

Abstract
Macular corneal dystrophy (MCD) is an autosomal recessive hereditary disease. In most cases, various mutations in carbohydrate sulfotransferase 6 (CHST6) gene are the main cause of MCD. These mutations lead to a defect in keratan sulfate synthesis. Retinitis pigmentosa (RP) is another eye disorder with nyctalopia as its common symptom. It has been shown that more than 65 genes have been implicated in different forms of RP. Herein, we report on a 9-member family with 2 girls and 5 boys. Both parents, one of the girls and one of the boys had normal eye vision and another boy had keratoconus. Other children (1 girl and 2 boys) suffered from both MCD and RP. Corneal transplantation and medical supplements were used for MCD and RP during the follow-up period, respectively. Based on the family tree, it seems that the inheritance of both diseases is autosomal recessive. Based on our search of databases, there is no report on the simultaneous presence of MCD and RP. To the best of our knowledge, the present article is the first case report on this topic. Molecular genetic investigation is needed to clarify the mechanism of concurrent MCD and RP.
Keywords: Macular corneal dystrophy, Retinitis pigmentosa, Pedigree, Genetic disease
What’s Known
Macular corneal dystrophy (MCD) and retinitis pigmentosa (RP) are two separate eye diseases.
There is no report on simultaneous presence of MCD and RP in a patient.
What’s New
Using clinical investigation, we report on three familial patients suffering from both MCD and RP simultaneously.
Based on the family tree, it seems that the inheritance of both diseases is autosomal recessive.
Introduction
Corneal dystrophies are a group of diverse bilateral genetic and non-inflammatory diseases limited to the cornea. Clinically, it is categorized into three groups; superficial corneal dystrophy, corneal stromal dystrophy, and posterior corneal dystrophy which are further subcategorized into other classes. Macular corneal dystrophy (MCD) is a subcategory of corneal stromal dystrophies.1 Mutation in carbohydrate (N-acetylglucosamine6-O) sulfotransferase 6 (CHST6) gene is usually responsible for MCD. However, all MCD cases cannot be explained by mutations in CHST6 coding region, deletion or replacement in the upstream region, or mutations in splice sites resulting in loss of splicing signal.2
Retinitis pigmentosa (RP) is a disease with a variety of disorders. Some patients show symptoms of vision loss during childhood while some others live without any symptoms until their middle age.3 Most cases present classical symptoms of difficulties with adapting to darkness and night blindness (nyctalopia) in old age and loss of vision in early adolescence. Following the disease progression, they lose their distant peripheral vision, tunnel vision, and finally central vision which usually occurs at the age of sixty. The reduction of ROD and CONE is similar in other types. Sometimes the decrease in CONE is greater than that in ROD which is then called cone-rod degeneration, a form of RP in which the loss of vision and defects in color vision are the predominant initial symptoms.4
Here, we present a case report study of a family with three children suffering from both MCD and RP diseases simultaneously. According to our search of databases, there has been no report regarding the co-occurrence of these diseases; hence, the present article is the first reported case on this topic.
Case Presentation
We present the case of a 9-member family that includes the parents (consanguineous marriage) and 7 children (2 girls and 5 boys). The parents had healthy eyes and among the children, only two had healthy vision. One male descendant had keratoconus while the other had MCD. Three children (two boys: 30 and 36-year-old man, one girl: 41 years old) showed the co-occurrence of both MCD and RP (figure 1). One of the patients, 36 years male, referred to the clinic with the complaint of difficulties in undertaking tasks at night or in dark places. His vision impairment had started from childhood but deteriorated over the past 10 years. Visual acuity of the patient was finger counting (CF) at 1.5 m and 1 m on the right and left eye, respectively. The use of eyeglasses had no effect on his vision. Written informed consent was obtained from all patients enrolled in this report.
Figure 1.
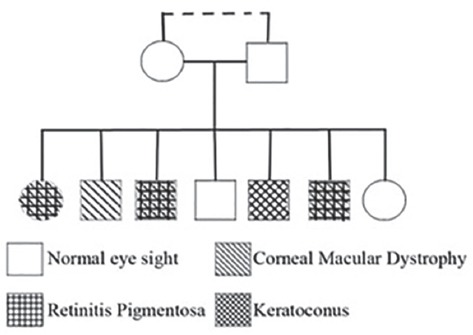
The genealogy of the family showed that two males and one female are suffering from macular corneal dystrophy and RP simultaneously.
MCD can be easily identified by clinical examination. Using a photoslit device, the patient’s cornea with MCD is shown in figure 2. Multiple gray-white patches are distributed over the cornea and all layers involved with peripheral expansion can be observed with the same pattern in both eyes. Histopathological evaluation confirmed MCD in this patient (figure 3). The diagnosis of RP is also clinical. Dilated fundus examination clearly showed blood vessels attenuation, boney spicules, and waxy disc pallor; all of which confirmed RP in this patient (figure 4). Conventional treatments were employed for these patients. Keratoplasty was performed for MCD due to the intensity of the disease. Serological analysis was performed before surgery and the results demonstrated that all patients were negative for infectious diseases such as syphilis. Since there is no effective treatment, supplementary drugs were used for RP.
Figure 2.
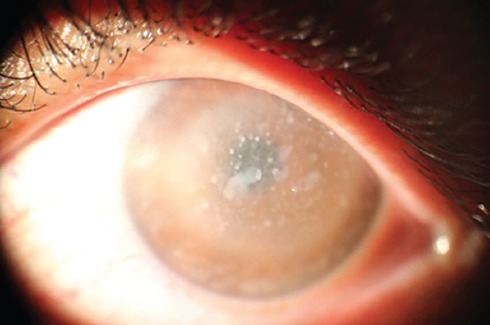
Gray-white patches are seen clearly that confirmed macular corneal dystrophy in this patient. This image is taken with photoslit device.
Figure 3.
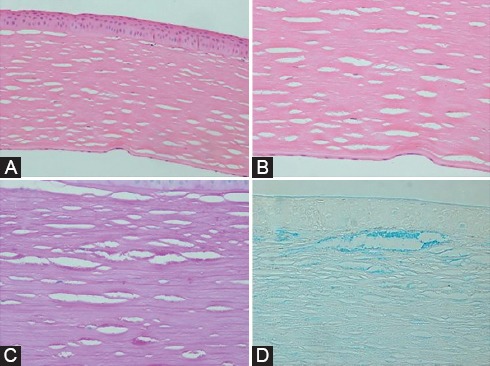
Histopathological examination of the recipient cornea discloses a full thickness cornea with fine inter lamellar stromal deposits (A and B, hematoxylin and eosin, magnification ×200 and ×400, respectively) that are PAS-positive on periodic acid-Schiff (C, magnification ×400) and stained blue with alcian blue staining method (D, magnification ×400).
Figure 4.
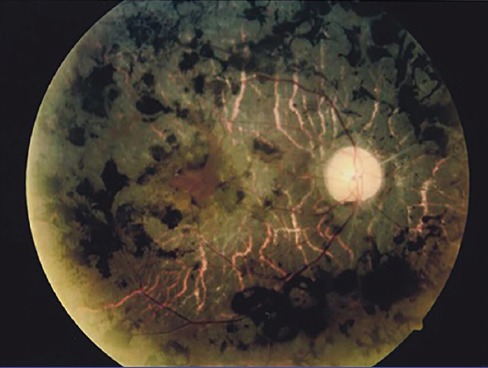
Appearance of the retina in one of the patients with RP is shown. The image is taken using optical corneal tomography (Optovue OCT, iFusion). Blood vessels attenuation, boney spicules, and waxy disc pallor is clearly seen in this image. All three patients showed these evidences in both eyes.
Discussion
The present case report describes a 9-member family in which 3 out of 5 children suffered from MCD and RP. Although Crawford5 had reported a case of concurrent granular corneal dystrophy (GCD) and RP with autosomal dominant inheritance in a 58-year-old Korean woman, we present the first report of simultaneous MCD and RP. MCD is inherited as recessive autosomal and it appears to be caused by abnormal structures of creatinine sulfate, which is triggered by a mutation in CHST6 gene.6 The most common disorder is mediated by missense or nonsense single nucleotide polymorphisms (SNPs) in CHST6, which causes a change in a protected amino acid. Heterozygous mutation in the exon 3 of CHST6 gene has been detected in most families, almost all of them are associated with another heterozygous mutation in the CHST6 coding region on the other chromosome.7 Other mutations causing MCD relate to the insertion or deletion of nucleotides in the CHST6 coding region, which alters frame-shift and also a few upstream deletion or substitution between CHST5 and CHST6.8
RP is a non-age-related disease and the symptoms can manifest in all ages.9 The inheritance of the disease has so far been reported as autosomal recessive and dominant, and X-linked forms. Studies have shown that there are at least 85 loci in mutagens that cause the disorder (mutation in 56 genes causes non-syndromic RP, mutation in 12 genes causes Usher syndrome, and mutation in 17 genes cause Bardet-Biedl syndrome). In general, mutations in RHO (rhodopsin), USH2A, and RPGR genes lead to 30% of all cases of RP. Of other genes that can cause autosomal dominant RP include RP1 [8q12.1], PRPF3 (pre-MRNA processing factor 3) [1q21.2], PRPF8 [17p13.3], and PRPF31 [19q13.42] have been identified to contribute 5.5%, 4%, 2%, and 5%, respectively. Mutations in genes such as RP1L1 [8q23.1], RLBP1 (retinaldehyde binding protein 1) [15q26.1], CRB1 (crumbs family member 1) [1q31.3], ABCA4 (ATP-binding cassette subfamily A member 4) [1p22.1], RPE65 (retinal pigment epithelium-specific protein 65kDa) [1p31.2], and CNGB1 (cyclic nucleotide gated channel beta 1) [16q21] also share 1%, 1%, 6.5%, 2%, and 4%, respectively, in autosomal recessive inheritance.9
As can be seen in figure 1, the inheritance of both diseases is recessive (lack of parental illness) and autosomal (not X-linked due to the occurrence of the disease in one of the girls and lack of paternal illness for any of the diseases). Considering that the parents’ werecousin marriage, the chance to inherit the defective gene from ancestors increases. However, in order to clarify and exclude the possibility of de novo dominant mutation in parents’ germ cells, further studies are required on both parents. According to the location of CHAT6 gene and also the major role of CNGB1 gene (with the frequency of 4% in patients with RP) located in 16q21 to develop RP, this gene can be used as one of the first candidates for genetic investigation with the chance of discovering a translocation mutation in these genes. On the other hand, Davidson et al.10 identified a homozygous frame-shifting mutation, c.601delG, p.Lys203Argfs*28, in RP1L1 in a consanguineous family with RP, while various mutations in this gene had been reported to be associated with occult macular dystrophy (OCMD). As a result of this case report, RP1L1 can be the second candidate for genetic investigations. Considering different mutations in CHST6 gene on the 16q23 region to create MCD as well as the role of different mutations (mutation in more than 40 genes) to generate RP disease, it is important to investigate both diseases genetically by molecular methods to discover the reason behind the association of these two diseases.
Conflict of Interest: None declared.
References
- 1.Sacchetti M, Macchi I, Tiezzi A, La Cava M, Massaro-Giordano G, Lambiase A. Pathophysiology of Corneal Dystrophies: From Cellular Genetic Alteration to Clinical Findings. J Cell Physiol. 2016;231:261–9. doi: 10.1002/jcp.25082. [DOI] [PubMed] [Google Scholar]
- 2.Klintworth GK. Advances in the molecular genetics of corneal dystrophies. Am J Ophthalmol. 1999;128:747–54. doi: 10.1016/S0002-9394(99)00358-X. [DOI] [PubMed] [Google Scholar]
- 3.Heckenlively JR, Yoser SL, Friedman LH, Oversier JJ. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 1988;105:504–11. doi: 10.1016/0002-9394(88)90242-5. [DOI] [PubMed] [Google Scholar]
- 4.Matsui R, McGuigan Iii DB, Gruzensky ML, Aleman TS, Schwartz SB, Sumaroka A, et al. SPATA7: Evolving phenotype from cone-rod dystrophy to retinitis pigmentosa. Ophthalmic Genet. 2016;37:333–8. doi: 10.3109/13816810.2015.1130154. [ PMC Free Article] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford C. Retinitis pigmentosa and granular dystrophy: A rare and unique combination in one patient. Binocul Vis Strabolog Q Simms Romano. 2013;28:36–8. [PubMed] [Google Scholar]
- 6.Aldave AJ, Yellore VS, Thonar EJ, Udar N, Warren JF, Yoon MK, et al. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am J Ophthalmol. 2004;137:465–73. doi: 10.1016/j.ajo.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 7.Vance JM, Jonasson F, Lennon F, Sarrica J, Damji KF, Stauffer J, et al. Linkage of a gene for macular corneal dystrophy to chromosome 16. Am J Hum Genet. 1996;58:757–62. [ PMC Free Article] [PMC free article] [PubMed] [Google Scholar]
- 8.Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy: A comprehensive molecular genetic review. Mol Vis. 2006;12:159–76. [PubMed] [Google Scholar]
- 9.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 10.Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–14. doi: 10.1002/humu.22264. [DOI] [PubMed] [Google Scholar]