung, brain (caudate nucleus), and spleen (5). In contrast, the mouse RDH11 protein appears to have a more limited tissue distribution, being primarily expressed in the liver and testis, with much lower protein levels present in brain, lung, spleen, and kidney (3). Both human and mouse proteins are also highly expressed in the eye retinal pigment epithelium (6). The latter observation together with the activity of RDH11 toward 11-cis–retinoids prompted a speculation that RDH11 might play a role in regeneration of retinoids during the visual cycle (6–8). However, analysis of the visual cycle in RDH11 knockout mice produced somewhat ambiguous results, with one study reporting that RDH11 has a measurable role in regenerating the visual pigment by complementing RDH5 as an 11-cis–RDH in RPE cells (7), and another claiming that the kinetics of 11-cis–retinaldehyde recycling during dark adaptation was not affected and thus, RDH11 is not involved in the visual cycle (8). Previous analysis has also shown that Rdh11 is expressed during mouse embryonic development but RDH11-null mice do not exhibit any consistent abnormalities in development, postnatal survival, or fertility when maintained on chow diet (9). The putative function of RDH11 in the reduction of lipid-derived aldehydes has not been supported by the currently available data (10). Thus, to date, the physiological role of RDH11 remains largely unknown.
Previously, we proposed that the retinaldehyde reductase activity of RDH11 could contribute to the conversion of dietary β-carotene to retinol via all-trans–retinaldehyde as intermediate (2). In the intestinal mucosa, the dietary β-carotene is cleaved into two molecules of all-trans–retinaldehyde by the β-carotene oxygenase type 1 (BCO1) (11, 12). All-trans–retinaldehyde is then reduced to all-trans–retinol by a yet unidentified microsomal retinaldehyde reductase activity (13). Retinol produced from retinaldehyde is esterified by lecithin–retinol acyltransferase (LRAT) and incorporated into the nascent chylomicrons (14). The retinyl esters associated with chylomicron remnants are cleared into hepatocytes where they are hydrolyzed and re-esterified for storage (14). It is generally believed that retinol secreted from the liver bound to plasma retinol-binding protein 4 (RBP4) serves as the major source of retinoids for peripheral tissues (15). However, there is growing evidence that a variety of different cell types are capable of utilizing β-carotene directly, supplementing their own retinoid stores, whereas the serum levels of retinol remain unchanged. For instance, in addition to the small intestine, β-carotene is converted to retinol in the liver (16, 17), human colon cancer cells (18), and human lung (19) and skin fibroblasts (20). In fact, human liver was shown to have 4 times the capacity for metabolizing β-carotene than the small intestine (16). Besides liver and intestine, the transcript encoding BCO1 is found in many other tissues such as kidney, brain, stomach, lung, testis, and prostate, suggesting that the dietary β-carotene can be absorbed and metabolized to retinol at multiple sites in the body, providing a tissue-specific vitamin A supply (21).
In humans about half of the dietary provitamin A carotenoids are converted to retinol in the intestine and about half are absorbed intact and can reach peripheral tissues (reviewed in Ref. 13). As described in Ref. 22, mice and rats efficiently convert β-carotene to vitamin A but absorb carotenoids intact only when they are provided in the diet at supraphysiologic levels. In mice, the mRNA encoding BCO1 is detectable in the intestine, liver, kidney, and testis (12, 23, 24), suggesting that mice might be able to supplement their tissue stores of vitamin A from β-carotene, at least to some extent. Currently, the data on the conversion of β-carotene to retinol in mouse tissues are very limited. Furthermore, the molecular identity of the enzyme involved in the reduction of retinaldehyde generated by BCO1 to retinol in the intestine and extraintestinal tissues remains obscure. RDH11 is co-expressed with BCO1 in several human and mouse tissues; and the retinaldehyde reductase activity of RDH11 is conserved in both human and mouse enzymes (3). Here, we used a combination of dietary approaches and gene knockout mouse models to investigate the role of RDH11 in retinoid metabolism in general, and in the conversion of β-carotene to retinol specifically.
Results
RDH11 contributes to the retinaldehyde reductase activity of mouse tissues and cells
Western blot analysis of microsomal fractions isolated from mouse tissues showed that RDH11 protein was most abundant in testis microsomes, with lower levels detectable in microsomes from liver, lung, and intestine (Fig. 1A). Although the protein levels of RDH11 in testis, liver, and lung microsomes were comparable across different animals (Fig. 1B), the amount of RDH11 protein in intestinal microsomes varied in samples from individual animals (Fig. 1, A and B). In agreement with the previous report (3), overnight starvation resulted in a decrease in the amount of RDH11 in livers of fasted mice (Fig. 1C).
Figure 1.

Distribution of RDH11 in mouse tissues. A, comparison of RDH11 protein levels in microsomes from various tissues. Samples of tissues from two individual animals were used. Immunoblotting was performed using 1:2,500 dilution of SCALD antibodies (3). Considering that it is difficult to find a reference protein that is expressed at equal levels in different tissues, we used loading based on protein amount (20 μg from each tissue) to compare RDH11 protein levels across tissues. In addition, staining for GAPDH was used as an independent loading control, with understanding that GAPDH levels can vary somewhat among tissues. GAPDH antibodies were from Sigma (catalog number G9545, lot 031M4817). B, analysis of RDH11 interindividual variability. Microsomes from tissues of three individual animals were analyzed for RDH11 knockout (KO) and wildtype (WT) mice. Note the absence of RDH11 protein in tissues from Rdh11−/− mice. Thirty μg of microsomal protein per lane was loaded for testis, and 50 μg for other tissues. For WT mice, the RDH11 protein band was identified using recombinant RDH11 expressed in Sf9 microsomes (10 μg) as a standard (not shown). For microsomes from the same tissue, CYP450 reductase (P450 red) was used a loading control. CYP450 reductase antibodies (1:2,500 dilution for testis and liver and 1:4,000 dilution for intestine and lung) were from Chemicon (catalog number AB1257, lot 23050777). The star indicates nonspecific bands recognized by RDH11 antibodies. Note the variability of RDH11 protein levels in the intestine (duodenum plus jejunum). C, changes in RDH11 expression in liver of WT mice after overnight fasting. Fifty μg of microsomal protein was loaded per lane; β-actin antibodies were from Abcam (catalog number ab8227, lot 951945).
To further define the localization of RDH11 in liver, we isolated primary hepatocytes and hepatic stellate cells (HSCs) and examined the expression pattern of Rdh11 transcript in these distinct populations of cells using Q-PCR (Fig. 2). To ascertain the purity of each fraction, the Rbp4 transcript was used as the marker of primary hepatocytes, and Lrat as the marker of HSCs. Q-PCR analysis showed that Rdh11 was abundantly expressed in hepatocytes, but was also present in HSCs, where it could support the LRAT activity by generating its substrate, retinol.
Figure 2.
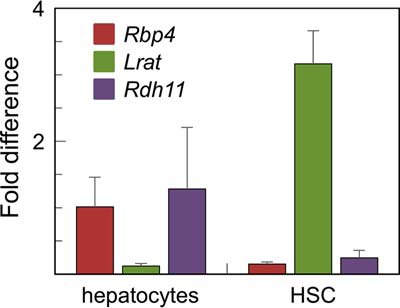
Expression of RDH11 in hepatocytes and HSCs. The purity of primary hepatocytes and HSCs was established using Rbp4 as the marker of hepatocytes, and Lrat as the marker of HSCs. The levels of Rdh11 were determined by Q-PCR. Similar results were obtained when normalizing Rdh11 expression per Gapdh, actin, or 18S RNA.
Activity assays with retinaldehyde and NADPH showed that the rate of retinaldehyde reduction to retinol by the microsomes isolated from RDH11-null testis was 3-fold lower compared with WT testis microsomes. At the same time, the rate of retinol oxidation to retinaldehyde in the presence of NAD+ was unchanged (Fig. 3A, inset), indicating that the oxidative NAD+-dependent retinol dehydrogenase activity was not affected by inactivation of the Rdh11 gene. Similarly to testis microsomes, liver microsomes lacking RDH11 showed a lower rate (1.7-fold) of retinaldehyde reduction (Fig. 3A). No differences were observed in the microsomal retinaldehyde reductase activities from livers of male versus female mice (data not shown).
Figure 3.

Analysis of the retinaldehyde reductase activity in tissues of RDH11 knockout (KO) mice versus wildtype (WT) littermates. A, the reduction of all-trans–retinaldehyde to all-trans–retinol by tissue microsomal fractions. Microsomes from testis (10 μg), liver (6 μg), lung (10 μg), and intestine (60 μg) were incubated with retinaldehyde (0.5 μm) and NADPH (1 mm) for 15 min, and the products of retinaldehyde metabolism were analyzed by normal-phase HPLC. Values represent mean ± S.D. of samples from three individual animals; **, p < 0.001. Inset, because testis microsomes showed the greatest difference in the rate of retinaldehyde reduction to retinol between WT and KO mice, testes microsomes (40 μg) were also incubated with all-trans–retinol (10 μm) in the presence of NAD+ (1 mm) to assess their retinol dehydrogenase activity. Note that the rate of retinol oxidation was ∼10-fold lower than the rate of retinaldehyde reduction, and there was no statistically significant difference (p = 0.28) between the rates of WT microsomes (0.039 ± 0.019 nmol × min−1 × mg−1, n = 5) versus KO microsomes (0.022 ± 0.018 nmol × min−1 × mg−1, n = 3). B, metabolism of retinaldehyde in living MEFs isolated from RDH11-null or WT mouse embryos. Freshly prepared E14.5 MEFs were seeded into 6-well plates and allowed to attach. On the next day, the cells were incubated with 5 μm all-trans–retinaldehyde for 1.5 h, and harvested for normal-phase HPLC analysis. Values represent mean ± S.D. of samples from three individual animals each performed in triplicates; **, p = 0.0009.
In lungs and intestines, the microsomal retinaldehyde reductase activities were comparable between RDH11-null mice and their WT littermates. Interestingly, the intestinal microsomes produced two products within the short 15-min incubations with retinaldehyde: retinol and retinyl esters. This suggested that, in the intestinal microsomes, the retinaldehyde reductase activity was coordinated with the retinol esterifying activity, possibly to ensure a highly efficient processing of retinaldehyde into retinyl esters for packaging into chylomicrons. The relative amounts of retinyl esters and retinol produced by the microsomal fractions in vitro ranged from 1:1 to 4:1, apparently reflecting the variability in the expression levels of the retinaldehyde reductase and LRAT in intestinal microsomes from individual animals. The overall rate of retinaldehyde conversion to retinol and retinyl esters exhibited high interindividual variability and did not correlate with the amount of RDH11 protein in these samples, which also varied widely (Fig. 1B). Taken together, these results indicated that RDH11 contributes to the conversion of retinaldehyde to retinol in liver and testis, but does not appear to be the major retinaldehyde reductase in the microsomes from intestine or lungs.
To determine whether native RDH11 plays a role in endogenous conversion of retinaldehyde to retinol in whole cells, we isolated mouse embryonic fibroblasts (MEFs) from E13–E14 Rdh11−/− embryos and their WT littermates and incubated the living MEFs with retinaldehyde. HPLC analyses consistently showed that the conversion of retinaldehyde to retinol in whole MEFs lacking RDH11 occurred at a slower rate than in WT MEFs (Fig. 3B). The conversion of retinaldehyde to retinoic acid was unaffected under these experimental conditions, because retinaldehyde was added in excess and retinaldehyde dehydrogenase did not have to compete with RDH11 for retinaldehyde.
RDH11 is essential for the maintenance of retinol levels in testis of mice on β-carotene diet
Retinaldehyde can be produced in the cells by the oxidation of retinol or by the cleavage of β-carotene at its central double bond (15,15′) catalyzed by cytosolic BCO1. In rodents, cleavage of β-carotene to retinaldehyde with subsequent conversion of retinaldehyde to retinol occurs mainly in the small intestine. In vitro activity assays suggested that the absence of RDH11 in the intestinal microsomes did not affect their retinaldehyde reductive activities, which varied over a wide range of values (Fig. 3A). To exclude the possibility that the enzymes were partially degraded during isolation of intestinal microsomes, we proceeded to perform in vivo studies on RDH11-null mice. To test whether RDH11 contributes to the reduction of retinaldehyde derived from the symmetrical cleavage of β-carotene in the small intestine, we placed a cohort of Rdh11+/− breeders on a vitamin A–deficient diet supplemented with β-carotene (BC diet) as the only source of dietary vitamin A. Rdh11+/+ and Rdh11−/− pups were placed on this diet after weaning. At 2 months of age, the mice were sacrificed and their tissues were collected for analyses.
Serum levels of retinol in mice on BC diet were comparable with those in fed mice on chow diet; however, the levels of retinol and retinyl esters in liver, testes, and lungs were dramatically reduced in mice on BC diet compared with chow diet (Table 1). No significant differences were observed in tissue and serum levels of retinoids between RDH11-null and WT mice (Table 1). Similarly, there was no difference in the kinetics of retinaldehyde conversion to retinol (Fig. 4A) or retinoic acid (Fig. 4B) after administration of retinaldehyde by gavage as indicated by HPLC analysis of serum and liver (Fig. 4C) retinoids in Rdh11−/− and WT male or female mice. These results indicated that RDH11 did not contribute to in vivo conversion of retinaldehyde to retinol in the small intestine, in agreement with the lack of differences in the microsomal retinaldehyde reductase activities of intestines from RDH11-null mice and their WT littermates.
Table 1.
Retinoid levels in tissues of Rdh11−/− and Rdh11−/−;Rbp4−/− mice versus their WT and Rbp4−/− littermates on β-carotene diet
For tissues, all values are expressed in nanogram/g wet weight; serum values are in nanogarm/ml. The concentration of β-carotene in serum of mice on β-carotene diet was 0.23 ± 0.2 nmol/ml. The same number of animals were used for measurements of retinol as indicated for retinyl esters. Animals were not fasted.

Figure 4.
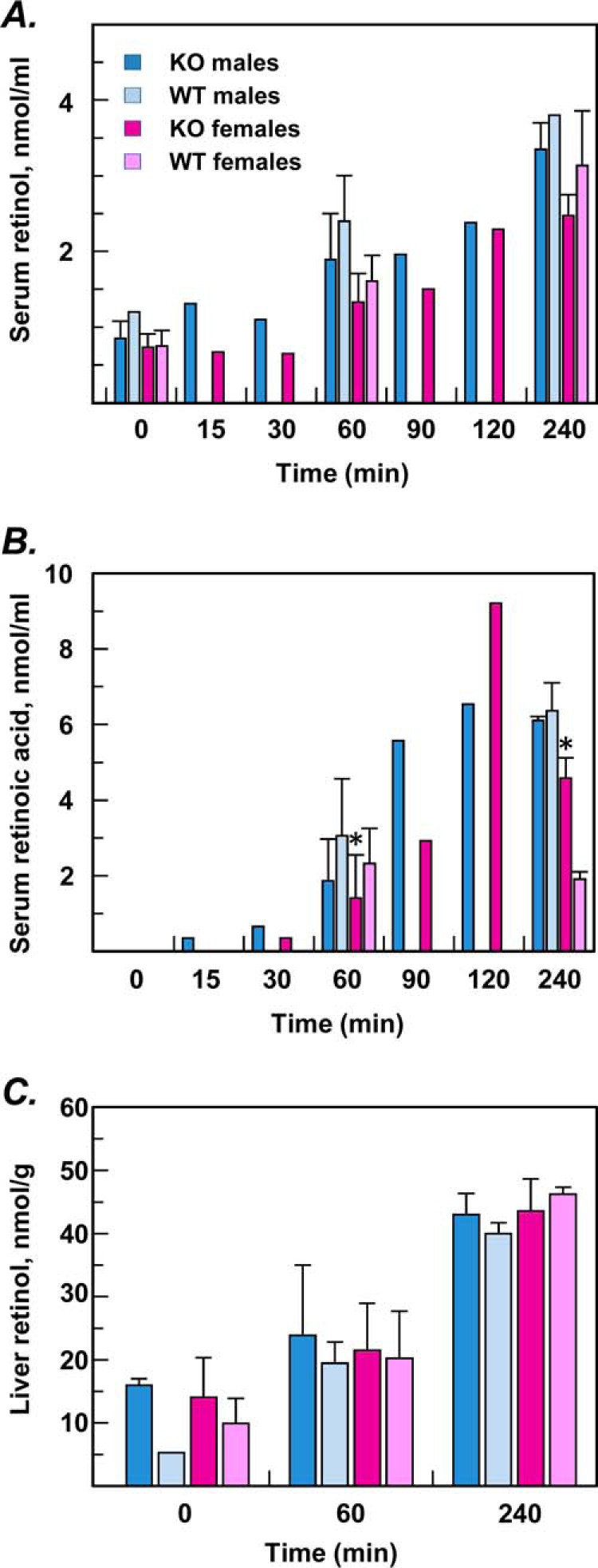
Metabolism of gavaged all-trans–retinaldehyde in RDH11 knockout (KO) mice versus wildtype (WT) littermates. Retinaldehyde (30 mg/kg body weight in corn oil:ethanol (85:15) was administered by gavage. Retinoids were extracted from serum collected at the indicated time points after gavage and analyzed by reversed-phase HPLC. Zero time point values reflect baseline retinoid levels after administration of vehicle. A–C, several animals were analyzed at 0-, 60-, and 240-min time points: KO males, n = 10, WT males, n = 6; KO females, n = 4–16, WT females, n = 3–12. Single animals were used for additional time points. A statistically significant decrease in retinoic acid levels was observed in serum of KO females relative to WT females at 60 min after gavage (*, p = 0.033; n = 16 (KO), n = 12 (WT)). However, at 240 min, serum retinoic acid levels were higher in KO mice (*, p = 0.0005; n = 4 (KO), n = 3 (WT)) (B). No significant differences were observed in serum retinoids of males as well as in liver retinol in males or females (C).
Although in rodents the bulk of dietary β-carotene is processed in the small intestine, BCO1 is also expressed in several extraintestinal tissues (12, 23, 24). To determine whether RDH11 contributes to the reduction of retinaldehyde derived from β-carotene in extraintestinal tissues, we generated a new mouse model by crossing Rdh11+/− mice with mice lacking RBP4 (15). This approach allowed us to exclude holo RBP4 as the source of retinol for peripheral tissues and to assess the role of RDH11 in the conversion of β-carotene to retinol by extrahepatic tissues. A cohort of Rdh11−/−;Rbp4−/− and Rdh11+/+;Rbp4−/− littermates were placed on BC diet described above. We chose to use a diet containing supraphysiologic levels of β-carotene (1.5 mg/g) to ensure that β-carotene was abundantly available in circulation for uptake by extraintestinal tissues (22). The concentration of β-carotene achieved in serum of mice on this diet was 0.23 ± 0.2 nmol/ml.
As expected, in the absence of holo RBP4, serum retinol in these mice was reduced by ∼36-fold compared with Rdh11−/−;Rbp4+/+ mice on the same diet. The liver stores of retinyl esters in Rdh11−/−;Rbp4−/− mice were elevated compared with Rdh11−/−;Rbp4+/+ mice on BC diet (Table 1). This observation was consistent with genetic ablation of Rbp4, which prevented the export of retinol from the liver to extrahepatic tissues. In the testis of Rdh11−/−;Rbp4+/+ mice on BC diet, the absence of holo RBP4 resulted in a 3–5-fold reduction in retinol and retinyl esters, suggesting that the conversion of β-carotene to retinol in testis was generally inefficient. However, the testes of Rdh11−/−;Rbp4−/− mice contained even lower levels of retinol (by ∼35%, p = 0.03) than the testes of their Rdh11+/+;Rbp4−/− littermates (Table 1). Considering that there was no difference in circulating serum retinol in these mice, this finding suggested that the absence of RDH11 impeded the generation of retinol via retinaldehyde from dietary β-carotene in testes.
RDH11 is essential for the maintenance of retinol levels in liver and testis of mice during dietary vitamin A deficiency
Experiments with mice on a BC diet suggested that the contribution of RDH11 to the conversion of retinaldehyde to retinol becomes evident when tissue stores of vitamin A are somewhat depleted because of inefficient utilization of β-carotene. To obtain further evidence that RDH11 is essential during vitamin A deficiency, we generated mice with partial depletion of vitamin A stores by placing a cohort of Rdh11−/− and Rdh11+/+ mice on a vitamin A–deficient (VAD) diet at 4 weeks of age and examined their serum and tissue retinoid levels by HPLC after 16 weeks (vitamin A reduced). This analysis showed that serum retinol levels in these mice were similar to mice on chow diet (Table 2). However, the liver stores of retinol and retinyl esters were reduced by at least 5- and 14-fold, respectively. In testis, the retinyl ester stores were unchanged, whereas retinol was reduced by ∼2-fold. Interestingly, even at this partial depletion of retinol in testis, there was an observable trend toward lower retinol levels in testis of Rdh11−/− mice versus Rdh11+/+ mice, although it did not quite reach statistical significance (p = 0.06).
Table 2.
Retinoid levels in tissues of Rdh11−/− and WT littermates during dietary vitamin A depletion
For tissues, all values are expressed in nanomole/g wet weight; serum values are in nanogram/ml. No retinyl esters were detected in serum, testis, liver, or lungs in mice on VAD diet. N.M., not measured. The same numbers of animals were used for measurements of retinol as indicated for retinyl esters. Animals were not fasted.

To achieve a more profound depletion of vitamin A stores, pregnant dams that were maintained on β-carotene diet for several months prior to pregnancy were placed on VAD diet midgestation. Rdh11−/− and Rdh11+/+ pups born from these dams were placed on a VAD diet after weaning and sacrificed 8 weeks later. HPLC analysis revealed that serum retinol in these mice was very low (∼0.05 nmol/ml) (Table 2) and comparable with that in Rbp4−/− mice on BC diet (Table 1). The retinol content in livers of mice on VAD diet was severely reduced compared with livers of mice on chow diet (∼100-fold) and BC diet (∼40-fold), and retinyl esters were undetectable. Importantly, livers of Rdh11−/− mice had ∼35% less retinol (p = 0.004) than livers of WT littermates. There was also a trend toward lower retinoic acid levels (7.87 ± 2.3 versus 10.44 ± 3.1 pmol/g in WT livers), with the p value of 0.05 (n = 10 for each group).
Notably, the testis of Rdh11−/− mice on a VAD diet also had ∼35% lower retinol (p = 0.007) than testis of Rdh11+/+ mice (Table 2). Thus, two different mouse models on two different dietary regiments consistently showed a reduction of steady-state levels of retinol in tissues of vitamin A-depleted mice in the absence of RDH11.
Gene expression pattern indicates a mild reduction in retinoic acid signaling in RDH11-null testis
The results described above suggested that RDH11 contributes to the maintenance of the retinol pool in testis and liver. Considering that the decrease in retinol levels in liver and testis of RDH11-null mice was relatively mild (∼35%), we wanted to identify the most sensitive markers of vitamin A status in these tissues to determine whether the mild decrease in retinol affected the retinoic acid signaling. Q-PCR analysis showed that in livers of mice on a VAD diet, the most down-regulated genes were Cyp26a1, Cyp26b1, and Lrat, followed by Rarβ (Fig. 5A). Interestingly, Bco1 and Sr-bI, the genes encoding the transporter of β-carotene (25), were both up-regulated in livers of VAD mice (Fig. 5A). In livers of mice on β-carotene diet, which had lower vitamin A stores than mice on chow diet, the most significant changes occurred in the expression of genes encoding CRBPI and BCO1, both of which were up-regulated, and in the Cyp26a1 gene, which was strongly down-regulated (Fig. 5A). The down-regulation of Cyp26a1 suggested reduced retinoic acid signaling, in agreement with the 5-fold lower levels of retinol in the livers of mice on BC diet versus chow diet (Table 1).
Figure 5.
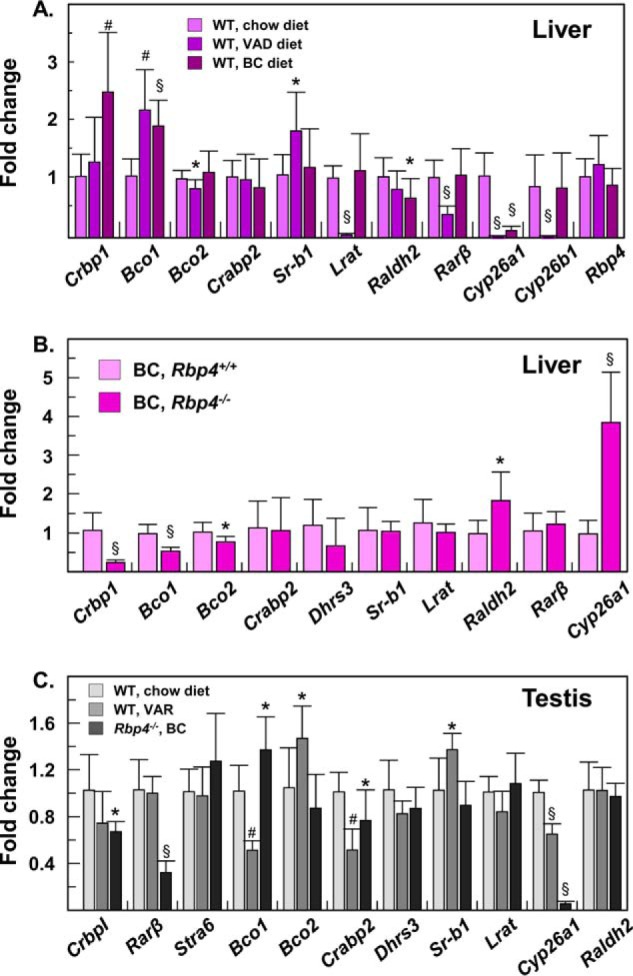
Effect of diets on expression levels of retinoic acid target genes. A, gene expression was compared in livers of WT mice maintained on chow diet (n = 9) versus WT mice maintained on a VAD diet (n = 10) or BC diet (n = 14). All mice had the same 129S4 genetic background. Note the increase in expression of Crbp1, Bco1, and Sr-b1, and a significant decrease in expression of Lrat, Rarβ, and Cyp26a1. B, gene expression was compared in livers of WT mice (129S4) on BC diet (n = 13) versus Rbp4−/− mice (mixed 129S4/C57) on BC diet (n = 8). Note that accumulation of retinoids in livers of Rbp4−/− mice results in a decreased expression of Crbp1, Bco1, and Bco2, but an increase in expression of Raldh2 and Cyp26a1. C, gene expression was compared in testes of WT mice on chow diet (n = 10) to that in testes of WT mice with reduced VA stores in liver (VAR) (n = 6), and Rbp4−/− mice on BC diet (n = 8). WT mice were on a 129S4 background and Rbp4−/− mice were on a 129S4/C57 mixed background. Note the reduced expression of Rarβ and Cyp26a1 in testes of Rbp4−/− mice on a BC diet. Values represent mean ± S.D.; *, p < 0.05; #, p < 0.001; §, p < 0.0001.
Remarkably, this expression pattern was reversed in the livers of RBP4-null mice on a BC diet (Fig. 5B), which had elevated levels of retinoids. The expression of genes encoding CRBPI and BCO1 was reduced, whereas Cyp26a1 expression was up-regulated by ∼4-fold. To determine whether the protein levels correlated with transcript levels, we utilized available CRBPI antibodies to perform side-by-side Western blot analysis of CRBPI expression in livers from Rbp4−/− mice on a BC diet, WT mice on BC diet, and WT mice on chow diet. As shown in Fig. 6A, CRBPI protein was reduced in livers of Rbp4−/− mice on the BC diet (high liver retinoids), but increased in livers of WT mice on the BC diet (low liver retinoids) relative to livers of WT mice on chow diet. Furthermore, two independent human HepG2 cell lines with stably silenced DHRS3, which have elevated steady-state levels of retinoic acid and enhanced retinoic acid signaling (26), also had reduced CRBPI protein (Fig. 6, B and C). These experiments confirmed that transcript levels of CRBPI in liver correlate with protein levels, and suggested that CRBPI expression in the liver is suppressed by chronically elevated retinoic acid.
Figure 6.

Western blot analysis of CRBPI expression in mouse livers and human HepG2 cells. The soluble fractions (105,000 g of supernatant) from (A) livers (50 μg each) or (B) total lysates of HepG2 cells (50 μg each) were subjected to immunoblotting with CRBPI antibodies (1:1,000 dilution, custom made by Cocalico Biologicals). Loading was normalized per GAPDH (A) or β-actin (B). DHRS3 antibodies (1:2,000 dilution) were from Proteintech. GAPDH (Ambion) and β-actin (Abcam) antibodies were each used at a 1:5,000 dilution. The intensity of bands was quantified by UN-SCANIT software (C).
In testis, the gene expression pattern was compared for mice on a chow diet to those with partial depletion of vitamin A stores (vitamin A reduced) and mice on β-carotene diet (Fig. 5C). Q-PCR analysis showed that, Cyp26a1 gene expression was again the most sensitive to vitamin A levels, followed by Rarβ and Crabp2 (illustrated in Fig. 7).
Figure 7.
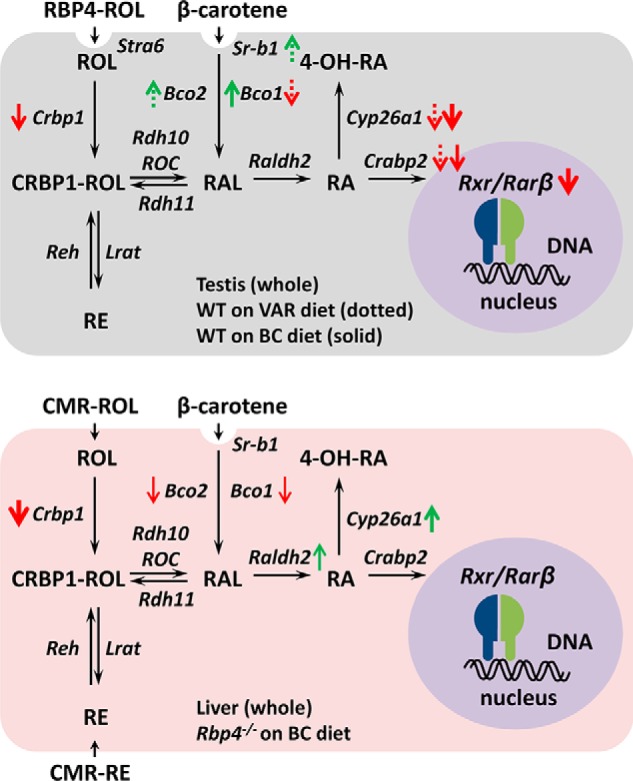
Gene expression changes in WT and RBP4-null mice on VAR or BC diets. Testis (upper), note that expression of retinoic acid (RA)-inducible genes, Cyp26a1 and Crabp2, was lower in mouse testis of mice on either VAR or BC diets, suggesting that the conversion of BC to retinoic acid in mouse testis is inefficient. At the same time, expression of genes encoding proteins involved in uptake and processing of β-carotene (Bco2 and Sr-b1, but not Bco1) was up-regulated on VAR, whereas Bco1 was up-regulated in testis of mice on a BC diet, possibly to maximize the uptake of carotenoids as the source of vitamin A. Liver (lower), in the liver of RBP4-null mice on a BC diet, expression of Cyp26a1 was up-regulated, suggesting increased production of retinoic acid from elevated liver retinoid stores. At the same time, expression of Crbp1, which is responsible for uptake of retinol, and Bco1 and Bco2, the enzymes that cleave β-carotene, was reduced, possibly to prevent further increase in cellular retinoids. The thickness of arrows reflects the magnitude of changes. ROC stands for retinoid oxidoreductase complex, which includes both RDH10 and DHRS3; CMR, chylomicron remnants.
Having identified the most sensitive marker genes, we compared their expression levels in livers and testes of mice that lacked RDH11. This analysis demonstrated that, in the liver, most of the marker genes were unaffected by the 35% reduction in liver retinol. On the other hand, in testis of Rdh11−/−;Rbp4−/− mice (n = 8) the expression level of Cyp26a1 was ∼20% lower (p = 0.038) than in Rdh11+/+;Rbp4−/− mice (n = 8). Thus, retinoic acid signaling appeared to be somewhat reduced in RDH11-deficient testis. Of note, we observed that Rdh11+/− mice did not breed well on a diet containing 4 IU of vitamin A/g of diet and had to be switched back to 25 IU/g of vitamin A diet to maintain the colony.
Discussion
This study presents the first evidence that RDH11 functions as an all-trans–retinaldehyde reductase in vivo and is essential for the maintenance of all-trans–retinol steady-state levels in mouse liver and testis. The role of RDH11 as an all-trans–retinaldehyde reductase is supported by experiments in two different mouse models on two different dietary regiments. In testis of Rdh11+/+;Rbp4−/− mice that receive β-carotene as the sole source of vitamin A, the levels of retinol are ∼35% lower than in testis of their WT littermates. Similar reduction in the testis retinol is also observed in the Rdh11−/− mouse model, in which vitamin A deficiency is achieved by placing mice on VAD diet. In liver, the role of RDH11 is evident in Rdh11−/− mice fed the VAD diet. The livers of these mice have one-third less retinol than livers of WT littermates. Thus, RDH11 appears to be critical for the maintenance of physiological levels of retinol under the conditions of dietary vitamin A deficiency. The role of RDH11 in promoting the biosynthesis of retinol, a polyunsaturated lipid, is consistent with up-regulation of its expression by SREBP-1c (3). The amount of SREBP-1c declines with fasting and rises with refeeding, to promote the NADPH-dependent biosynthesis of fatty acids and triglycerides. Thus, it is fitting that RDH11, an NADPH-dependent enzyme up-regulated by SREBP-1c during refeeding, contributes to biosynthesis of retinol rather than oxidation of retinol.
Our data also support the notion that mouse extraintestinal tissues can utilize β-carotene as a source of vitamin A and that RDH11 contributes to this conversion. For example, the circulating levels of retinol in serum of Rbp4−/− mice on BC diet (∼0.02 nmol/ml) are ∼40-fold lower than in serum of Rbp4+/+ mice on BC diet (∼0.8 nmol/ml). One would expect that at such low levels of retinol in serum, peripheral tissues are depleted of retinoids. However, retinyl esters in testis of Rbp4−/− mice on BC diet (∼0.2 nmol/g wet weight) are only 3-fold lower than in testis of Rbp4+/+ mice on the same diet (∼0.6 nmol/g wet weight) (Table 1). This suggests that testis is able to utilize the circulating dietary β-carotene to supplement its local retinoid stores. As another example, Rbp4+/+ mice on VAD diet have 2-fold higher serum retinol (∼0.04 nmol/ml) than Rbp4−/− mice on BC diet (∼0.02 nmol/ml) (Table 2), but their testis has no detectable retinyl esters. In contrast, testis of Rbp4−/− mice on BC diet contains measurable retinyl esters (∼0.2 nmol/g wet weight) (Table 1). Thus, retinyl esters present in the testis of RBP4-null mice must be generated from the β-carotene taken up from serum. The role of RDH11 in the conversion of β-carotene to retinol in testis is supported by the observation that Rdh11−/−;Rbp4−/− mice on BC diet contain serum retinol levels similar to control mice, but their testis retinol levels are ∼35% lower than in control mice.
A new finding of this study is that in the liver vitamin A deficiency results in increased expression of Bco1 and Sr-b1, the gene encoding the β-carotene transporter, which might imply a coordinated increase in uptake and processing of β-carotene in the liver. The sensitivity of Bco1 expression to retinoic acid levels in the liver is also supported by the fact that Bco1 transcript levels in livers of Rbp4−/− mice on BC diet (high retinoid stores) are lower than in livers of Rbp4+/+ mice on BC diet (lower retinoid stores). It is well-established that Bco1 is highly sensitive to vitamin A status in the intestine, and that the mechanism of molecular regulation of Bco1 involves the retinoic acid-dependent induction of the intestinal transcription factor, ISX, and the subsequent ISX-induced repression of Bco1 expression (27). The mechanism of Bco1 and Sr-b1 regulation in the liver remains to be elucidated.
We have also observed that CRBPI expression is decreased at both transcript level and protein level in livers of Rbp4−/− mice on BC diet compared with livers of Rbp4+/+ mice on BC diet (illustrated in Fig. 7). Because the livers of Rbp4−/− mice have 4-fold higher levels of Cyp26a1 transcript, this may be taken to indicate that retinoic acid signaling is increased in Rbp4−/− livers, and that the chronically increased retinoic acid signaling suppresses CRBPI expression in Rbp4−/− livers. Similarly, CRBPI expression is reduced in human HepG2 cells that have chronically enhanced retinoic acid signaling due to silencing of dehydrogenase reductase 3 (DHRS3) (26). Previous studies demonstrated that CRBPI gene expression can be induced in some tissues by treatment with retinoic acid (28–30). The gene encoding CRBPI was shown to contain a retinoic acid response element (31, 32). At the same time, CRBPI levels did not change significantly in livers of rats that were fed a retinoid-deficient diet and were then supplemented with retinoic acid (28, 33), and liver CRBPI levels were also quite stable in retinoid-deficient compared with retinol-repleted rats (29). We speculate that the suppression of CRBPI expression observed in Rbp4−/− livers is due to sustained exposure of the liver cells to elevated levels of retinoic acid as opposed to transient treatments with retinoic acid. The down-regulation of CRBPI in liver loaded with retinoids seems like a reasonable mechanism to prevent further uptake of retinol. At this point, it is unclear whether the changes in CRBPI expression occur in hepatocytes or in stellate cells because both types of liver cells express CRBPI (34–36).
In mouse testis, RDH11 was reported to show a highly restricted pattern of expression in pachytene spermatocytes, but not in progenitor spermatogonia or in mature sperm (Ref. 3, and Table S1 in Ref. 37). Vitamin A is required for spermatogonia differentiation and treatment of vitamin A–deficient males with either retinol or retinoic acid results in the complete recovery of spermatogenesis (38). The presence of RDH11 all-trans–retinaldehyde reductase in pachytene spermatocytes may be necessary for the maintenance of sufficient levels of retinol at this stage during differentiation. Coincidentally, pachytene spermatocytes also robustly express retinaldehyde dehydrogenase Aldh1a2 (Raldh2) mRNA, the enzyme that converts retinaldehyde to retinoic acid (39). Thus, as proposed recently (40, 41), pachytene spermatocytes might serve as a source of retinoic acid. The identity of the retinol dehydrogenase that catalyzes the rate-limiting step in retinoic acid biosynthesis, the oxidation of retinol to retinaldehyde in pachytene spermatocytes, remains to be established.
Finally, it is important to note that the role of RDH11 in all-trans–retinoid metabolism was not immediately evident, because RDH11-null mice are viable and breed normally when maintained on 24 IU/g of vitamin A. In contrast, a gene knockout of DHRS3 (SDR16C9 in mice, SDR16C1 in humans) (26), a retinaldehyde reductase that belongs to a different branch of SDR superfamily than RDH11 (SDR7C9 in mice, SDR7C1 in humans), results in late embryonic lethality, consistent with the critical role of this enzyme in retinoid metabolism. However, as suggested by our recent study (42), the primary function of DHRS3 is to control the activity of retinol dehydrogenase 10 (RDH10) in the retinoid oxidoreductase complex composed of RDH10 and DHRS3 rather than to serve as a general purpose retinaldehyde reductase, because DHRS3 is inactive in the absence of RDH10. Here, we demonstrate that, in contrast to DHRS3, RDH11 acts as an independent all-trans–retinaldehyde reductase the function of which is to maintain the levels of retinol rather than to control the rate of retinoic acid biosynthesis as does DHRS3. RDH11 is responsible for about two-thirds of the retinaldehyde reductase activity of microsomes isolated from testis and about one-third of microsomal activity of the liver. On the other hand, RDH11 does not play a major role in the retinaldehyde reductive activities of lung and intestinal microsomes. As shown by our previous studies (4, 43, 44), RDH11 is only one of a group of phylogenetically related SDRs with the NADPH-dependent retinaldehyde reductase activities. Besides RDH11, this group of enzymes includes RDH12 (4), which exhibits a photoreceptor-specific expression pattern (6), and two other members, the mitochondrial low-efficiency RDH13, which is abundantly expressed in human kidney, heart, and lung (43), and the microsomal RDH14 (also known as PAN2) (44), which has ubiquitous tissue distribution and a higher catalytic efficiency than RDH11. The two latter enzymes could be responsible for the reduction of retinaldehyde to retinol in the intestine and lungs. The discovery that DHRS3 (together with RDH10) is involved primarily in the control of retinoic acid homeostasis, whereas RDH11 appears to be required for homeostasis of retinol in testis and liver advances our understanding of the molecular mechanisms responsible for the overall retinoid homeostasis. Future studies will likely uncover additional retinaldehyde reductases and retinol dehydrogenases that are critical for fine-tuning retinol and retinoic acid levels in a tissue-specific manner.
Experimental procedures
Animals
Rdh11−/− mice were generated by deleting exons 2 and 3 through homologous recombination as described previously (7). Rbp4−/− mice were a generous gift of Dr. William Blaner at Columbia University. Rdh11−/−;Rbp4−/− and Rdh11+/+;Rbp4−/− littermates used for experiments were obtained from Rdh11−/+;Rbp4−/− parents, which were derived by cross-breeding Rdh11−/− and Rbp4−/− animals. RBP4-null phenotype was confirmed using rabbit anti-mouse RBP antiserum provided by Dr. Blaner. Genotyping for Rdh11 was carried out using primers mPSDR_4897, 5′-ACT ATG GCG TGC ATG TGG AAG T-3′, and mPSDR_5285, 5′-TCT CCT TCC CAA TGC CTG TG-3′, to identify the WT allele and mPSDR_4897 and Neo5207, 5′-GCT AAA GCG CAT GCT CCA GA-3′, to identify the disrupted Rdh11 gene. Mouse strains with RBP4-null background were maintained on a vitamin A-supplemented diet (VAS, 28 IU Vitamin A/g) (catalog number 1813123, TestDiet, St. Louis, MO) unless indicated otherwise. All animal experiments employed procedures approved by the University of Alabama Animal Care Committee, and conformed to recommendations of the American Veterinary Medical Association Panel on Euthanasia.
Diets
To deplete vitamin A storage in experimental mice, Rdh11 heterozygous parents were fed β-carotene-supplemented diet, which was custom made by adding β-carotene (DSM Nutritional Products AG Kaiseraugst, Switzerland) to 15 g/kg of vitamin A-free diet (1.5 mg/g) (TD.08459, Harlan Teklad). After weaning, Rdh11−/− and Rdh11+/+ littermates were placed either on a VAD diet (TD.86143, Harlan Teklad) for 4 weeks or on a β-carotene-supplemented diet for 8 weeks as indicated. Rdh11 heterozygous breeders with Rbp4-deficient genetic background were maintained on VAS diet and their Rdh11−/− and Rdh11+/+ offspring were placed on BC diet after weaning. To achieve partial depletion of vitamin A stores (vitamin A-reduced), Rdh11−/− and Rdh11+/+ littermates fed a chow diet were placed on VAD diet for 16 weeks at 2 months of age.
Isolation of primary hepatocytes and hepatic stellate cells
Mice were anesthetized with a mixture consisting of 1 ml of Ketaset, 0.5 ml of xylazine, and 8.5 ml of PBS given at a dose of 10 μl/g body weight. The abdomen was opened to allow access to the liver. With the organs pulled to the side, a catheter was angled parallel to the mouse's body and was inserted into the vein that connects the kidney to the inferior vena cava.
Primary mouse hepatocytes were isolated through in situ perfusion of the liver with collagenase type IV (0.75 mg/ml, Worthington Biochemical Corp.). The liver was first perfused at a flow rate of 5 ml/min with Hanks' balanced salt solution (without Ca2+) followed by Hanks' balanced salt solution (with Ca2+) containing collagenase, for 5 and 15 min, respectively. Immediately after the first solution began to enter the liver, the portal vein was cut and the suprahepatic inferior vena cava was clamped. After perfusion, the partially digested liver was excised, the digest filtered through 100-μm nylon mesh to remove undigested material, and resuspended in Dulbecco's modified Eagle's medium (DMEM) containing 1% (w/v) penicillin/streptomycin. Isolated hepatocytes were separated from the nonparenchymal cells and debris by centrifugation; twice for 5 min at 4 °C and 20 × g, once for 10 min at 4 °C and 50 × g, and twice again for 5 min at 4 °C and 20 × g. The supernatant was aspirated and the hepatocytes present in the pellet were resuspended in DMEM.
Primary mouse HSC were isolated by in situ liver perfusion with a solution containing Pronase E (EMD Chemicals Inc., Gibbstown, NJ) and collagenase type IV (Worthington Biochemical Corp.). The liver was first perfused at a flow rate of 5 ml/min with a solution containing EGTA, followed by perfusions with solutions containing Pronase E (0.4 mg/ml) and collagenase IV (0.5 mg/ml) solution, for 5 and 8 min, respectively. Immediately after the first solution began to enter the liver, the portal vein was cut and the suprahepatic inferior vena cava was clamped. After perfusion, the partially digested liver was excised, the digest passed through a 100-μm nylon mesh to remove undigested materials, and resuspended in a solution containing Pronase E (0.5 mg/ml), collagenase IV (0.5 mg/ml), and DNase I (2 mg/ml). The suspension was incubated in a beaker placed on a stirring platform for 25 min at 37 °C. HSCs were purified from the remainder of nonparenchymal cells and hepatocyte-derived debris by floatation through 9% (w/v) Nycodenz (Accurate Chemical and Scientific Corp., Westbury, NY) in Gey's balanced salt solution. The yield of cells from each isolation was determined by counting using a hemacytometer.
Microsomal retinaldehyde reductase activity assays
Mouse tissue samples were homogenized on ice using a Dounce homogenizer in PBS with 0.25 m sucrose and protease inhibitors. Microsomal fractions from mouse tissue homogenates were isolated by differential centrifugation essentially as described previously (4), and resuspended in the reaction buffer (40 mm potassium chloride, 90 mm potassium phosphate, pH 7.4) supplemented with 20% (v/v) glycerol, 1 mm DTT, and 0.5 mm EDTA to the final concentration of 1 μg/μl. Six to 60 μg of microsomal protein were incubated with 0.5 μm all-trans–retinaldehyde and 1 mm NADPH for 15 min in a 0.5-ml reaction volume. All-trans–retinaldehyde was solubilized with equimolar BSA prior to the addition (45). Reactions were terminated with 0.5 ml of ice-cold methanol, extracted twice with 2 ml of hexane, and analyzed by normal-phase HPLC at 0.7 ml/min using Spherisorb S3W (Waters, Milford, MA) and isocratic mobile phase consisting of hexane:ethyl acetate (95:5).
Immunoblotting
Samples of microsomal fractions from different tissues (30–50 μg of protein) were separated in 12% SDS-PAGE and analyzed by Western blotting using 1:2,500 dilution of RDH11 (anti-SCALD) polyclonal rabbit antibody (3).
Analyses of retinoid content in tissues
Flash-frozen tissue samples from fed animals were weighed and homogenized in PBS in the dark. Liver samples were homogenized in 0.9 ml. For analysis of retinoic acid, 0.9-ml aliquots of the homogenate were mixed with 1.5 ml of ethanol containing 0.025 n KOH and extracted twice with 5 ml of hexane (46). The organic phase was discarded; aqueous phase was acidified by the addition of 0.045 ml of 4 n HCl and extracted with 5 ml of hexane. The hexane layer was collected, dried, and the residue was dissolved in 0.2 ml of a 80:20 mixture of solvent A (acetonitrile, 2% (v/w) ammonium acetate, glacial acetic acid, methanol, 79:16:3:2) and solvent B (acetonitrile). For analysis of retinol and retinyl esters, two 0.05-ml aliquots of liver homogenates were diluted with 0.3 ml of PBS, mixed with 1 ml of ethanol, and each aliquot was extracted twice with 3 ml of hexane. The dry residue was dissolved in 0.2 ml of a 80:20 mixture of solvent A and solvent B for analysis of retinol, or in a 70:30 mixture of solvent B and dichloromethane for analysis of retinyl esters and β-carotene.
Tissues other than liver were homogenized in 1 ml of PBS and 0.45-ml aliquots were mixed with 1 ml of ethanol. For analysis of retinol and retinoic acid, these tissue samples were extracted with 3 ml of hexane, acidified with 0.045 ml of 4 n HCl, and re-extracted with 3 ml of hexane. Extracts were pooled, dried, and dissolved in 0.2 ml of a 80:20 mixture of solvent A:solvent B. For analysis of retinyl esters and β-carotene, samples were extracted twice with 3 ml of hexane and dry residue was dissolved in 70:30 solvent B:dichloromethane.
For analysis of retinol and retinoic acid, samples were separated using reversed-phase HPLC with SUPELCOSILTM SuplexTM pKb-100 column (Sigma) as a stationary phase and an isocratic mobile phase consisting of solvent A:solvent B (80:20). Retinyl esters and β-carotene were separated by a gradient mobile phase at 0.7 ml/min as follows: 0–15 min, 100% solvent A; 15–16 min, change to 100% solvent B; 16–40 min, 100% solvent B; 40–41 min, change to 100% solvent A; 41–45 min, 100% solvent A.
Separation was performed using Waters Alliance 2695 Separation Module and 2996 Photodiode Array Detector. Retinoids were identified by reference absorbance spectra and co-elution with standards. Absorbance peak areas for each retinoid (extracted at 325 nm for retinol and retinyl esters, 357 nm for retinoic acid, 450 nm for β-carotene, and 367 nm for retinaldehyde in normal-phase or 383 nm in reversed phase) were converted to picomole amounts using linear regression of peak areas obtained by injections of serial dilutions of retinoid standards. The internal standard used for retinoic acid extraction was acitretin, for retinyl esters and retinol-retinyl acetate.
Real-time quantitative PCR
RNA from mouse tissues was extracted with TRIzol (Invitrogen), treated with DNase I (Promega), and re-extracted with TRIzol according to the manufacturer's protocols. Five μg of RNA was reverse-transcribed using Superscript III kit (Invitrogen). Real-time PCR was performed in duplicates for each sample in a LightCycler® 480 instrument (Roche Diagnostics) using LightCycler® 480 SYBR Green I Master Mix (Roche Applied Science) with 0.5-μm primers and 5 μl of 5- or 15-fold dilution of RT reactions in the final volume of 20 μl. Levels of transcripts were determined using a relative quantification method (47) and normalized to the geometric mean of transcript levels of three reference genes (β-Actin, Gapdh, and Hprt). Sequences of the primers are available by request. PCR without cDNA templates did not produce significant amplification products. Specificity of the primers was verified by amplification of a single PCR product, which was determined by observing a single dissociation curve from each tissue.
Total RNA from primary hepatocytes and hepatic stellate cells was isolated using a E.Z.N.A. Total RNA Kit I (Omega Bio-tek) according to the manufacturer's protocol, and quantified at 260 nm using Nanodrop spectrophotometer. For cDNA synthesis, 2 μg of total RNA (in the final volume of 10 μl) was employed. cDNA synthesis was carried out for 10 min at 25 °C and 120 min at 37 °C employing High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The reaction was stopped at 85 °C for 5 min, using thermal cycler (Mastercycler Pro Thermal Cycler, Eppendorf). The primers employed for Q-PCR analyses of target genes are available upon request. Q-PCR was performed in a total volume of 20 μl, including cDNA template, forward and reverse primers (100 nm each), and PerfeCTa SYBR Green FastMix (QuantaBio) using a LightCycler 480 instrument (Roche). After the initial enzyme activation (95 °C for 10 min), 40 cycles (94 °C for 10 s, at 55 °C for 30 s, 72 °C for 30 s) were performed for anneal/extension steps, and fluorescence was measured. A dissociation curve program was performed after each reaction. Relative quantification of target genes was calculated based on the efficiency of each reaction and the crossing point deviation of each sample versus control, and expressed compared with the reference gene (18S RNA).
Isolation of MEFs and activity assays
Embryos isolated from 14.5-day pregnant mice were washed with Hanks' balanced salt solution. The head and visceral tissues were removed from isolated embryos. Fetal tails were saved for genotyping. The remaining bodies were washed in fresh Hanks' balanced salt solution, minced using a pair of scissors, transferred into 5 ml/embryo of 0.25% (w/v) trypsin, 1 mm EDTA solution (Invitrogen, catalog number 25200), and incubated at room temperature for 45 min with gentle shaking to help with tissue dissociation. To neutralize trypsin, 10 ml/embryo of DMEM containing 10% FBS was added. Undigested tissue chunks were filtered out and the supernatant was transferred into a new tube. Cells were collected by centrifugation and resuspended in fresh medium. After plating onto 100-mm culture dishes, cells were incubated at 37 °C with 5% CO2. For activity assay, MEF cells were trypsinized and plated (1 × 106/well) onto 6-well plates. After overnight incubation at 37 °C with 5% CO2, the cells were treated with either 5 μm all-trans–retinaldehyde or 10 μm all-trans–retinol (Sigma) for 3 or 24 h, respectively. Media and cells were collected separately under dimmed light. Retinoids were extracted into hexane and separated by normal phase high performance LC (HPLC) using Waters Alliance Separation Module and 2996 Photodiode Array Detector. Peaks were identified by comparison to retention times of retinoid standards and evaluation of wavelength maxima and quantified as described previously (4).
Statistical analysis
Unpaired t test was used to test for statistical significance. Q-PCR data are presented as the mean ± S.E.
Author contributions
O. V. B. and N. Y. K. conceptualization; O. V. B., L. W., I. S., P. S. N., and N. Y. K. data curation; O. V. B., L. W., I. S., P. S. N., and N. Y. K. formal analysis; O. V. B. and N. Y. K. supervision; O. V. B., L. W., and I. S. validation; O. V. B., L. W., I. S., and P. S. N. investigation; O. V. B., L. W., I. S., and P. S. N. visualization; O. V. B., L. W., and I .S. methodology; O. V. B., L. W., I. S., and N. Y. K. writing-original draft; O. V. B., L. W., I. S., P. S. N., and N. Y. K. writing-review and editing; I. S., P. S. N., and N. Y. K. resources; N. Y. K. funding acquisition; N. Y. K. project administration.
Acknowledgments
We are grateful to Dr. Max Gottesman (Columbia University) for sharing the RBP4-null mice and Drs. Joseph Goldstein and Michael Brown (University of Texas Southwestern Medical Center) for providing SCALD antibodies.
This work was supported by United States Public Health Services, National Institutes of Health Grants R01AA012153, R01DK068437, and R01DK101251. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RDH
- retinol dehydrogenase
- RBP
- retinol-binding protein
- SDR
- short-chain dehydrogenase reductase
- RalR1
- retinaldehyde reductase 1
- SCALD
- short-chain aldehyde reductase
- SREBP
- sterol regulatory element-binding protein
- BCO
- β-carotene oxygenase
- LRAT
- lecithin-retinol acyltransferase
- HSC
- hepatic stellate cells
- MEFs
- mouse embryonic fibroblasts
- BC
- β-carotene
- VAD
- vitamin A–deficient
- VAR
- vitamin A reduced
- SR-B1
- scavenger receptor class B type I
- CYP
- cytochrome P450
- CRBPI
- cellular retinol-binding protein type I
- DHRS3
- dehydrogenase reductase 3
- VAS
- vitamin A-supplemented
- Q-PCR
- quantitative PCR
- DMEM
- Dulbecco's modified Eagle's medium.
References
- 1. Lin B., White J. T., Ferguson C., Wang S., Vessella R., Bumgarner R., True L. D., Hood L., and Nelson P. S. (2001) Prostate short-chain dehydrogenase reductase 1 (PSDR1): a new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res. 61, 1611–1618 [PubMed] [Google Scholar]
- 2. Kedishvili N. Y., Chumakova O. V., Chetyrkin S. V., Belyaeva O. V., Lapshina E. A., Lin D. W., Matsumura M., and Nelson P. S. (2002) Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1). J. Biol. Chem. 277, 28909–28915 10.1074/jbc.M202588200 [DOI] [PubMed] [Google Scholar]
- 3. Kasus-Jacobi A., Ou J., Bashmakov Y. K., Shelton J. M., Richardson J. A., Goldstein J. L., and Brown M. S. (2003) Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 278, 32380–32389 10.1074/jbc.M304969200 [DOI] [PubMed] [Google Scholar]
- 4. Belyaeva O. V., Korkina O. V., Stetsenko A. V., Kim T., Nelson P. S., and Kedishvili N. Y. (2005) Biochemical properties of purified human retinol dehydrogenase 12 (RDH12): catalytic efficiency toward retinoids and C9 aldehydes and effects of cellular retinol-binding protein type I (CRBPI) and cellular retinaldehyde-binding protein (CRALBP) on the oxidation and reduction of retinoids. Biochemistry 44, 7035–7047 10.1021/bi050226k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belyaeva O. V., Stetsenko A. V., Nelson P., and Kedishvili N. Y. (2003) Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry 42, 14838–14845 10.1021/bi035288u [DOI] [PubMed] [Google Scholar]
- 6. Haeseleer F., Jang G. F., Imanishi Y., Driessen C. A. G. G., Matsumura M., Nelson P. S., and Palczewski K. (2002) Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 277, 45537–45546 10.1074/jbc.M208882200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim T. S., Maeda A., Maeda T., Heinlein C., Kedishvili N., Palczewski K., and Nelson P. S. (2005) Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J. Biol. Chem. 280, 8694–8704 10.1074/jbc.M413172200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kasus-Jacobi A., Ou J., Birch D. G., Locke K. G., Shelton J. M., Richardson J. A., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., and Edwards A. O. (2005) Functional characterization of mouse RDH11 as a retinol dehydrogenase involved in dark adaptation in vivo. J. Biol. Chem. 280, 20413–20420 10.1074/jbc.M413789200 [DOI] [PubMed] [Google Scholar]
- 9. Kanan Y., Wicker L. D., Al-Ubaidi M. R., Mandal N. A., and Kasus-Jacobi A. (2008) Retinol dehydrogenases RDH11 and RDH12 in the mouse retina: expression levels during development and regulation by oxidative stress. Invest. Ophthalmol. Vis. Sci. 49, 1071–1078 10.1167/iovs.07-1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marchette L. D., Thompson D. A., Kravtsova M., Ngansop T. N., Mandal M. N., and Kasus-Jacobi A. (2010) Retinol dehydrogenase 12 detoxifies 4-hydroxynonenal in photoreceptor cells. Free Radic. Biol. Med. 48, 16–25 10.1016/j.freeradbiomed.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. von Lintig J., and Vogt K. (2000) Filling the gap in vitamin A research: molecular identification of an enzyme cleaving β-carotene to retinal. J. Biol. Chem. 275, 11915–11920 10.1074/jbc.275.16.11915 [DOI] [PubMed] [Google Scholar]
- 12. Paik J., During A., Harrison E. H., Mendelsohn C. L., Lai K., and Blaner W. S. (2001) Expression and characterization of a murine enzyme able to cleave β-carotene: the formation of retinoids. J. Biol. Chem. 276, 32160–32168 10.1074/jbc.M010086200 [DOI] [PubMed] [Google Scholar]
- 13. Harrison E. H. (2012) Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim. Biophys. Acta 1821, 70–77 10.1016/j.bbalip.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y., Wongsiriroj N., and Blaner W. S. (2014) The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg. Nutr. 3, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., and Gottesman M. E. (1999) Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633–4644 10.1093/emboj/18.17.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. During A., Smith M. K., Piper J. B., and Smith J. C. (2001) β-Carotene 15,15′-dioxygenase activity in human tissues and cells: evidence of an iron dependency. J. Nutr. Biochem. 12, 640–647 10.1016/S0955-2863(01)00184-X [DOI] [PubMed] [Google Scholar]
- 17. During A., Nagao A., Hoshino C., and Terao J. (1996) Assay of beta-carotene 15,15′-dioxygenase activity by reverse-phase high-pressure liquid chromatography. Anal. Biochem. 241, 199–205 10.1006/abio.1996.0400 [DOI] [PubMed] [Google Scholar]
- 18. During A., Albaugh G., and Smith J. C. (1998) Characterization of β-carotene 15,15′-dioxygenase activity in TC7 clone of human intestinal cell line Caco-2. Biochem. Biophys. Res. Commun. 249, 467–474 10.1006/bbrc.1998.9160 [DOI] [PubMed] [Google Scholar]
- 19. Scita G., Aponte G. W., and Wolf G. (1993) Uptake and cleavage of β-carotene by cultures of rat small intestinal cells and human lung fibroblasts. Methods Enzymol. 214, 21–32 10.1016/0076-6879(93)14050-S [DOI] [PubMed] [Google Scholar]
- 20. Wei R. R., Wamer W. G., Lambert L. A., and Kornhauser A. (1998) β-Carotene uptake and effects on intracellular levels of retinol in vitro. Nutr. Cancer 30, 53–58 10.1080/01635589809514640 [DOI] [PubMed] [Google Scholar]
- 21. Lindqvist A., and Andersson S. (2002) Biochemical properties of purified recombinant human β-carotene 15,15′-monooxygenase. J. Biol. Chem. 277, 23942–23948 10.1074/jbc.M202756200 [DOI] [PubMed] [Google Scholar]
- 22. Lee C. M., Boileau A. C., Boileau T. W., Williams A. W., Swanson K. S., Heintz K. A., and Erdman J. W. Jr. (1999) Review of animal models in carotenoid research. J. Nutr. 129, 2271–2277 10.1093/jn/129.12.2271 [DOI] [PubMed] [Google Scholar]
- 23. Wyss A., Wirtz G. M., Woggon W. D., Brugger R., Wyss M., Friedlein A., Riss G., Bachmann H., and Hunziker W. (2001) Expression pattern and localization of β,β-carotene 15,15′-dioxygenase in different tissues. Biochem. J. 354, 521–529 10.1042/0264-6021:3540521,10.1042/bj3540521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Redmond T. M., Gentleman S., Duncan T., Yu S., Wiggert B., Gantt E., and Cunningham F. X. Jr. (2001) Identification, expression, and substrate specificity of a mammalian β-carotene 15,15′-dioxygenase. J. Biol. Chem. 276, 6560–6565 10.1074/jbc.M009030200 [DOI] [PubMed] [Google Scholar]
- 25. van Bennekum A., Werder M., Thuahnai S. T., Han C. H., Duong P., Williams D. L., Wettstein P., Schulthess G., Phillips M. C., and Hauser H. (2005) Class B scavenger receptor-mediated intestinal absorption of dietary β-carotene and cholesterol. Biochemistry 44, 4517–4525 10.1021/bi0484320 [DOI] [PubMed] [Google Scholar]
- 26. Adams M. K., Belyaeva O. V., Wu L., and Kedishvili N. Y. (2014) The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis. J. Biol. Chem. 289, 14868–14880 10.1074/jbc.M114.552257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lobo G. P., Hessel S., Eichinger A., Noy N., Moise A. R., Wyss A., Palczewski K., and von Lintig J. (2010) ISX is a retinoic acid-sensitive gatekeeper that controls intestinal β,β-carotene absorption and vitamin A production. FASEB J. 24, 1656–1666 10.1096/fj.09-150995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blaner W. S., Das K., Mertz J. R., Das S. R., and Goodman D. S. (1986) Effects of dietary retinoic acid on cellular retinol- and retinoic acid-binding protein levels in various rat tissues. J. Lipid Res. 27, 1084–1088 [PubMed] [Google Scholar]
- 29. Rajan N., Blaner W. S., Soprano D. R., Suhara A., and Goodman D. S. (1990) Cellular retinol-binding protein messenger RNA levels in normal and retinoid-deficient rats. J. Lipid Res. 31, 821–829 [PubMed] [Google Scholar]
- 30. Okuno M., Caraveo V. E., Goodman D. S., and Blaner W. S. (1995) Regulation of adipocyte gene expression by retinoic acid and hormones: effects on the gene encoding cellular retinol-binding protein. J. Lipid Res. 36, 137–147 [PubMed] [Google Scholar]
- 31. Husmann M., Hoffmann B., Stump D. G., Chytil F., and Pfahl M. (1992) A retinoic acid response element from the rat CRBPI promoter is activated by an RAR/RXR heterodimer. Biochem. Biophys. Res. Commun. 187, 1558–1564 10.1016/0006-291X(92)90480-9 [DOI] [PubMed] [Google Scholar]
- 32. Smith W. C., Nakshatri H., Leroy P., Rees J., and Chambon P. (1991) A retinoic acid response element is present in the mouse cellular retinol binding protein I (mCRBPI) promoter. EMBO J. 10, 2223–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato M., Blaner W. S., Mertz J. R., Das K., Kato K., and Goodman D. S. (1985) Influence of retinoid nutritional status on cellular retinol- and cellular retinoic acid-binding protein concentrations in various rat tissues. J. Biol. Chem. 260, 4832–4838 [PubMed] [Google Scholar]
- 34. Blaner W. S., Hendriks H. F., Brouwer A., de Leeuw A. M., Knook D. L., and Goodman D. S. (1985) Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J. Lipid Res. 26, 1241–1251 [PubMed] [Google Scholar]
- 35. Shmarakov I., Fleshman M. K., D'Ambrosio D. N., Piantedosi R., Riedl K. M., Schwartz S. J., Curley R. W. Jr, von Lintig J., Rubin L. P., Harrison E. H., and Blaner W. S. (2010) Hepatic stellate cells are an important cellular site for β-carotene conversion to retinoid. Arch. Biochem. Biophys. 504, 3–10 10.1016/j.abb.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Ambrosio D. N., Walewski J. L., Clugston R. D., Berk P. D., Rippe R. A., and Blaner W. S. (2011) Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS ONE 6, e24993 10.1371/journal.pone.0024993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tong M. H., Yang Q. E., Davis J. C., and Griswold M. D. (2013) Retinol dehydrogenase 10 is indispensible for spermatogenesis in juvenile males. Proc. Natl. Acad. Sci. U.S.A. 110, 543–548 10.1073/pnas.1214883110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hogarth C. A., and Griswold M. D. (2010) The key role of vitamin A in spermatogenesis. J. Clin. Invest. 120, 956–962 10.1172/JCI41303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghyselinck N. B., Vernet N., Dennefeld C., Giese N., Nau H., Chambon P., Viville S., and Mark M. (2006) Retinoids and spermatogenesis: lessons from mutant mice lacking the plasma retinol binding protein. Dev. Dyn. 235, 1608–1622 10.1002/dvdy.20795 [DOI] [PubMed] [Google Scholar]
- 40. Raverdeau M., Gely-Pernot A., Féret B., Dennefeld C., Benoit G., Davidson I., Chambon P., Mark M., and Ghyselinck N. B. (2012) Retinoic acid induces Sertoli cell paracrine signals for spermatogonia differentiation but cell autonomously drives spermatocyte meiosis. Proc. Natl. Acad. Sci. U.S.A. 109, 16582–16587 10.1073/pnas.1214936109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Endo T., Freinkman E., de Rooij D. G., and Page D. C. (2017) Periodic production of retinoic acid by meiotic and somatic cells coordinates four transitions in mouse spermatogenesis. Proc. Natl. Acad. Sci. U.S.A. 114, E10132–E10141 10.1073/pnas.1710837114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Belyaeva O. V., Adams M. K., Wu L., and Kedishvili N. Y. (2017) The antagonistically bifunctional retinoid oxidoreductase complex is required for maintenance of all-trans-retinoic acid homeostasis. J. Biol. Chem. 292, 5884–5897 10.1074/jbc.M117.776914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Belyaeva O. V., Korkina O. V., Stetsenko A. V., and Kedishvili N. Y. (2008) Human retinol dehydrogenase 13 (RDH13) is a mitochondrial short-chain dehydrogenase/reductase with a retinaldehyde reductase activity. FEBS J. 275, 138–147 10.1111/j.1742-4658.2007.06184.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Belyaeva O. V., and Kedishvili N. Y. (2002) Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Lett. 531, 489–493 10.1016/S0014-5793(02)03588-3 [DOI] [PubMed] [Google Scholar]
- 45. Gallego O., Belyaeva O. V., Porté S., Ruiz F. X., Stetsenko A. V., Shabrova E. V., Kostereva N. V., Farrés J., Parés X., and Kedishvili N. Y. (2006) Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem. J. 399, 101–109 10.1042/BJ20051988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Napoli J. L., and Horst R. L. (1998) Quantitative analyses of naturally occurring retinoids. Methods Mol. Biol. 89, 29–40 [DOI] [PubMed] [Google Scholar]
- 47. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]