

Abstract
Pompe disease (PD), also known as “glycogen storage disease type II (OMIM # 232300)” is a rare autosomal recessive disorder characterized by progressive glycogen accumulation in cellular lysosomes. It ultimately leads to cellular damage. Infantile-onset Pompe disease (IOPD) is the most severe type of this disease and is characterized by severe hypertrophic cardiomyopathy and generalized hypotonia. Mutations in the acid alpha-glucosidase (GAA) gene, located at locus 17q25.3, are responsible for the disease leading to reduced activity of the acid alpha-glucosidase enzyme. To date, approximately 400 pathogenic mutations have been reported in the GAA gene. The aim of this study is to report a novel nonsense mutation in exon 4 of the GAA gene in an Iranian child suffering from IOPD. The patient was a female neonate with hypertrophic cardiomyopathy and a positive family history of IOPD. After definite diagnosis, enzyme-replacement therapy (ERT) was started for the patient, who was 2 months old. Now at the age of 20 months, she has had good growth and development and her echocardiographic parameters are within the normal range. This report shows that IOPD patients with this mutation can be treated with ERT successfully.
Keywords: Enzyme replacement therapy, Glycogen storage disease type II, GAA protein, Human, Cardiomyopathy, Hypertrophy
What’s Known
Infantile-onset Pompe disease is a rare genetic disorder that is life-threatening. Enzyme-replacement therapy (ERT) can be lifesaving in some of these patients.
What’s New
We have discovered a new homozygous mutation in exon 4 of the acid alpha-glucosidase gene.
The patient had a good response to ERT. Thus, patients with this mutation may respond to ERT.
Introduction
Originally described in 1932, Pompe disease (PD), also known as “glycogen storage disease type II (OMIM #232300)”, is a rare autosomal recessive disorder characterized by progressive glycogen accumulation in cellular lysosomes ultimately leading to cellular damage. Mutations in the acid alpha-glucosidase (GAA) gene, located at locus 17q25.3, are responsible for the disease leading to reduced activity of the acid alpha-glucosidase enzyme.1
Generally, the lesser the enzyme activity, the greater the disease severity with an earlier age of onset.2 Infantile-onset Pompe disease (IOPD) is the most severe type of this disease. The main clinical manifestations of IOPD include cardiomegaly, severe hypertrophic cardiomyopathy, profound hypotonia, and generalized muscle weakness.2
Recently, enzyme-replacement therapy (ERT) has improved the outcome of these patients by helping not only to reverse cardiomyopathy but also to achieve normal motor milestones.3
Thus far, approximately 400 pathogenic mutations have been reported in the GAA gene,4 of which the c.-32-13T>G mutation is the most common among Caucasians.5
We report a novel nonsense mutation in exon 4 of the GAA gene in an Iranian child suffering from PD and her good response to ERT.
Case Presentation
The patient was referred with the chief complaint of hypertrophic cardiomyopathy, which was detected on echocardiography.
She was an Iranian female neonate, whose brother was a known case of IOPD. Her brother expired at the age of 5 months because at that time ERT had not been started in Iran. Given this positive family history, echocardiography was performed for the patient at 1 month of age while she was asymptomatic. The echocardiography showed hypertrophic cardiomyopathy. The results of echocardiography at that age were ejection fraction of 69%, interventricular septum diameter in systole of 8 mm (normal value=3.5–5 mm), interventricular septum diameter in diastole of 7 mm (normal value=3.5–5 mm), and posterior wall diameter of 9 mm in systole and 7 mm in diastole (normal value=3.5–5 mm).
In physical examination, muscular tone was normal, the tongue was not enlarged, and the facial picture was normal. Heart examination was normal, and she did not have organomegaly.
The diagnosis was confirmed through enzyme assay on a dried blood specimen, which revealed decreased alpha-glucosidase activity. A definite diagnosis was made through molecular analysis. The GAA gene was analyzed by polymerase chain reaction (PCR) and sequencing of both the DNA strands of the entire coding region and the highly conserved exon-intron splice junctions. In addition, a specific PCR to screen for the common deletion of exon 18 was also performed. The reference sequence of the GAA gene is NM_000152.3. The results of the molecular analysis are demonstrated in table 1. The electropherogram of the sequence (coding positions) and the discovered mutation are depicted in figure 1.
Table 1.
Results of the molecular analysis of the acid alpha-glucosidase (GAA) gene of the patient (with the GAA gene reference sequence: NM_000152.3)
| E×03 | c. 596A>G (homo) | p.H199R | rs1042393 | SNP Site |
|---|---|---|---|---|
| E×03 | c. 642C>T (homo) | p.S214S | rs1800301 | SNP site |
| E×03 | c. 668G>A (homo) | p.R223H | rs1042395 | SNP site |
| E×04 | c. 730C>T (homo) | p.Q244* | none | Disease-causing |
| Int04 | c. 858+8ins7bp (homo) | rs35373675 | SNP site | |
| Int04 | c. 858+30T>C (homo) | rs2304845 | SNP site | |
| Int05 | c. 955+12G>A (homo) | rs2252455 | SNP site | |
| E×08 | c. 1203G>A (homo) | p.Q401Q | rs1800304 | SNP site |
| Int08 | c. 1327-18A>G (homo) | rs2278619 | SNP site | |
| Int09 | c. 1438-19G>C (homo) | rs2304844 | SNP site | |
| E×15 | c. 2133A>G (homo) | p.T711T | rs1800310 | SNP site |
| E×17 | c. 2338G>A (homo) | p.V780I | rs1126690 | SNP site |
| E×18 | c. 2553G>A (homo) | p.G851G | rs1042397 | SNP site |
| 3’ UTR | c.*223C>T (homo) | - | rs8132 | SNP site |
Figure 1.
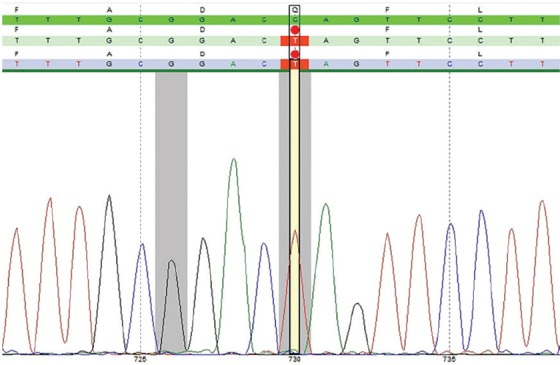
GAA gene analysis; electropherogram of the sequence.
As is seen in table 1, we found an undescribed homozygous mutation in exon 4 of the GAA gene (c.730C>T p.Q244*), creating a premature stop codon and leading to a truncated protein or loss of protein production. This mutation is compatible with the result of the enzyme assay, which showed decreased activity of the alpha-glucosidase enzyme.
As soon as the diagnosis was confirmed, Myozyme® (alglucosidase alfa, Sanofi Genzyme, USA) was initiated for the patient (20 mg/kg, every 2 wk). At 20 months of age, after about 18 months of ERT (without any side effects), echocardiography was repeated and demonstrated ejection fraction of 75%, interventricular septum diameter in systole of 7 mm (normal value=5–7 mm), interventricular septum diameter in diastole of 6.5 (normal value=4.5–6.5 mm), and posterior wall diameter of 7 mm (normal value=4.5–7 mm).
Since the initiation of ERT, the patient’s condition has significantly improved and currently, at 20 months of age, her weight is 11 kg (50th percentile) and her height is 80 cm (25–50th percentile). She has also reached her age-specific normal motor and cognitive milestones.
To the best of our knowledge, this mutation is a novel, previously undetected, pathogenic nonsense mutation, associated with severe, classical IOPD with a good response to ERT (Database: www.pompecenter.nl).
Written consent was taken from the parents for the presentation of this case.
Discussion
We identified a novel disease-causing nonsense mutation in exon 4 of the GAA gene. This mutation led to a premature stop codon, which very likely resulted in a truncated protein or loss of protein production. Consequently, as was expected, the disease outcome was very severe and had an early infantile onset. This finding supports the previous view that IOPD is a result of truncating mutations.1
Currently as many as 400 pathogenic mutations have been detected.4 Most mutations are unique to each patient; nevertheless, some of them are more prevalent in certain populations. For example among Caucasians, the c.-32-13T>G mutation is the most common.1 This mutation has never been reported in Asians and, thus, indicates an ethnic-specific mutation.6 The c.1935C>A mutation (p.Asp645Glu) is seen in up to 80% of infantile cases in Taiwan and China, proposing a possible founder effect.7 Another possible founder effect is observed in individuals of African descent with the mutation c.2560C>T (p.Arg854*), seen in up to 60% of cases.8 In Dutch individuals, the c.2481+102_2646+31del (exon 18 deletion) and p.T525del mutations are frequently found.9 In 2013, Esmer et al.1 reported 2 unrelated Mexican cases of IOPD with the same novel homozygous frameshift mutation “c.1987delC”. They were both related to a small region from Central Mexico, thus suggesting a possible founder effect.
As was mentioned, apart from these population-specific mutations, the vast majority of mutations are unique to each individual.1
In a previously published case report in 2013, a 10-month-old Iranian boy affected with PD was introduced by Galehdari et al.,5 whose molecular analysis revealed a new homozygous mutation in the GAA gene, as of a single adenine insertion at codon 693 of exon 15. This is an insertion which will cause an anticipated premature stop codon at codon 736. Our report seems to be the 2nd molecular report from Iran.
Our case has responded well to ERT so far; therefore, if another patient with this mutation is found, we can be hopeful that the patient will most probably respond to ERT.
Several articles have been published on response to ERT among IOPD patients. A review article performed by Chien YH et al.10 showed that ERT was overall beneficial. The authors reported that ERT was able to cause a gradual decrease in heart size 3 months after treatment commencement. Additionally, mortality in their report was also decreased up to 99%. However, response to treatment was variable among the patients, with the treatment being more effective if started early, before the destruction of the muscle architecture.
Another study on patients with advanced disease, who needed ventilator support with markedly delayed motor development and abnormal left ventricular mass indices, showed that ERT even on these patients was effective insofar as their cardiac function and motor function gradually improved, although only one of these patients was able to walk by the end of that study.11
As much as the early initiation of treatment is a good prognostic factor for ERT among patients with IOPD, some other factors, including the genetic type and the titer of anti-rh GAA antibodies, are also very important. Some studies have indicated that the baseline clinical status of patients is a more reliable index of potential response to ERT than age alone.11 Consequently, the good response of our patient to ERT can be due to the early start of treatment or the genetic type or both.
We treated our patient with a dose of 20 mg/kg every other week. There are some controversies surrounding the dose of the drug. The standard dose is 20 mg/kg every other week, but many centers treat their patients with higher doses such as 40 mg/kg every other week or 20 mg/kg every week.10
Although higher doses may confer a better response in some patients, they can be associated with a higher risk of anaphylactic reactions, infusion-associated reactions, and antibody production.
Due to inadequate facilities, we did not determine cross-reactive immunologic material (CRIM) status and nor did we check the anti-rh GAA antibody titer.
The height and weight of our patient were within the normal range, but failure to thrive is a typical finding in untreated patients with IOPD. Accordingly, the good growth of our patient is also another indicator of good response to ERT.
Cognitive development in our patient has been normal up to now. There are few data on cognitive development among untreated IOPD patients because of early mortality in these patients.
In addition to extending our information about disease-causing mutations in PD, the result of this report could be drawn upon for the molecular screening of PD, a disease in which timing is gold given the irreversible nature of its complications.
However, a comprehensive screening requires that further investigations be undertaken in other GSD II patients in order to find mutation diversity and frequency in the region, especially in Iran.
Conclusion
A female patient with a new homozygous mutation in exon 4 of the GAA gene (c.730C>T p.Q244*) was herein introduced. She had a good response to ERT. As a result, future possible patients with this mutation may be managed with ERT successfully.
Acknowledgement
The authors would like to thank Centogene AG Germany Company for its accurate genetic analysis of our patient. Many thanks are also due to the Center for the Development of Clinical Research of Nemazee Hospital in Shiraz University of Medical Sciences for editing this article.
Conflict of Interest: None declared.
References
- 1.Esmer C, Becerra-Becerra R, Pena-Zepeda C, Bravo-Oro A. A novel homozygous mutation at the GAA gene in Mexicans with early-onset Pompe disease. Acta Myol. 2013;32:95–9. [ PMC Free Article] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirschhorn R, Reuser AJ. Glucogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet A, Sly WS, Valle D, editors. The metabolic and Molecular Bases of Inherited Disease. New York; NY: McGraw-Hill; 2001. pp. 3389–420. [Google Scholar]
- 3.Prater SN, Banugaria SG, DeArmey SM, Botha EG, Stege EM, Case LE, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14:800–10. doi: 10.1038/gim.2012.44. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galehdari H, Emami M, Mohammadian G, Khodadadi A, Azmoon S, Baradaran M. Detection of a novel mutation in the GAA gene in an Iranian child with glycogen storage disease type II. Arch Iran Med. 2013;16:126–8. doi: 10.3162/AIM.0015. [DOI] [PubMed] [Google Scholar]
- 6.Wan L, Lee CC, Hsu CM, Hwu WL, Yang CC, Tsai CH, et al. Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J Neurol. 2008;255:831–8. doi: 10.1007/s00415-008-0714-0. [DOI] [PubMed] [Google Scholar]
- 7.Shieh JJ, Lin CY. Frequent mutation in Chinese patients with infantile type of GSD II in Taiwan: Evidence for a founder effect. Hum Mutat. 1998;11:306–12. doi: 10.1002/(SICI)1098-1004(1998)11:4<306:AID-HUMU8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 8.Becker JA, Vlach J, Raben N, Nagaraju K, Adams EM, Hermans MM, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet. 1998;62:991–4. doi: 10.1086/301788. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van der Kraan M, Kroos MA, Joosse M, Bijvoet AG, Verbeet MP, Kleijer WJ, et al. Deletion of exon 18 is a frequent mutation in glycogen storage disease type II. Biochem Biophys Res Commun. 1994;203:1535–41. doi: 10.1006/bbrc.1994.2360. [DOI] [PubMed] [Google Scholar]
- 10.Chien YH, Hwu WL, Lee NC. Pompe disease: Early diagnosis and early treatment make a difference. Pediatr Neonatol. 2013;54:219–27. doi: 10.1016/j.pedneo.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–9. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]