

Abstract
Little is known about the prevalence of HBV genotypes/sub-genotypes in Jeddah province, although the hepatitis B virus (HBV) was identified as the most predominant type of hepatitis in Saudi Arabia. To characterize HBV genotypes/sub-genotypes, serum samples from 15 patients with chronic HBV were collected and subjected to HBsAg gene amplification and sequence analysis. Phylogenetic analysis of the HBsAg gene sequences revealed that 11 (48%) isolates belonged to HBV/D while 4 (18%) were associated with HBV/C. Notably, a HBV/D sub-genotype phylogenetic tree identified that eight current isolates (72%) belonged to HBV/D1, whereas three isolates (28%) appeared to be more closely related to HBV/D5, although they formed a novel cluster supported by a branch with 99% bootstrap value. Isolates belonging to D1 were grouped in one branch and seemed to be more closely related to various strains isolated from different countries. For further determination of whether the three current isolates belonged to HBV/D5 or represented a novel sub-genotype, HBV/DA, whole HBV genome sequences would be required. In the present study, we verified that HBV/D1 is the most prevalent HBV sub-genotype in Jeddah, and identified novel variant mutations suggesting that an additional sub-genotype designated HBV/DA should be proposed. Overall, the results of the present HBsAg sequence analyses provide us with insights regarding the nucleotide differences between the present HBsAg/D isolates identified in the populace of Jeddah, Saudi Arabia and those previously isolated worldwide. Additional studies with large numbers of subjects in other areas might lead to the discovery of the specific HBV strain genotypes or even additional new sub-genotypes that are circulating in Saudi Arabia.
Abbreviations: HBV, hepatitis B virus; HAV, hepatitis A virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma; P, HBV polymerase gene; C/pre C, HBV core/pre Core gene; X, HBV X gene; HBsAg, HBV surface antigen; IFN, interferon; Pre S1/Pre S2/S, HBsAg genes; PCR, polymerase chain reaction; DDBJ, DNA Data Bank of Japan; EMBL, European Molecular Biology Laboratory
Keywords: Hepatitis B virus, HBV sub-genotypes, HBV/D, HBsAg, Viral isolates, Population studies
1. Introduction
Many nations have a heavy financial load from the high percentage of both individuals with chronic hepatitis B virus (HBV) infection and the approximately 400 million HBV carriers worldwide. HBV infection leads to the development of severe liver diseases including cirrhosis and hepatocellular carcinoma (HCC) (Norder et al., 1992, Norder et al., 1994). Human HBV is the prototype member of the family Hepadnaviridae that contains a circular, partially double-stranded DNA genome of about 3200 bp. HBV DNA contains four overlapping open reading frames encoding the pre S1/pre S2/S, pre C/C, pol, and X viral proteins (Fig. 1) (Magnius and Norder, 1995).
Figure 1.

Genome organization of HBV. The colored arrows indicate overlapping genes: (ORF P) polymerase gene; (ORF C/pre C) core/pre core gene; (ORF X) X gene and (ORF Pre S1/ORF Pre S2/ORF S) HBsAg gene (Jalali and Alavian, 2006).
Compared to most DNA viruses, HBV has a high rate of nucleotide substitution although this is lower than the mutation rate of RNA viruses (Okamoto et al., 1987). Previously, HBV genomes were classified into four genotypes, but recently have been categorized into eight genotypes designated as A-H. This categorization was based on inter-genotype divergence of at least 8% of the complete nucleotide sequence or on more than 4% divergence in the HBsAg gene (Arauz-Ruiz et al., 1997, Okamoto et al., 1987). These genotypes possess different geographical distributions as illustrated in Fig. 2 (Arauz-Ruiz et al., 1997, Chu et al., 2003, Miyakawa and Mizokami, 2003). Recently, a complex recombinant of genotypes A, C, and G has been referred to as representing a new genotype (I), which was described and sequenced in northwestern China, Vietnam, and Brazil (Santos et al., 2010). In addition, sub-genotypes have been described in certain HBV genotypes; these are A1-A6 in genotype A (HBV/A), and B1-B8, C1-C16, and D1-D8 in HBV/B, C, and D, respectively (Abdou Chekaraou et al., 2010, Mulyanto et al., 2009).
Figure 2.

Worldwide distribution of HBsAg genotypes (A-I) and some subgenotypes. Dark blue represents regions where the genotypes are highly endemic (>8%), whereas blue and light blue represent regions with intermediate and low endemicity (2.7% and <2%), respectively. Saudi Arabia was identified as an HBsAg high endemic region (Hou et al., 2005).
HBV genotypes show a direct correlation with the severity of liver disease. HBV/C, HVB/D, and HBV/B are associated with severe liver cirrhosis (Banerjee et al., 2006, Huy et al., 2006, Kramvis et al., 2008, Sakamoto et al., 2006) and appear to have a higher incidence of HCC (Chan and Sung, 2006, Liu et al., 2007, Liu et al., 2011, Sumi et al., 2003, Yuen et al., 2003, Yuen et al., 2009). Additionally, patients with either HBV/C or HBV/D have a lower response rate to treatment with interferon (IFN)γ when compared with those carrying other HBV genotypes (Zöllner et al., 2001). The HBV genotype might also influence the emergence of lamivudine resistance mutations, which appear to be more strongly associated with HBV/A than HBV/D (Wen, 2004).
HBV surface antigen (HBsAg) can induce protection against HBV infection as it is related directly to B-cell epitopes and is considered as the major target of neutralizing antibodies; therefore, it is used an HBV vaccine (Kramvis et al., 2005). The complete HBsAg gene consists of three regions: large S, Pre S2, and Pre S1, which share their C-terminal 226 amino acid residues (Carman, 1997, Szmuness et al., 1981). Mutant HBsAg nucleotides might cause amino acid substitutions (EL Hadad et al., 2013), which could affect the binding of specific anti-HB antibodies and the detection by conventional diagnostic assays (Torresi, 2002). In addition, a relationship has been observed between low antigenicity of HBV (leading to HBV reinfection) and the increased incidence of HCC in Egyptian patients with chronic HBV (Tian et al., 2007). Furthermore, correlations between mutations in the HBsAg gene, particularly in the Pre S regions, and the development of HCC have been verified in patients with chronic HBV (Yang et al., 2010).
The prevalence of HBV infection is generally high in Asian and African countries (Lee, 1997). Saudi Arabia reported HBV as the most predominant type (53%) of hepatitis infection followed by HCV (30%) and HAV (17%) (Alshabanat et al., 2013). Furthermore, the Jeddah province of Saudi Arabia hosts large numbers of HBV infected individuals originating from countries with a high-HBV burden because Jeddah is a main entry point as well as being the largest commercial port in the country. For these reasons and because little is known regarding the HBsAg genotypes/sub-genotype sequences circulating in Saudi Arabia, and in particular in the Jeddah region (Al-Faleh et al., 1992; 1999), the present study was conducted to provide sequence data of the most common HBsAg genotypes/subgenotypes circulating in Jeddah for subsequent phylogenetic analysis.
2. Materials and methods
2.1. Patient samples
Serum samples were collected from 17 Saudi patients with chronic HBV (5 women and 12 men, mean age 32.7 years) and were randomly selected as they became available from different hospitals in Jeddah; review of the medical records confirmed that all samples were positive for HBsAg with no co infection with either HIV or HCV. The samples were divided into aliquots and stored at −70 °C until used.
The study protocol was reviewed and approved by the Deanship of Scientific Research Ethics Committee of King Abdulaziz University and the King Abdulaziz Hospital Ethics Committee. Written consent was obtained from all patients after full explanation of the purpose of the study.
2.2. HBV-DNA extraction, amplification, and HBsAg genotype/sub-genotype determination
HBV DNA was obtained from 17 patient sera samples using the Mini Elute viral Extraction Kit (QIAGEN, Inc., Valencia, CA, USA) according to the manufacturer’s instructions. The resultant HBV-DNA samples were stored at −70 °C until use (McElhinney et al., 2011). The presence of HBV DNA was determined by nested PCR using a Hot start Taq plus PCR Master Mix Kit (QIAGEN) using the primers reported in Table 1. The first round PCR amplification reaction was performed according to the manufacturer’s instructions using 10 μl HBV-DNA and 50 pmol each of primers SBFO10 and SBRO20. The cycling conditions were performed with an initial 5 min of preheating at 95 °C, followed by 35 cycles of denaturing for 30 s at 95 °C, annealing for 30 s at 52 °C, and an elongation step for 1 min at 72 °C, with a final extension period of 10 min at 72 °C. Nested PCR using the SBFI30 and SBRI40 primers was performed on 10 μl of the samples negative for PCR products in the first round amplification. The second round of amplification was performed with an initial 5 min preheating step at 95 °C, followed by 35 cycles of denaturing for 30 s at 95 °C, annealing for 30 s at 55 °C, and elongation for 1 min at 72 °C, with a final extension period of 10 min at 72 °C. All PCR contamination precautions were observed and negative controls using sera from healthy individuals who were not positive for any HBV markers were included (EL Hadad et al., 2013, Mulyanto et al., 2011).
Table 1.
Primers for full length HBsAg amplification and their nucleotide position.
| Primer code | Primer sequence | Primer length | Nucleotide position |
|---|---|---|---|
| SBFO10 | 5′ GGGTCACCATATTCTTGG 3′ | 19 | 2859–2878 |
| SBRO20 | 5′ CCCACCTTAGAGTCCAAGG 3′ | 19 | 873–892 |
| SBFI30 | 5′ GAACAAGAGCTACCGCATGGG 3′ | 21 | 2877–2898 |
| SBRI40 | 5′ CAAGAGACAAAAGAAAATTGG 3′ | 21 | 810–789 |
Nucleotide positions are based on the HBV reference sequence (contained in 3221 bp) that was retrieved from DDBJ/EMBL/GenBank.
2.3. Interpretation of HBsAg nucleotide sequences and phylogenetic analysis
Purified PCR products were sequenced in both directions using a Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems (ABI), Foster City, CA, USA). The ABI Prism 310 genetic analyzer was used for electrophoresis and data collection according to the manufacturer’s protocol. All isolate sequences were assembled using SeqMan II software (DNAStar Inc., Madison, WI, USA) and multiple alignments with the reference sequences of HBsAg genotypes and sub-genotypes (A-I) were confirmed using CLUSTAL W and MEGA 5.2.2 software. Phylogenetic trees were constructed using the Tamura-Nei model of evolutionary distance, and the topology was evaluated by bootstrap analysis (1000 replicates) using the neighbor joining method (Saitou and Nei, 1987, Tamura and Nei, 1993).
2.4. Amino acid composition analysis
The protein coding regions of the HBsAg (pre S1, Pre S2, and large S) gene were translated into amino acid sequences using the standard and universal genetic codes, respectively, and were compared to the peptide sequences of the surface antigens of other HBsAg strains that had been retrieved from the DNA Data Bank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), and GenBank databases (Tallo et al., 2008).
2.5. Nucleotide sequence submission
The base sequence data reported in this study has been submitted to DDBJ/EMBL/GenBank and assigned accession numbers KP191641–KP191650 for the 1.2 Kb partial sequences.
3. Results
3.1. Amplification of the HBsAg gene using nested PCR
Nested PCR confirmed the presence of HBV-DNA in 15 (88.3%) out of 17 HBsAg positive samples whereas in the remaining 2 (11.7%), the absence of HBV DNA was verified even after the second round of PCR amplification. All positive PCR products were at the expected size of approximately 1.2 Kb that included almost the entire Pre S1/Pre S2 and S genomic region (Fig. 3).
Figure 3.

PCR amplification of the HBsAg gene using nested PCR. “L” represents the 100 bp DNA ladder ranging from 100 to 1200 bp. Lanes 1, 2, and 5 contain DNA fragments of the HBsAg gene, whereas lanes 3 and 4 show negative results.
3.2. Identification of HBV genotypes and sub-genotypes based on the complete nucleotide sequence of the HBsAg gene
Phylogenetic analysis of the 1.2 Kb genomic regions from each sample, corresponding to the complete HBsAg gene and containing approximately the entire Pre S1/Pre S2/S genomic locus was performed in order to analyze the nucleotide heterogeneity of the present isolates with respect to the reference HBV genotypes (A-I). The completed tree demonstrated nine distinct clusters comparable to the known HBV genotypes (A-I); 11 (73%; S028, S118S, S18014, S318S, S140014, S20014, S140914, S02E12, S2312E, S07E, and S10612E) out of the 15 new isolates seemed to be more closely related to HBV/D by nucleotide distance identity, means ±0.034, whereas 4 (27%) isolates (S4S214, S5S014, S34014, and S04S) were identified as HBV/C with a nucleotide distance means ±0.036 that verified this relationship (Fig. 4).
Figure 4.

Phylogenetic tree constructed using the neighbor-joining method, based on nine 1.3-kb, full length HBsAg reference sequences retrieved from the DDBJ/EMBL/GenBank Database representing all HBV genotypes (A-I). All the reference isolates are indicated with their accession nos. In addition, the 15 Saudi isolates whose nucleotide sequences were determined in the present study (indicated with a blue closed triangle) are shown; 11 isolates clustered with HBV/D and are represented by the red branches, while 4 isolates were grouped with HBV/C and are represented by the green branches. Bootstrap values indicate the major nodes as a percentage of the data obtained from 1000 replications.
Whole HBsAg reference sequences of 117 HBV/D sub-genotype strains including the present 11 isolates were used to generate another phylogenetic tree. The reference sequences were retrieved from the DDBJ/EMBL/GenBank databases along with their accession number and country of origin for identification. All 117 isolates were grouped into clusters that represented the nine sub genotypes of HBV/D (D1–D8 and D10). An exclusive subset of eight HBV/D isolates (S18014, S140014, S20014, 140914, S02E12, S2312E, S07E, and S10612E) (72%) were observed to be closest to reported HBV/D1 strains with identities of 97.5–99%, indicating that these eight HBV/D isolates belonged to sub-genotype D1. The three remaining isolates (S028, S118S, and S318S) formed a particular cluster that belonged to subtype DA, despite segregating into a larger cluster related D5 with identities of 94–96% (Fig. 5).
Figure 5.

Phylogenetic tree constructed by the neighbor-joining method, based on the entire nucleotide sequences of the 106 reported HBsAg genes of sub genotypes D1-D10. The gene sequences are represented by different colored branches as indicated by the accession no., followed by the country of isolation. The 11 Saudi HBV/D isolates whose entire pre/S1, pre/S2, and S gene sequences were determined in the present study are indicated with black closed triangles. The HBV/C isolate (X01587) was used as an out group and was indicated with a brown closed circle. Bootstrap values are indicated for the major nodes as a percentage of the data obtained from 1000 replicates.
3.3. Determination of the amino acids specific for HBV/D1
In the present study, all eleven HBV/D isolates exhibited a common 11 amino acid deletion in the pre S1 region consistent with the categorization into genotype D. The amino acid sequence of S18014, S140014, S20014, S140914, S02E12, S2312E, S07E, and S10612E isolates were compared against those of HBV/D1 reference sequences retrieved from the DDBJ/EMBL/GenBank databases representing strains from different countries such as Egypt, Turkey, Germany, Uzbekistan, India, USA, Iran, and China. As indicated in Table 2, the lack of any amino acid insertion or deletion was confirmed in all eight HBV/D1 isolates, despite the identification of substitutions established across the entire Pre S1/Pre S2/S region, and particularly within the Pre S1 region. Therein, one novel amino acid substitution was demonstrated in S01612E, S07S, and S140014 isolates. Two amino acid substitutions were found to be specific to S02E12 and S07S isolates. Four and five amino acid substitutions were characterized in S2312E and S140914 isolates, respectively, while S18014 and S20014 showed six amino acid substitutions. In the Pre S2 region, one amino acid residue substitution was verified in both S07S and S140914, and the S20014 isolate showed two amino acid residue changes in the S region.
Table 2.
Conserved amino acid residues in HBV/D1 isolates.
 |
Conserved amino acid residues in the HBsAg region (Pre S1, Pre S2, S) were derived from the different HBV/D reference sequences retrieved from DDBJ/EMBL/GenBank and from the eight isolates described in the present study (S18014, S140014, S20014, S140914, S02E12, S2312E, S07E, and S10612E). The amino acid sequence was derived from the nucleotide sequence.
n = No. of sub-genotypes isolates.
Subgenotype-specific substitutions are shown in red.
3.4. Amino acid changes of HBV/DA isolates in comparison with other isolates of HBV/D5
Comparison of the amino acid sequences among the complete HBsAg genes of two references isolates (DQ315779 India and AB033558 Indonesia) belonging to D5 with the present HBV/DA isolates verified 10 DA-specific amino acid substitutions in all HBV/DA strains. In the Pre S1 region, these were Arg28Ala, Pro81Ala, and Gln89Lys; in the Pre S2 region, Pro162Leu; and in the S region, Phe171Leu, Met288Thr, Thr290Pro, Gly348Glu, Leu349Val, and Met361Arg were found. In addition, two unique amino acid residues; Pro149His in the Pre/S2 gene and Pro377Leu in the S gene, were observed in the S318S and S118S strains, respectively (Table 3).
Table 3.
Conserved amino acid residues in HBV/D5 isolates.
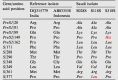 |
Conserved amino acid residues in HBsAg (Pre S1, Pre S2, S) were derived from 2 references sequences of HBV/D5 indicated by their DDBJ/EMBL/GenBank Accession numbers and 3 isolates of the present study (S028, S118S, and S318S). The amino acid sequence was derived from the nucleotide sequence.
Sub-genotype-specific substitutions are italicized.
Sub-genotype-unique amino acids are shown in red.
4. Discussion
HBV has drawn attention for the peculiar geographic distribution of its nine distinctive immunologic genotypes (Kao et al., 2003, Liu et al., 2002). Saudi Arabia, and especially Jeddah, receives millions of pilgrims from various countries annually because of the Hajj and Umrah seasons. However, despite the import of the infection, molecular epidemiological data of the specific HBV genotype and sub-genotype strains circulating in Jeddah are limited (Al-Faleh et al., 1992, Al-Faleh et al., 1999). Phylogenetic analysis of 15 current isolates demonstrated that HBV/D is the most dominant HBV genotype (73%) among Saudi patients with chronic HBV, followed by HBV/C (27%). This result is consistent with many previous observations that postulated that the HBV genotype D appeared to be predominant in the Mediterranean basin, including the Middle Eastern countries (EL Hadad et al., 2013, Norder et al., 2004, Saudy et al., 2003), whereas HBV/C was dominant in Southeast Asian countries (Lusida et al., 2008).
Recently, ten sub-genotypes of HBV/D (D1 to D10) have been reported, each showing distinct geographical clustering (Tran et al., 2014). In the present work, eight isolates belonging to HBV/D (S18014, S140014, S20014, S140914, S02E12, S2312E, S07E, and S10612E) segregated with sub-genotype D1 reference isolates, and the inter distances equaling 0.006 verified this effect. This result is in agreement with recent studies that reported the prevalence of HBV/D1 in regions of the Middle East through North Africa (EL Hadad et al., 2013, Tran et al., 2014). Additionally, a possible new HBV sub-genotype (DA) was suggested by 3 isolates, S028, S318S, and S118S, which showed identity similarities of 94−96%.
Many conflicting reports have arisen in the past few years regarding the identification of novel HBV/D-sub-genotypes. Recently, new HBV/B3 sub-genotypes have been sorted out using partial sequence of the S gene (Ghosh et al., 2013). In addition, HBV/D9 has been classified from isolates showing recombination between genotypes D and C in the pre core/core regions, although the pre-S/S open reading frame did not possess any unique motifs that could distinguish HBV/D9 isolates from the other eight sub genotypes of D. Furthermore, the frequent practice of sequencing only the pre-S/S region for defining genotype/sub genotype can lead to their improper classification within sub-genotype D3 or D2 (Ghosh et al., 2013, Tran et al., 2014). The former phenomenon was confirmed by our present phylogenetic tree, wherein current Pre S/S sequence analysis of the JN664922 India isolate placed it as belonging to HBV/D3 instead of as an HBV/D9 subtype, as characterized by previous analysis. In the present study, three current isolates (S028, S318S, and S118S) were clustered with reference strains representing HBV/D5. However, this observation did not affect the proposed establishment of a novel HBV/DA sub-genotype, as the sub-genotype HBV/D6 strains clustered within the HBV/D3 branch. In addition, the phylogenetic analysis provided corroboration of both HBV/D9 and HBV/D10 (Yousif and Kramvis, 2013). Thus, whole genome sequence analysis is required to substantiate whether the present isolates actually belong to HBV/D5 or represent a novel sub-genotype, despite that HBV classification was initially built on the premise of inter-genotype divergence of at least 8% of the complete nucleotide sequence or more than 4% in the HBsAg gene, which stringencies these isolates meet (Magnius and Norder, 1995).
The demonstrated importance of illustrating the difference in the HBsAg nucleotide sequences of the present isolates urges further study on the differences in the sequences of amino acids. No alterations in amino acid sequence result from “nonsense” nucleotide mutations (Rodriguez-Frias et al., 1999, Thuy le et al., 2005), whereas “sense” mutations might lead to alterations in the amino acid sequence either by substitution, insertion, or deletion (Weinberger et al., 2000). Therefore, sense mutations might lead to the creation of mutants with the specific ability to escape antibody detection and neutralization. These mutations thus might also lead to re-infection because HBV replicates through an RNA intermediate synthesized by the reverse transcriptase of viral genomes (Kfoury Baz et al., 2001, Kohno et al., 1996, Kreutz, 2002, Miyake et al., 1996, Ohishi et al., 2004).
As shown in Table 2, the present HBV/D1 isolates (S18014, S140014, S20014, 140914, S02E12, S2312E, S07E, and S10612E) showed the presence of 1–8 unique amino acid residues out of the 390 that represent the entire Pre S/S region. Amino acid sequence alignments were also performed between the three HBV/DA isolates (S028, S318S, and S118S) and two reported reference sequences related to HBV/D5 (DQ315779 India and AB033558 Indonesia) to ensure that the proposed sub-genotype harbored nucleotide and amino acid motifs that were specific to the novel sub-genotype. The current HBV/DA isolates harbored 3, 1, and 6 amino acid residues, respectively in the Pre S1/Pre S2/S locus that were unique to the proposed sub-genotype. In addition, two unique amino acid residues, Pro149His in the Pre/S2 gene and Pro377Leu in the S gene, were observed in the S318S and S118S strains, respectively (Table 3). The relatively large number of sense mutations identified in HBV suggests that this virus might be better at avoiding elimination than HAV and HCV; in addition, HBV might be more stable than the latter, which are RNA viruses. Therefore, our result might explain one of the reasons why in Saudi Arabia; the incidence of viral hepatitis is decreasing for both HAV and HCV, but not for HBV, which showed a minimal increase. Overall, between hepatitis A, B, and C, HBV was the most predominant type, accounting for 53% of identified cases, followed by HCV (30%) and HAV (17%) (Alshabanat et al., 2013).
5. Conclusion
This study highlighted the sequence analysis of HBsAg genes isolated from Saudi patients with chronic HBV. The sequence results obtained from these isolates verified that HBV/D and HBV/C are the most common genotypes in this population. This result confirmed previously published findings demonstrating that HBV/D was the dominant type in the Middle East (EL Hadad et al., 2013, Liu et al., 2002). In addition, the current study revealed the presence of multiple sub-genotypes of HBV within the D genotype, with the dominant distribution of the seemingly indigenous sub-genotype D1. Therefore, the availability of complete HBsAg sequences might serve as a reference for future epidemiological studies of HBV viruses. Additionally, this study identified a new sub-genotype, tentatively designated DA in Jeddah, KSA, which meets the proposed rules for HBV/D classification (Norder et al., 2004). For further confirmation whether the three current isolates belong to HBV/D5 or represent a novel sub-genotype, HBV/DA, whole HBV genome sequences would be required. Similarly, additional studies with large numbers of cases in previously examined and unexamined areas will likely lead to the discovery of the HBV strains and even possibly additional new sub-genotype strains circulating in Saudi Arabia.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah (Grant No. 462/247/1433). The authors acknowledge with thanks the DSR technical and financial support.
Footnotes
Peer review under responsibility of King Saud University.
References
- Abdou Chekaraou M., Brichler S., Mansour W., Le Gal F., Garba A., Dény P., Gordien E. A novel hepatitis B virus (HBV) subgenotype D (D8) strain, resulting from recombination between genotypes D and E, is circulating in Niger along with HBV/E strains. J. Gen. Virol. 2010;91:1609–1620. doi: 10.1099/vir.0.018127-0. [DOI] [PubMed] [Google Scholar]
- Al-Faleh F.Z., Al-Jeffri M., Ramia S., Al-Rashed R., Arif M., Rezeig M., Al-toraif I., Bakhsh M., Mishkkhas A., Makki O., Al-Freihi H., Mirdad S., AlJuma A., Yasin T., Al-Swallem A., Ayoola A. Seroepidemiology of hepatitis B virus infection in Saudi children 8 years after a mass hepatitis B vaccination programme. J. Infect. 1999;38:167–170. doi: 10.1016/s0163-4453(99)90245-1. [DOI] [PubMed] [Google Scholar]
- Al-Faleh F.Z., Ayoola E.A., Arif M., Ramia S., Al-Rashad R., Al-Jeffry M., Al-Mofarreh M., Al-Karawi M., Al-Shabrawy M. Seroepidemiology of hepatitis B virus infection in Saudi Arabian children: a baseline survey for mass vaccination against hepatitis B. J. Infect. 1992;24:197–206. doi: 10.1016/0163-4453(92)93006-c. [DOI] [PubMed] [Google Scholar]
- Alshabanat A.A., Albacker R.B., Basalama A.A., Bin Salamah A.A., Alfrayh A.S. Profile of viral hepatits in Saudi Arabia. Biomed. Res. 2013;24:396–399. [Google Scholar]
- Arauz-Ruiz P., Norder H., Visona K.A., Magnius L.O. Genotype F prevails in HBV infected patients of hispanic origin in Central America and may carry the precore stop mutant. J. Med. Virol. 1997;51:305–312. [PubMed] [Google Scholar]
- Banerjee A., Kurbanov F., Datta S., Chandra P.K., Tanaka Y., Mizokami M., Chakravarty R. Phylogenetic relatedness and genetic diversity of hepatitis B virus isolates in Eastern India. J. Med. Virol. 2006;78:1164–1174. doi: 10.1002/jmv.20677. [DOI] [PubMed] [Google Scholar]
- Carman W.F. The clinical significance of surface antigen variants of hepatitis B virus. J. Viral Hepat. 1997;4:11–20. doi: 10.1111/j.1365-2893.1997.tb00155.x. [DOI] [PubMed] [Google Scholar]
- Chan H.L., Sung J.J. Hepatocellular carcinoma and hepatitis B virus. Semin. Liver Dis. 2006;26:153–161. doi: 10.1055/s-2006-939753. [DOI] [PubMed] [Google Scholar]
- Chu C.J., Keeffe E.B., Han S.H., Perrillo R.P., Min A.D., Soldevilla-Pico C., Carey W., Brown R.S., Jr., Luketic V.A., Terrault N., Lok A.S. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology. 2003;125:444–451. doi: 10.1016/s0016-5085(03)00895-3. [DOI] [PubMed] [Google Scholar]
- EL Hadad S.R., Alakilli S.Y., Ramadan H.A., Baeshen N.A. Novel mutations of hepatitis B virus surface antigen genotype D among chronic Egyptian patients. Afr. J. Microbiol. Res. 2013;7:814–824. [Google Scholar]
- Ghosh S., Banerjee P., Deny P., Mondal R.K., Nandi M., Roychoudhury A., Das K., Banerjee S., Santra A., Zoulim F., Chowdhury A., Datta S. New HBV subgenotype D9, a novel D/C recombinant, identified in patients with chronic HBeAg-negative infection in Eastern India. J. Viral Hepat. 2013;20:209–218. doi: 10.1111/j.1365-2893.2012.01655.x. [DOI] [PubMed] [Google Scholar]
- Hou J., Liu Z., Gu F. Epidemiology and prevention of hepatitis b virus infection. Int. J. Med. 2005;2:50–57. doi: 10.7150/ijms.2.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huy T.T., Ushijima H., Sata T., Abe K. Genomic characterization of HBV genotype F in Bolivia: genotype F subgenotypes correlate with geographic distribution and T1858 variant. Arch. Virol. 2006;151:589–597. doi: 10.1007/s00705-005-0671-1. [DOI] [PubMed] [Google Scholar]
- Jalali M.V., Alavian S. Hepatitis B e antigen-negative chronic hepatitis B. Hepat. Mon. 2006;6(1):31–35. [Google Scholar]
- Kao J.H., Chen P.J., Lai M.Y., Chen D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327–334. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- Kfoury Baz E.M., Zheng J., Mazuruk K., Van Le A., Peterson D.L. Characterization of a novel hepatitis B virus mutant: demonstration of mutation-induced hepatitis B virus surface antigen group specific a determinant conformation change and its application in diagnostic assays. Transfus. Med. 2001;11:355–362. doi: 10.1046/j.1365-3148.2001.00323.x. [DOI] [PubMed] [Google Scholar]
- Kohno H., Inoue T., Tsuda F., Okamoto H., Akahane Y. Mutations in the envelope gene of hepatitis B virus variants co-occurring with antibody to surface antigen in sera from patients with chronic hepatitis B. J. Gen. Virol. 1996;77:1825–1831. doi: 10.1099/0022-1317-77-8-1825. [DOI] [PubMed] [Google Scholar]
- Kramvis A., Arakawa K., Yu M.C., Nogueira R., Stram D.O., Kew M.C. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J. Med. Virol. 2008;80:27–46. doi: 10.1002/jmv.21049. [DOI] [PubMed] [Google Scholar]
- Kramvis A., Kew M., François G. Hepatitis B virus genotypes. Vaccine. 2005;23:2409–2423. doi: 10.1016/j.vaccine.2004.10.045. [DOI] [PubMed] [Google Scholar]
- Kreutz C. Molecular, immunological and clinical properties of mutated hepatitis B virus. J. Cell Mol. Med. 2002;6:113–143. doi: 10.1111/j.1582-4934.2002.tb00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.M. Hepatitis B virus infection. N. Engl. J. Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- Liu C.J., Kao J.H., Shau W.Y., Chen P.J., Lai M.Y., Chen D.S. Naturally occurring hepatitis B surface gene variants in chronic hepatitis B virus infection: correlation with viral serotypes and clinical stages of liver disease. J. Med. Virol. 2002;68:50–59. doi: 10.1002/jmv.10169. [DOI] [PubMed] [Google Scholar]
- Liu W.C., Phiet P.H., Chiang T.Y., Sun K.T., Hung K.H., Young K.C., Wu I.C., Cheng P.N., Chang T.T. Five subgenotypes of hepatitis B virus genotype B with distinct geographic and virological characteristics. Virus Res. 2007;129:212–223. doi: 10.1016/j.virusres.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Liu S., Xie J., Yin J., Zhang H., Zhang Q., Pu R., Li C., Ni W., Wang H., Cao G. A matched case-control study of hepatitis B virus mutations in the preS and core promoter regions associated independently with hepatocellular carcinoma. J. Med. Virol. 2011;83:45–53. doi: 10.1002/jmv.21829. [DOI] [PubMed] [Google Scholar]
- Lusida M.I., Nugrahaputra V.E., Soetjipto Handajani.R., Nagano-Fujii M., Sasayama M., Utsumi T., Hotta H. Novel subgenotypes of hepatitis B virus genotypes C and D in Papua, Indonesia. J. Clin. Microbiol. 2008;46:2160–2166. doi: 10.1128/JCM.01681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnius L.O., Norder H. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology. 1995;38:24–34. doi: 10.1159/000150411. [DOI] [PubMed] [Google Scholar]
- McElhinney L.M., Marston D.A., Freuling C.M., Cragg W., Stankov S., Lalosevic D., Lalosevic V., Müller T., Fooks A.R. Molecular diversity and evolutionary history of rabies virus strains circulating in the Balkans. J. Gen. Virol. 2011;92:2171–2180. doi: 10.1099/vir.0.032748-0. [DOI] [PubMed] [Google Scholar]
- Miyakawa Y., Mizokami M. Classifying hepatitis B virus genotypes. Intervirology. 2003;46:329–338. doi: 10.1159/000074988. [DOI] [PubMed] [Google Scholar]
- Miyake Y., Oda T., Li R., Sugiyama K. A comparison of amino acid sequences of hepatitis B virus S gene in 46 children presenting various clinical features for immunoprophylaxis. Tohoku J. Exp. Med. 1996;180:233–247. doi: 10.1620/tjem.180.233. [DOI] [PubMed] [Google Scholar]
- Mulyanto, De pamede S.N., Surayah K., Tsuda F., Ichiyama K., Takahashi M., Okamoto H. A nationwide molecular epidemiological study on hepatitis B virus in Indonesia: identification of two novel subgenotypes, B8 and C7. Arch. Virol. 2009;154:1047–1059. doi: 10.1007/s00705-009-0406-9. [DOI] [PubMed] [Google Scholar]
- Mulyanto Depamede.S.N., Wahyono A., Jirintai Nagashima.S., Takahashi M., Okamoto H. Analysis of the full-length genomes of novel hepatitis B virus subgenotypes C11 and C12 in Papua, Indonesia. J. Med. Virol. 2011;83:54–64. doi: 10.1002/jmv.21931. [DOI] [PubMed] [Google Scholar]
- Norder H., Couroucé A.M., Coursaget P., Echerarria J.M., Lee S.D., Mushahwar I.K., Robertson B.H., Locarnini S., Magnius L.O. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes and HBsAg subtypes. Intervirology. 2004;47:289–309. doi: 10.1159/000080872. [DOI] [PubMed] [Google Scholar]
- Norder H., Couroucé A.M., Magnius L.O. Molecular basis of hepatitis B virus serotype variations within the four major subtypes. J. Gen. Virol. 1992;73:3141–3145. doi: 10.1099/0022-1317-73-12-3141. [DOI] [PubMed] [Google Scholar]
- Norder H., Couroucé A.M., Magnius L.O. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology. 1994;198:489–503. doi: 10.1006/viro.1994.1060. [DOI] [PubMed] [Google Scholar]
- Ohishi W., Shirakawa H., Kawakami Y., Kimura S., Kamiyasu M., Tazuma S., Nakanishi T., Chayama K. Identification of rare polymerase variants of hepatitis B virus using a two-stage PCR with peptide nucleic acid clamping. J. Med. Virol. 2004;72:558–565. doi: 10.1002/jmv.20026. [DOI] [PubMed] [Google Scholar]
- Okamoto H., Imai M., Tsuda F., Tamaka T., Miyakawa Y., Mayumi M. Point mutation in the S gene of hepatitis B virus for ad/y or w/r subtypic change in two blood donors carrying a surface antigen of compound subtype adyr or adwr. J. Virol. 1987;61:3030–3034. doi: 10.1128/jvi.61.10.3030-3034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Frias F., Buti M., Jardi R., Vargas V., Quer J., Cotrina M., Martell M., Esteban R., Guardia J. Genetic alterations in the S gene of hepatitis B virus in patients with acute hepatitis B, chronic hepatitis B and hepatitis B liver cirrhosis before and after liver transplantation. Liver. 1999;19:177–182. doi: 10.1111/j.1478-3231.1999.tb00032.x. [DOI] [PubMed] [Google Scholar]
- Sakamoto T., Tanaka Y., Orito E., Co J., Clavio J., Sugauchi F., Ito K., Ozasa A., Quino A., Ueda R., Soliano J., Mizokami M. Novel subtypes (subgenotypes) of hepatitis B virus genotypes B and C among chronic liver disease patients in the Philippines. J. Gen. Virol. 2006;87:1873–1882. doi: 10.1099/vir.0.81714-0. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Santos A.O., Alvarado-Mora M.V., Botelho L., Vieira D.S., Pinho J.R., Carrilho F.J., Honda E.R., Salcedo J.M. Characterization of hepatitis B virus (HBV) genotypes in patients from Rondônia. Brazil. Virol. J. 2010;7:315. doi: 10.1186/1743-422X-7-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudy N., Sugauchi F., Tanaka Y., Suzuki S., Aal A.A., Zaid M.A., Agha S., Mizokami M. Genotypes and phylogenetic characterization of hepatitis B and delta viruses in Egypt. J. Med. Viol. 2003;70:529–536. doi: 10.1002/jmv.10427. [DOI] [PubMed] [Google Scholar]
- Sumi H., Yokosuka O., Seki N., Arai M., Imazeki F., Kurihara T., Kanda T., Fukai K., Dato M., Saisho H. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology. 2003;37:19–26. doi: 10.1053/jhep.2003.50036. [DOI] [PubMed] [Google Scholar]
- Szmuness W., Stevens C.E., Zang E.A., Harley E.J., Kellner A. A controlled clinical trial of the efficacy of the hepatitis B vaccine (Heptavax B): a final report. Hepatology. 1981;1:377–385. doi: 10.1002/hep.1840010502. [DOI] [PubMed] [Google Scholar]
- Tallo T., Tefanova V., Priimägi L., Schmidt J., Katargina O., Michallov M., Mukomolov S., Magnius L., Norder H. D2: Major subgenotype of hepatitis B virus in Russia and the Baltic region. J. Gen. Virol. 2008;89:1829–1839. doi: 10.1099/vir.0.83660-0. [DOI] [PubMed] [Google Scholar]
- Tamura K., Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Thuy le T.T., Thi L., Ryo H., Van Phung L., Furitsu K., Nomura T. Distribution of genotype/subtype and mutational spectra of the surface gene of hepatitis B virus circulating in Hanoi, Vietnam. J. Med. Virol. 2005;76:161–169. doi: 10.1002/jmv.20337. [DOI] [PubMed] [Google Scholar]
- Tian Y., Xu Y., Zhang Z., Meng Z., Qin L., Lu M., Yang D. The amino acid residues at positions 120 to 123 are crucial for the antigenicity of hepatitis B surface antigen. J. Clin. Microbiol. 2007;45:2971–2978. doi: 10.1128/JCM.00508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torresi J. The virological and clinical significance of mutations in the overlapping envelope and polymerase genes of hepatitis B virus. J. Clin. Virol. 2002;25:97–106. doi: 10.1016/s1386-6532(02)00049-5. [DOI] [PubMed] [Google Scholar]
- Tran H., Yu M.L., Dai C.Y., Lin I.L., Yeh M.L., Chuang W.L., Abe K. Novel quasi-subgenotypeD2 of hepatitis B virus identified in Taianese aborigines. Virus Genes. 2014;49:30–37. doi: 10.1007/s11262-014-1072-x. [DOI] [PubMed] [Google Scholar]
- Weinberger K.M., Bauer T., Böhm S., Jilg W. High genetic variability of the group-specific a-determinant of hepatitis B virus surface antigen (HBsAg) and the corresponding fragment of the viral polymerase in chronic virus carriers lacking detectable HBsAg in serum. J. Gen. Virol. 2000;81:1165–1174. doi: 10.1099/0022-1317-81-5-1165. [DOI] [PubMed] [Google Scholar]
- Wen Y.M. Structural and functional analysis of full-length hepatitis B virus genomes in patients: implications in pathogenesis. J. Gastroenterol. Hepatol. 2004;19:485–489. doi: 10.1111/j.1440-1746.2003.03158.x. [DOI] [PubMed] [Google Scholar]
- Yang H.I., Sherman M., Su J., Chen P.J., Liaw Y.F., Iloeje U.H., Chen C.J. Nomograms for risk of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. J. Clin. Oncol. 2010;28:2437–2444. doi: 10.1200/JCO.2009.27.4456. [DOI] [PubMed] [Google Scholar]
- Yousif M., Kramvis A. Genotype D of hepatitis B virus and its subgenotypes: an update. Hepatol. Res. 2013;43:355–364. doi: 10.1111/j.1872-034X.2012.01090.x. [DOI] [PubMed] [Google Scholar]
- Yuen M.F., Sablon E., Yuan H.J., Wong D.K., Hui C.K., Wong B.C., Chan A.O., Lai C.L. Significance of hepatitis B genotype in acute exacerbation, HBeAg seroconversion, cirrhosis-related complications and hepatocellular carcinoma. Hepatology. 2003;37:562–567. doi: 10.1053/jhep.2003.50098. [DOI] [PubMed] [Google Scholar]
- Yuen M.F., Tanaka Y., Fong D.Y., Jung J., Wong D.K., Yuen J.C., But D.Y., Chan A.O., Wong B.C., Mizokami M., Lai C.L. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol. 2009;50:80–88. doi: 10.1016/j.jhep.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Zöllner B., Petersen J., Schröter M., Laufs R., Schoder V., Feucht H.H. 20-fold increase in risk of lamivudine resistance in hepatitis B virus subtype adw. Lancet. 2001;357:934–935. doi: 10.1016/S0140-6736(00)04219-7. [DOI] [PubMed] [Google Scholar]