

Abstract
The flow coupling of epichlorohydrin with substituted phenols, while efficient, limits the nature of the epoxide available for the development of focused libraries of β-amino alcohols. This limitation was encountered in the production of analogues of 1-(4-nitrophenoxy)-3-((2-((4-(trifluoromethyl)pyrimidin-2-yl)amino)ethyl)amino)propan-2-ol 1, a potential antibiotic lead. The in situ (flow) generation of dimethyldoxirane (DMDO) and subsequent flow olefin epoxidation abrogates this limitation and afforded facile access to structurally diverse β-amino alcohols. Analogues of 1 were readily accessed either via (i) a flow/microwave hybrid approach, or (ii) a sequential flow approach. Key steps were the in situ generation of DMDO, with olefin epoxidation in typically good yields and a flow-mediated ring opening aminolysis to form an expanded library of β-amino alcohols 1 and 10a–18g, resulting in modest (11a, 21%) to excellent (12g, 80%) yields. Alternatively flow coupling of epichlorohydrin with phenols 4a–4m (22%–89%) and a Bi(OTf)3 catalysed microwave ring opening with amines afforded a select range of β-amino alcohols, but with lower levels of aminolysis regiocontrol than the sequential flow approach.
Keywords: dimethyl dioxirane, epoxide, flow chemistry, β-amino alcohols, microwave
1. Introduction
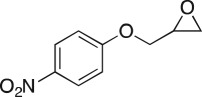
As part of our medicinal chemistry programme we identified β-amino alcohol 1 as a lead compound of interest. Analogues of this nature are known to be biologically active and have been used as key intermediates in the synthesis of natural products [1–5]. β-Amino alcohols are typically accessed through amine-mediated ring opening of epoxides, catalysed by Lewis acids [6–9]. In keeping with an epoxide ring opening strategy, 1 can be accessed via aminopyrimidine 2, racemic epichlorohydrin 3 and 4-nitrophenol 4, enabling robust focused library development (figure 1).
Figure 1.
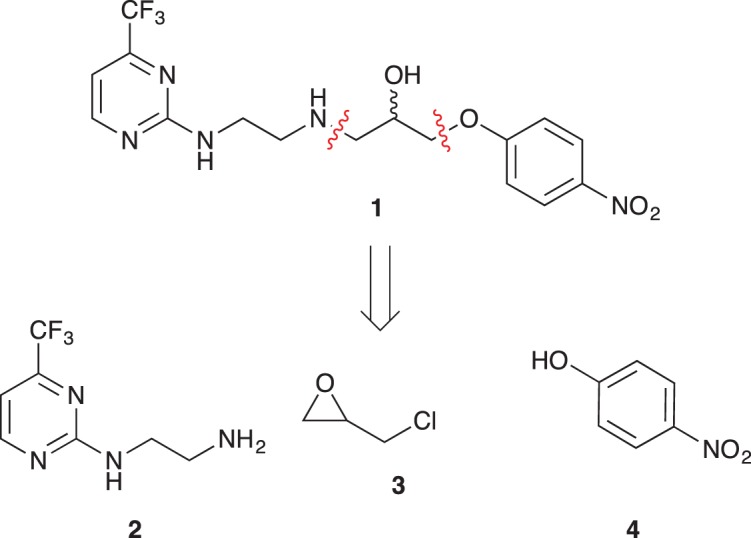
Chemical structure of lead 1 and the identification of three readily accessible fragments: aminopyrimidine 2, epichlorohydrin 3 and 4-nitrophenol 4 for library development.
We like others have developed an interest in process intensification and streamlining of reaction optimization using flow chemistry approaches. Arguably, flow chemistry can now be considered a mature technology with considerable progress in the use of flow chemistry approaches in multi-step synthesis [10–20], the synthesis of drug like molecules [21–26], selective hydrogenations [27] and in the use of unstable and/or dangerous reagents [16–18,27,28].
To date, while the flow coupling of epichlorohydrin with phenols has been reported [29], the scope of this addition has been limited to a few examples only; as was the subsequent epoxide ring opening to form β-amino alcohols, a key constituent of active pharmaceutical ingredients (APIs) [23,30–36]. Critical to our proposed pathway was epoxide 3, and for analogue development, related systems. Epoxides are known versatile intermediates giving rise to drugs such as pioglitazone, metoprolol and levofloxacin; or chemical probes such as wiskostatin [23,31,37,38]. Epoxide installation is typically by direct coupling of an epihalohydrin [37–40] or through m-chloroperoxybenzoic acid, hydrogen peroxide or tert-butyl hydroperoxide olefin epoxidation [41–43].
Herein we report a flow chemistry approach to epoxide installation using epichlorohydrin and the in situ epoxidation of olefins with dimethyldioxirane (DMDO) and the subsequent epoxide aminolysis, facilitating access to a focused library of β-amino alcohols based on 1 [44].
2. Results and discussion
Method development commenced with the use of a flow instrument equipped with peristaltic pumps; passing two reagent streams, epichlorohydrin 3 (neat) and 4-nitrophenol 4 (0.1 M) in dimethylformamide (DMF), through an Omnifit® column packed with Cs2CO3 and acid washed sand (1 : 1) at 75°C, increasing in 10°C increments to 105°C, with the reaction monitored by UPLC-MS (scheme 1; table 1). The system pressure was maintained at 5 bar using the third pump in an active back pressure regulation mode, to circumvent the blockage issues caused by the precipitation of the product we had previously observed. This screen identified an enhanced coupling efficiency with 3 and 4 at higher temperatures, with DMF affording 97% at 105°C. Solvent switching to CH3CN/Cs2CO3 or DMF/diisopropylethyl amine (DIPEA) returned only unreacted 3 and 4. DMF/1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and DMF/N(Bu)4OAc gave 90% and 100% conversion to 5a, respectively. With DMF/N(Bu)4OAc, poor product recovery was a function of difficulties observed in isolating the desired product in the presence of N(Bu)4OAc; thus Cs2CO3 was used for on-going optimizations. Despite the ready recycling of epichlorohydrin, the use of reagents as solvents seriously limited future library generation and poses a greater environmental impact. Thus the effect of reducing the concentration of 3 on the conversion to 5a was examined (table 2).
Scheme 1.

Reagents and conditions: (i) varying concentrations of 3 and 0.05 M 4 (DMF or CH3CN), base, 75–105°C, 1 ml min−1.
Table 1.
Flow optimization of the coupling of epichlorohydrin 3 with 4-nitrophenol 4 at 0.5 ml min−1.
| solvent | base | temp (°C) | ratio (4 : 5a)a,b |
|---|---|---|---|
| DMF | Cs2CO3 | 75 | 50 : 50 |
| DMF | Cs2CO3 | 85 | 20 : 80 |
| DMF | Cs2CO3 | 95 | 5 : 95 |
| DMF | Cs2CO3 | 105 | 3 : 97 |
| CH3CN | Cs2CO3 | 105 | 100 : 0 |
| DMF | DIPEA | 105 | 100 : 0 |
| DMF | DBU | 105 | 10 : 90 |
| DMF | N(Bu)4OAc | 105 | 0 : 100 |
aReaction conditions: (i) 0.1 M 4-nitrophenol (4), neat epichlorohydrin (3), anhydrous DMF, residence time (tr) = 10 min, 5 bar.
bRatios calculated by ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS) analysis.
Table 2.
Flow optimization of the coupling of epichlorohydrin 3, with 0.1 M 4-nitrophenol (4) at 1 ml min−1.
| epichlorohydrin 3 (M) | residence time (min) | temp (°C) | ratioa,b (4 : 5a : 6a) |
|---|---|---|---|
| 6 | 10 | 105 | 16 : 84 : 0 |
| 5 | 10 | 105 | 34 : 66 : 0 |
| 2.5 | 10 | 105 | 61 : 39 : 0 |
| 0.5 | 10 | 105 | 87 : 13 : 0 |
| 2.5 | 20 | 105 | 30 : 70 : 0 |
| 2.5 | 20 | 115 | 54 : 0 : 46 |
| 2.5 | 20 | 125 | 5 : 0 : 95 |
| 5 | 20 | 105 | 0 : 100 : 0 |
a Reaction conditions: tr = 10 min (10 ml loop) or 20 min (2 × 10 ml loop), 5 bar.
bRatios calculated by UPLC-MS analysis.
Use of 0.1 M of 4-nitrophenol 4 and 5, 2.5 and 0.5 M 3 in DMF gave 66%, 39% and 13% conversion to 5a, respectively. At 2.5 M of 3, a 20 min residence time saw increased conversion to 5a from 39% to 70%. At 115°C, unwanted diol 6a was evident, but this was reduced through the use of a 4 Å sieves/acid washed sand/Cs2CO3 (2 : 1 : 1 v/v/v) loaded Omnifit® column. Quantitative conversion to epoxy ether 5a was noted at 105°C, with 5 M 3 and a 20 min residence time (scheme 2).
Scheme 2.

Reagents and conditions: anhydrous DMF, tr = 20 min, 105°C, 5 bar, 1.0 ml min−1.
With phenols 4a–4m a range of conversion rates to the desired epoxy ether 5a–5m from quantitative (5a, 5b and 5d) to modest (5j, 28%) were observed (table 3). Isolated yields in most cases were excellent (5a, 5d, both 89%) with the exception of 5j (22%, from 28% conversion). The remaining analogues showed consumption of phenol 4 but the presence of the undesired diol (5c, 5e and 5m), incomplete consumption of 4 (5j, 5k and 5l) or incomplete consumption of 4 and undesired diol (5f–5i) (table 3). In our hands the use of DMF and solid Cs2CO3 bypassed any potential issue with bi-phasic solutions, and the boiling point difference between DMF and epichlorohydrin facilitated easy removal and recycling of the latter reagent [29]. Racemic epichlorohydrin was used; while moderately inexpensive, the recycling procedure was considered necessary to reduce the environmental effects of excess epichlorohydrin, as well as demonstrate the use of the protocol if enantiomerically pure products were required.
Table 3.
Flow synthesis of aromatic epoxy ethers 5a–5m.
| compounda | phenol (4a–m) | conversion (4a–m : 5a–m: 6) | yield (%) |
|---|---|---|---|
| 5a | 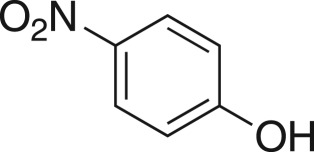 |
0 : 100 : 0 | 84–89 (n = 4) |
| 5b | 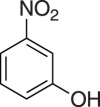 |
0 : 100 : 0 | 88 |
| 5c | 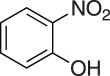 |
0 : 70 : 30 | 67 |
| 5d | 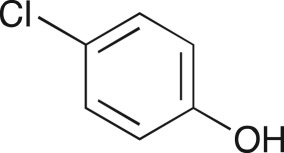 |
0 : 100 : 0 | 89 |
| 5e | 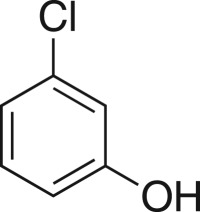 |
0 : 76 : 24 | 74 |
| 5f | 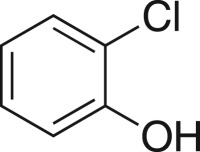 |
10 : 76 : 14 | 70 |
| 5g | 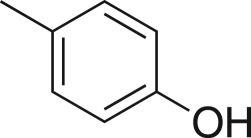 |
32 : 65 : 3 | 60 |
| 5h |  |
29 : 64 : 3 | 56 |
| 5i |  |
17 : 76 : 4 | 61 |
| 5j |  |
71 : 28 : 0 | 22 |
| 5k |  |
22 : 78 : 0 | 62 |
| 5l | 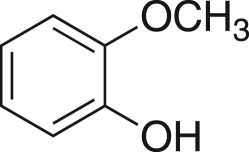 |
16 : 84 : 0 | 70 |
| 5 m | 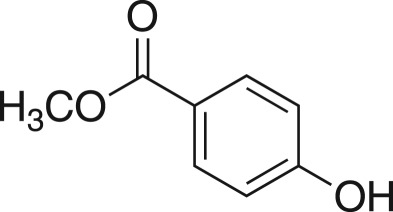 |
0 : 81 : 19 | 62 |
aFor reagents and conditions, refer to scheme 2.
Concerned about the possible depletion of the Cs2CO3 from the Omnifit® column, we scaled up the synthesis of 5a, with a column packed with 16 g 1 : 1 w/w clean sand : Cs2CO3, examining the conversion rates as a function of time with analysis conducted at 30 min time points (figure 2). From this data, we were able to generate 1.48 g 5a in 3 h (84% isolated yield; 0.49 g h−1). During the course of this continuous production the observed conversion rate remained essentially constant for the first 2.5 h, only showing a slight diminution after 3 h, to 94%. There was no evidence of channels of favoured flow paths developing within the Omnifit® column.
Figure 2.
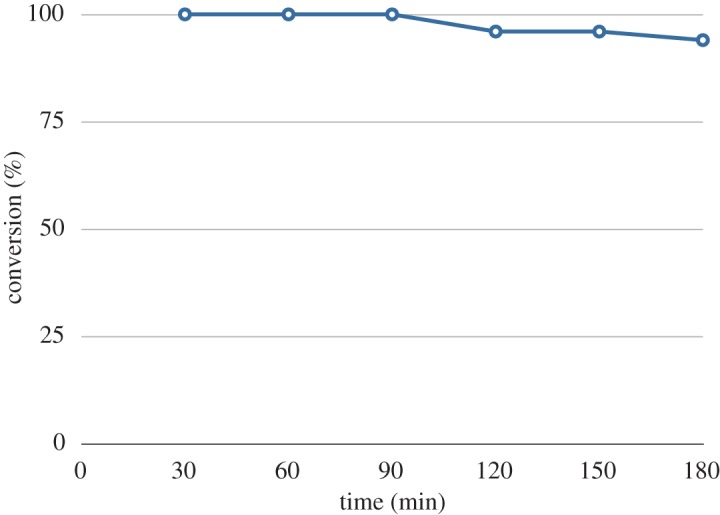
The percentage conversion of 5 M 3 and 0.1 M 4 (both in DMF) to 5a at 1 ml min−1, 105°C as a function of time. Data collected at 30 min intervals, with conversions based on UPLC-MS analysis with ultraviolet-visible spectrometry detection at 320 and 255 nm.
To nullify the limited availability of novel epoxides, we explored the in situ epoxidation capability of DMDO with selected aromatic and aliphatic olefins (table 4) [45,46]. The production of water-soluble by-products, as well as leveraging the known advantage of flow approaches in handling potentially sensitive reagents, made this method more attractive than traditional epoxidizing approaches [31,41–43]. No epoxidation of 4-allylanisole 7 with NaHCO3/Oxone® (1 : 1; in situ production of DMDO) was observed at 19°C, but at 30°C 16% conversion to epoxide 8a was noted by gas chromatography mass spectrometry (GCMS) analysis (scheme 3). This increased to 61% at 60°C. Quantitative conversion to epoxide 8a was accomplished using 2 equivalents of Oxone® at 60°C (electronic supplementary material, table S1). To further improve the safety of this reaction, the optimized reaction was repeated with the addition of a 0.4 M sodium sulfite(aq) stream introduced after the back pressure regulator to quench any unreacted DMDO. Analysis and subsequent yield calculation showed no reduction in conversion or isolated yields.
Table 4.
DMDO-mediated flow epoxidation of unsaturated scaffolds 8a–8k.
| alkene | product | conversiona (7 : 8) | yield (%) |
|---|---|---|---|
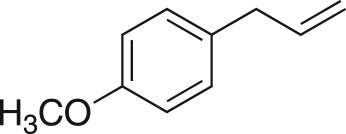 |
 |
0 : 100 | 93 |
| 7a | 8a | ||
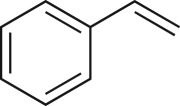 |
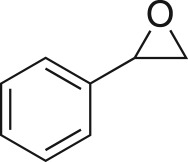 |
0 : 100 | 98 |
| 7b | 8b | ||
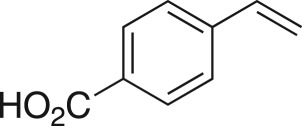 |
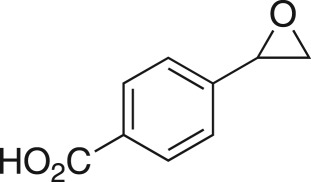 |
30 : 70 | 65 |
| 7c | 8c | ||
 |
 |
20 : 80 | 74 |
| 7d | 8d | ||
 |
 |
30 : 70 | 60 |
| 7e | 8e | ||
 |
 |
no reaction | — |
| 7f | 8f | ||
 |
 |
no reaction | — |
| 7g | 8g | ||
 |
 |
95 : 5 | n.d.b |
| 7h | 8h | ||
 |
 |
no reaction | — |
| 7i | 8i | ||
 |
 |
0 : 100 | 90 |
| 7j | 8j | ||
 |
 |
21 : 79 | 63 |
| 7k | 8k |
aConversion determine by 1H nuclear magnetic resonance spectroscopy (NMR).
bn.d., not determined.
Scheme 3.

Reagents and conditions: 0.05 M 7 (acetone; 333 µl min−1), 0.315 M NaHCO3 (H2O, 333 µl min−1), Oxone® (H2O, 333 µl min−1), 60°C, 5 bar.
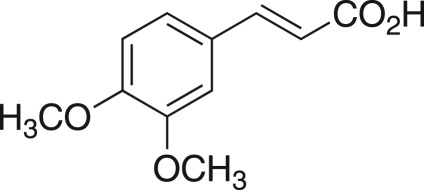
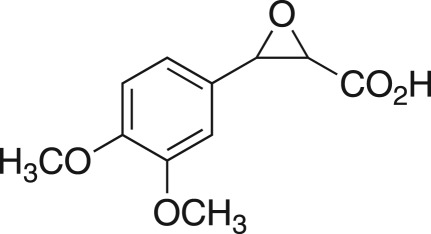
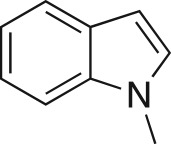
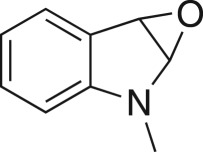
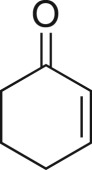
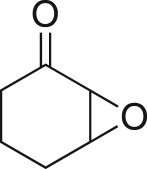
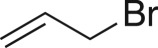
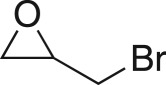
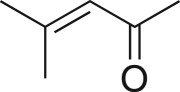
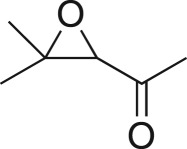
The DMDO flow epoxidation protocol was explored with an auto-sampler and fraction collector equipped flow system and provided an effective and robust approach to the semi-automated synthesis and collection of epoxides via aromatic and aliphatic olefins (scheme 3 and table 4). Simple aromatic vinyl scaffolds showed good conversion (greater than 70%), with excellent isolated yields of benzyl epoxide 8a, styrene oxides 8b and 8c, as did naphthoquinone and cinnamyl alcohol, yielding 8d and 8e (80% and 70%, respectively). By contrast, cinnamic acid 7f, chromeone 7g, indole 7h and cyclohex-2-en-one 7i scaffolds were resistant to DMDO epoxidation, with epoxides 8f–8i at best observed at low levels (5% or less). Allyl bromide 7j showed quantitative conversion to 8j, while the highly substituted mesityl oxide 7k showed a 79% conversion to mesityl epoxide 8 k (table 4).
Having established two robust flow protocols to install epoxides via epichlorohydrin and DMDO, we next examined the amine-mediated ring opening of the freshly installed epoxide moiety. As we had previously reported similar amine mediated ring openings using bismuth (III) chloride as catalyst [8], we explored this approach; but to suppress the formation of chlorinated side products that can occur with this catalyst, bismuth (III) triflate (Bi(OTf)3) was examined. However, under flow conditions, employing 30 mol% Bi(OTf)3, 120°C, 5 bar and a residence time of 20 min, only a small amount (17%) of the amino alcohol was detected. Increasing the amount of bismuth catalyst to 50% increased the formation of product to 75%; however, as excess catalyst is undesirable for purification, we shifted focus to the application of microwave approaches (scheme 4) [44,47,48].
Scheme 4.

Reagents and conditions: (i) microwave irradiation, 1.1 equiv. aniline, 15% Bi(OTf)3, CH3CN, 10 min.
Coupling of epoxide 5a with aniline and Bi(OTf)3 (15% in CH3CN) under microwave irradiation (60°C, 10 min) gave 57% conversion to the β-amino alcohol 10a (electronic supplementary material, table S2). The optimal conditions were identified as 10 min microwave irradiation at 140°C with 15% Bi(OTf)3 in CH3CN, affording 96% conversion and an 83% isolated yield of 10a. However, limited regiocontrol with other amines of interest was observed under these conditions (for ratio of regioisomers, see Experimental section).
While the Bi-catalysed approach did allow access to the desired amino alcohols, the poor regioselectivity observed severely hampered compound access. We turned our attention to a recent report from the Seeberger laboratory that reported the model epoxide aminolysis of styrene with t-butyl and isopropyl amine by flow, [29] as well as a previous publication by Jamison and coworkers. [30]. While Seeberger's work reported the successful synthesis of a number of APIs under catalyst free conditions, the aminolysis step was limited to the aforementioned amines and the scope of the reaction was not defined. We endeavoured to extend the reach of these approaches. Application of the former protocols in our systems revealed that the optimum epoxide aminolysis conditions for aryl substituted epoxides ranged from 5–7 equivalents of t-butylamine, 120–150°C, 5–10 bar back pressure (scheme 5) and 1 : 1 v/v mixture of toluene : ethanol. These conditions afforded predominately the desired secondary amino alcohols (table 5).
Scheme 5.

Reagents and conditions: epoxide 5a, 8a or 8b (0.5 M in toluene) 0.5 ml min−1, t-butylamine (0.5 M in ethanol) 0.5 ml min−1, 120–150°C, 5–10 bar.
Table 5.
Optimized reaction conditions for flow aminolysis of epoxides 5a, 8a and 8b with t-butylamine.
| epoxide | t-butylamine (equiv.) | temp (°C) | pressure (bar) | conversion |
|---|---|---|---|---|
| 5a | 5 | 120 | 5 | 100 : 0 : 0 |
| 10a : 13a : 16a | ||||
| 8a | 5 | 150 | 10 | 80 : 0 : 20 |
| 11a : 14a : 17a | ||||
| 8b | 7 | 150 | 10 | 85 : 15: 0 |
| 12a : 15a : 18a |
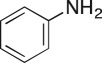
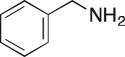
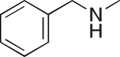
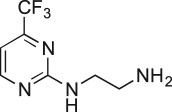
Having established standard reaction conditions for aryl, aryl methyl and aryloxy epoxides, we applied this methodology across a range of primary, secondary and tertiary amines, as well as aniline, benzyl and N-benzylmethyl amines. Regioselective conversion to secondary alcohols 10b–10g was observed with epoxide 5a and amines 9b–9g (table 6), as with the microwave protocol, and flow aminolysis yields ranged from modest to excellent (figure 2, 36%–80%). The exceptions to this were reactions of 5a with 9a and 9b. Reaction of N-propylamine 9b afforded a mixture of primary (10b), secondary alcohols (13b) and bis-alkylation (16b), while reaction of epoxide 5a and t-butylamine 9a furnished a 20% impurity of the bis-alkylated by-product (16a). Treatment of 5a with N1-(4-(trifluoromethyl)pyrimidin-2-yl)ethane-1,2-diamine 2 regioselectively afforded exclusively 1 in a 65% isolated yield. This protocol delivered higher selectivity than microwave and batch methods, with no evidence of bis-alkylated side-product; [9] it has been reported that flow methodologies offer better selectivity over other methods owing to improved mixing [49].
Table 6.
Flow aminolysis 5a, 8a and 8b leading to the formation of amino alcohols 1 and (10a–g)–(18a–g).
| flow chemistry | microwave | ||||||
|---|---|---|---|---|---|---|---|
| epoxide | amine | 2° alcohol (%) | 1° alcohol (%) | bis-alkyl (%) | conversion (%) | isolated yield of 2° alcohol (%) | isolated yield of 2° alcohol (%) |
 |
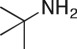 |
80 | — | 20 | >99 | 66 | — |
| 5a | 9a | 10a | 13a | 16a | 10a | ||
 |
68 | 17 | 15 | >99 | not isolated | — | |
| 9b | 10b | 13b | 16b | — | |||
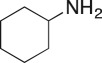 |
100 | — | — | >99 | 75 | — | |
| 9c | 10c | 13c | 16c | 10c | |||
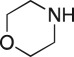 |
100 | — | — | >99 | 36 | 40 | |
| 9d | 10d | 13d | 16d | 10d | |||
 |
81 | — | — | 81 | 56 | 83 | |
| 9e | 10e | 13e | 16e | 10e | |||
 |
89 | — | — | 89 | 73 | — | |
| 9f | 10f | 13f | 16f | 10f | |||
 |
100 | — | — | >99 | 80 | 53 | |
| 9g | 10g | 13g | 16g | 10g | |||
 |
100 | — | — | >99 | 63 | 45 | |
| 2 | 1 | 13h | 16h | 1 | |||
 |
92 | 8 | — | >99 | 21 | — | |
| 8a | 9a | 11a | 14a | 17a | 11a | ||
| — | >99 | not isolated | — | ||||
| 9b | 11b | 14b | 17b | — | |||
| 100 | — | — | >99 | 49 | — | ||
| 9c | 11c | 14c | 17c | 11c | |||
| 100 | — | — | >99 | 79 | 23 | ||
| 9d | 11d | 14d | 17d | 11d | |||
| 100 | — | — | >99 | 59 | IMa 2 : 3 | ||
| 9e | 11e | 14e | 17e | 11e | 11e : 14eb | ||
| 90 | — | 10 | >99 | 50 | — | ||
| 9f | 11f | 14f | 17f | 11f | |||
| 100 | — | — | >99 | 66 | 4 : 1a | ||
| 9g | 11g | 14g | 17g | 11g | 11g : 14gb | ||
 |
85 | 15 | — | >99 | 49 | — | |
| 8b | 9a | 12a | 15a | 18a | 12a | ||
| 11 | 65 | 24 | >99 | not isolated | — | ||
| 9b | 12b | 15b | 18b | — | |||
| 82 | 18 | — | >99 | 58 | — | ||
| 9c | 12c | 15c | 18c | 12c | |||
| 100 | — | — | >99 | 78 | 64 | ||
| 9d | 12d | 15d | 18d | 12d | |||
| 49 | 51 | — | >99 | 35 | 32 | ||
| 9e | 12e | 15e | 18e | 12e | |||
| 85 | 12 | 3 | >99 | 76 | — | ||
| 9f | 12f | 15f | 18f | 12f | |||
| 88 | 12 | — | >99 | 60 (5 : 1a 12g : 15g) | |||
| 9g | 12g | 15g | 18g |
aIM, inseparable mixture.
bSee the electronic supplementary material for experimental details.
Aminolysis of epoxide 8a with amines 9a–9g furnished 11a–11g with ≥90% regioselectivity for the secondary alcohol, in principle simplifying purification of the desired regioisomer compared to the microwave methodology. However, the isolated yields of 11a–11g ranged from 21% to 79%. LCMS analysis revealed product contamination with residual amine, and this was most apparent with aliphatic amines 9a and 9c.
Reaction of styrene epoxide 8b with amines 9a–9g showed reduced regioselectivity. The coupling of styrene epoxide 8b with 9a and 9f afforded mixtures of primary (12a and 12f) and secondary alcohols (15a and 15f), as well as bis-alkylation (18f). Aniline 9e afforded a 1 : 1 mixture of primary (12e) and secondary alcohols (15e), while the secondary amines 9b and 9c, as well as morpholine 9d, benzyl amine 9f and N-benzylmethylamine 9 g, furnished the secondary alcohol in ≥82% regioselectivity. Yields of 12a–12f ranged from modest to excellent (35–78%). As seen with epoxides 5a and 8a, reaction of N-propylamine 9b resulted in a mixture of primary (12b), secondary (15b) and bis-alkyated products (18b). Compared with the microwave protocol, purification was improved because of improved regioselectivity (see Experimental section for regioisomer ratios). The notable exception to this proved to be 12g, which was inseparable from 15g by column chromatography, but offered comparable selectivity to the desired secondary alcohol as the microwave protocol.
3. Conclusion
Herein we have demonstrated that the flow coupling of phenols with epichlorohydrin provides a facile and highly scalable route to a wide variety of epoxides in moderate to excellent yields (22–89%). This access is scalable with multi-gram quantities of aryloxy epoxides accessible, e.g. 5a (0.49 g h−1; with our three hour flow synthesis realizing 1.48 g of material). A microwave/Bi(OTf)3-mediated epoxide opening aminolysis gave moderate control over β-amino alcohol regiochemistry. However, our refinement of previously published aminolysis protocols [29,30] enabled development of a robust flow protocol with aryl, arylmethyl and aryloxy epoxides and a range of primary, secondary and tertiary amines, as well as aniline, benzyl and N-benzylmethyl amines. With this approach we were able to access a diverse range of β-amino alcohols with high regioselectivity. Further, we abrogated the limitations of this protocol, i.e. the use of epichlorohydrin, through the first application of flow DMDO olefin epoxidation, which was applicable to a wide range of aromatic and aliphatic olefins with isolated epoxide yields of 60–98%. The in situ generation of the reactive DMDO species improves the safety of the reaction compared with traditional batch methodologies, and semi-automation of this protocol using an auto-sampler and fraction collector facilitates rapid library generation. The in-flow epoxidation of olefins and flow epoxide aminolysis protocols developed herein offer the medicinal chemist a simple pathway to novel epoxides and, consequently, highly decorated β-amino alcohols of potential biological importance.
4. Experimental
4.1. General methods
All reagents were purchased from Sigma-Aldrich, AK Scientific, Matrix Scientific or Lancaster Synthesis and were used without purification. All solvents were used as supplied.
1H and 13C NMR spectra were recorded on a Brüker Advance™ AMX 400 MHz NMR spectrometer at 400.1 and 100.6 MHz, respectively or Brüker Advance™ AMX 600 MHz NMR spectrometer at 600.2 and 150.9 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) measured relative to the internal standards. Coupling constants (J) are expressed in hertz (Hz). Mass spectra were recorded on a Shimadzu LCMS 2010 EV or Agilent 6100 series single quadrupole LCMS using a mobile phase of 1 : 1 acetonitrile : H2O with 0.1% formic acid. The University of Wollongong, Australia, Mass Spectrometry User resource & Research Facility analysed samples for high resolution mass spectrometry (HRMS). All samples returned satisfactory analyses. Flow reactions were carried out using a Vapourtec RS-400 equipped with stainless steel pump module, fraction collection kit and auto-sampler; Vapourtec RS-200 equipped with V3 pumps, fraction collection kit and auto-sampler; and a Vapourtec easy-MedChem equipped with V3 pumps. Compound purity was confirmed by a combination of LCMS (UPLC), and HRMS and NMR analysis. All compounds are ≥95% purity.
4.2. Recycling procedure for epichlorohydrin
Epichlorohydrin was recovered and recycled by collecting post-reaction under vacuum (30 mbar, 50°C). The resulting crude mixture was then diluted with CH2Cl2 (100 ml). The solution was washed with water (3 × 150 ml) and saturated brine (150 ml). The organic layers were separated, dried over Mg2SO4 and concentrated in vacuo (40°C, 240 mbar) to afford a colourless oil.
4.3. General procedure 1
The Vapourtec easy-MedChem was charged with a 0.1 M solution of phenol (2 mmol) in anhydrous DMF (20 ml) at a flow rate of 0.5 ml min−1. The solution was then passed through a 7.85 ml Omnifit column packed with a 1 : 1 mixture of Cs2CO3/acid washed sand (2.30 g, total weight) and mixed with 5 M solution of epichlorohydrin in anhydrous DMF (0.5 ml min−1) through a T-piece. The resulting mixture was subsequently flowed through two 10 ml perfluroalkoxy (PFA) reaction coils set up in series at 105°C, 5 bar and affording a total of 20 min residence time. The resulting reaction mixture was collected, concentrated in vacuo and subject to column chromatography. The up-scaled reaction was carried out with the above concentration and an Omnifit column packed with a 1 : 1 mixture of Cs2CO3/acid (1 M HCl) washed sand (16 g, total mass).
4.4. 2-((4-Nitrophenoxy)methyl)oxirane (5a)
Compound 5a was prepared using general procedure 1 and 4-nitrophenol (0.278 g, 2 mmol). The product was obtained as a light yellow solid (347 mg, 89%), m.p.: 66–69°C. 1H NMR (400 MHz, acetone-d6) δ 8.38–8.13 (m, 2H), 7.31–7.11 (m, 2H), 4.55 (dd, J = 11.4, 2.5 Hz, 1H), 4.05 (dd, J = 11.4, 6.5 Hz, 1H), 3.38 (ddt, J = 5.1, 4.4, 2.5 Hz, 1H), 2.91–2.84 (m, 1H), 2.76 (dd, J = 5.1, 2.5 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 164.8, 142.6, 126.6 (2C), 115.8 (2C), 71.0, 50.2, 44.3; IR υmax/cm−1 1504 (C–NO2), 1329 (C–NO2), 1250 (epox.) 1107 (C–O–C), 843 (arom.), 814 (epox.); low resolution mass spectrometry (LRMS) electron impact (EI) m/z: 195 (M+, 50%), 109 (35) 57 (100) [50].
4.5. 2-((3-Nitrophenoxy)methyl)oxirane (5b)
Compound 5b was prepared using general procedure 1 and 3-nitrophenol (0.278 g, 2 mmol). The product was obtained as a light yellow solid (345 mg, 88%), m.p.: 55–58°C. 1H NMR (400 MHz, acetone-d6) δ 7.87 (ddd, J = 8.1, 2.1, 0.8 Hz, 1H), 7.81 (dd, J = 2.4, 2.1 Hz, 1H), 7.61 (t, J = 8.2 Hz, 1H), 7.45 (ddd, J = 8.3, 2.5, 0.7 Hz, 1H), 4.56 (dd, J = 11.4, 2.4 Hz, 1H), 4.05 (dd, J = 11.4, 6.5 Hz, 1H), 3.39 (ddt, J = 5.0, 4.3, 2.5 Hz, 1H), 2.89 (dd, J = 5.0, 4.3 Hz, 1H), 2.79 (dd, J = 5.1, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 160.3, 150.2, 131.4, 122.6, 116.6, 109.8, 71.0, 50.4, 44.3; IR υmax/cm−1: 1520 (C–NO2), 1346 (C–NO2), 1248 (epox.) 1034 (C–O–C), 813 (epox.), 797 (arom.); LRMS (EI) m/z: 195 (M+, 50%), 92 (35) 57 (100) [50].
4.6. 2-((2-Nitrophenoxy)methyl)oxirane (5c)
Compound 5c was prepared using general procedure 1 and 2-nitrophenol (0.278 g, 2 mmol). The product was obtained as a yellow oil (125 mg, 67%). 1H NMR (400 MHz, acetone-d6) δ 7.84 (d, J = 8.1 Hz, 1H), 7.63 (dd, J = 11.5, 4.4 Hz, 1H), 7.37 (d, J = 8.5 Hz, 1H), 7.15 (t, J = 7.7 Hz, 1H), 4.55 (dd, J = 11.5, 2.2 Hz, 1H), 4.13 (dd, J = 11.4, 5.9 Hz, 1H), 3.42–3.27 (m, 1H), 2.87–2.83 (m, 1H), 2.78 (dd, J = 5.1, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 152.3, 141.3, 134.9, 125.8, 121.8, 116.2, 71.2, 50.2, 44.3; IR υmax/cm−1: 1521 (C–NO2), 1355 (C–NO2), 1259 (epox.) 1150 (C–O–C), 818 (epox.), 737 (arom.); LRMS (EI) m/z: 195 (M+, 10%), 57 (100) [50].
4.7. 2-((4-Chlorophenoxy)methyl)oxirane (5d)
Compound 5d was prepared using general procedure 1 and 4-chlorophenol (0.257 g, 2 mmol). The product was obtained as a white solid (187 mg, 89%), m.p.: less than 40°C. 1H NMR (400 MHz, acetone-d6) δ 7.36–7.27 (m, 2H), 7.05–6.94 (m, 2H), 4.36 (dd, J = 11.3, 2.6 Hz, 1H), 3.89 (dd, J = 11.3, 6.4 Hz, 1H), 3.32 (ddt, J = 5.2, 4.2, 2.6 Hz, 1H), 2.85 (dt, J = 8.4, 4.2 Hz, 1H), 2.72 (dd, J = 5.1, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 158.6, 130.1 (2C), 126.1, 117.1 (2C), 70.5, 50.5, 44.3; IR υmax/cm−1: 1490 (arom.), 1239 (epox.) 1093 (C–O–C), 863 (arom.), 821 (epox.), 665 (C–Cl); LRMS (EI) m/z: 184 (M+, 80%), 128 (100) [51].
4.8. 2-((3-Chlorophenoxy)methyl)oxirane (5e)
Compound 5e was prepared using general procedure 1 and 3-chlorophenol (0.257 g, 2 mmol). The product was obtained as a clear oil (228 mg, 74%). 1H NMR (400 MHz, acetone-d6) δ 7.29 (t, J = 8.1 Hz, 1H), 7.01 (t, J = 2.2 Hz, 1H), 6.98 (ddd, J = 8.1, 2.4, 0.7 Hz, 1H), 6.93 (dd, J = 8.4, 2.4 Hz, 1H), 4.37 (dd, J = 11.3, 2.5 Hz, 1H), 3.90 (dd, J = 11.3, 6.4 Hz, 1H), 3.31 (ddt, J = 5.1, 4.2, 2.6 Hz, 1H), 2.84 (dd, J = 4.9, 4.4 Hz, 1H), 2.72 (dd, J = 5.1, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 160.7, 135.3, 131.6, 121.8, 115.7, 114.4, 70.5, 50.4, 44.3; IR υmax/cm−1: 1475 (arom.), 1246 (epox.) 1072 (C–O–C), 858 (epox.), 767 (arom.), 679 (C–Cl); LRMS (EI) m/z: 184 (M+, 80%), 128 (100); HRMS electrospray ionisation (ESI) calcd for C9H9ClO2 (M+), 184.0291; no mass observed, material degraded.
4.9. 2-((2-Chlorophenoxy)methyl)oxirane (5f)
Compound 5f was prepared using general procedure 1 and 2-chlorophenol (0.258 g, 2 mmol). The product was obtained as a clear oil (258 mg, 70%). 1H NMR (400 MHz, acetone-d6) δ 7.41 (dd, J = 7.9, 1.6 Hz, 1H), 7.30 (td, J = 7.9, 1.6 Hz, 1H), 7.16 (dd, 8.3, 0.9 Hz, 1H) 6.98 (td, J = 7.8, 1.4 Hz, 1H), 4.44 (dd, J = 11.4, 2.6 Hz, 1H), 4.02 (dd, J = 11.4, 6.1 Hz, 1H), 3.37 (ddt, J = 5.2, 4.2, 2.6 Hz, 1H), 2.87 (dd, J = 5.1, 4.3 Hz, 1H), 2.78 (dd, J = 5.2, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 155.2, 131.0, 129.0, 123.2, 122.7, 114.9, 70.9, 50.5, 44.3; IR υmax/cm−1: 1509 (arom.), 1238 (epox.) 1112 (C–O–C), 812 (epox.), 742 (arom.); LRMS (EI) m/z: 184 (M+, 40%), 128 (100) [41].
4.10. 2-((4-Tolyloxy)methyl)oxirane (5g)
Compound 5g was prepared using general procedure 1 and p-cresol (210 µl, 2 mmol). The product was obtained as a clear oil (196 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.09 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 4.18 (dd, J = 11.0, 3.3 Hz, 1H), 3.95 (dd, J = 11.0, 5.6 Hz, 1H), 3.37–3.32 (m, 1H), 2.98–2.81 (m, 1H), 2.75 (dd, J = 4.9, 2.7 Hz, 1H), 2.29 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 156.5, 130.6, 130.1 (2C), 114.6 (2C), 69.0, 50.4, 44.9, 20.6; IR υmax/cm−1: 1600, 1582, 1491 (arom.), 1260 (epox.) 1154 (C–O–C), 857 (epox.), 770 (arom.); LRMS (EI) m/z: 164 (M+, 100%), 108 (90) [51].
4.11. 2-((3-Tolyloxy)methyl)oxirane (5h)
Compound 5h was prepared using general procedure 1 and m-cresol (0.210 µl, 2 mmol). The product was obtained as a clear oil (184 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 7.17 (t, J = 7.8 Hz, 1H), 6.79 (d, J = 7.5 Hz, 1H), 6.77–6.70 (m, 2H), 4.19 (dd, J = 11.0, 3.3 Hz, 1H), 3.96 (dd, J = 11.0, 5.6 Hz, 1H), 3.39–3.32 (m, 1H), 2.92–2.88 (m, 1H), 2.76 (dd, J = 4.9, 2.6 Hz, 1H), 2.33 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 159.8, 140.2, 130.1, 122.5, 116.1, 112.3, 70.0, 50.6, 44.4, 21.5; IR υmax/cm−1: 2882 (O–CH3), 1505 (arom.), 1221 (epox.) 1035 (C–O–C), 849 (arom.); LRMS (EI) m/z: 164 (M+, 100%), 108 (90) [50].
4.12. 2-((2-Tolyloxy)methyl)oxirane (5i)
Compound 5i was prepared using general procedure 1 and o-cresol (0.210 µl, 2 mmol). The product was obtained as a clear oil (200 mg, 61%). 1H NMR (400 MHz, acetone-d6) δ 7.13 (bt, J = 6.7 Hz, 2H), 6.91 (d, J = 8.4 Hz, 1H), 6.84 (t, J = 7.4 Hz, 1H), 4.31 (dd, J = 11.3, 2.7 Hz, 1H), 3.91 (dd, J = 11.3, 6.0 Hz, 1H), 3.33 (ddd, J = 8.7, 4.2, 2.6 Hz, 1H), 2.83 (dd, J = 5.1, 4.3 Hz, 1H), 2.73 (dd, J = 5.2, 2.6 Hz, 1H), 2.20 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 157.8, 131.4, 127.8, 127.2, 121.5, 112.3, 70.0, 50.7, 44.4, 16.3; IR υmax/cm−1: 1497 (arom.), 1242 (epox.) 1121 (C–O–C), 837 (epox.), 749 (arom.); LRMS (EI) m/z: 164 (M+, 90%), 108 (100) [52].
4.13. 2-((4-Methoxyphenoxy)methyl)oxirane (5j)
Compound 5j was prepared using general procedure 1 and 4-methoxyphenol (0.248 g, 2 mmol). The product was obtained as a white solid (79 mg, 22%), m.p.: 48 – 48°C. 1H NMR (400 MHz, CDCl3) δ 6.96–6.75 (m, 4H), 4.17 (dd, J = 11.0, 3.2 Hz, 1H), 3.92 (dd, J = 11.1, 5.6 Hz, 1H), 3.77 (s, 3H), 3.36–3.31 (m, 1H), 2.91–2.88 (m, 1H), 2.74 (dd, J = 4.9, 2.7 Hz, 1H); 13C NMR (101 MHz, dimethylsulfoxide (DMSO)-d6) δ 153.6, 152.3, 115.4 (2C), 114.6 (2C), 69.4, 55.3, 49.8, 43.7; IR υmax/cm−1: 2882 (O–CH3), 1505 (arom.), 1221 (epox.) 1035 (C–O–C), 849 (arom.), 823 (epox.); LRMS (EI) m/z: 180 (M+, 80%), 124 (100) [51].
4.14. 2-((3-Methoxyphenoxy)methyl)oxirane (5k)
Compound 5k was prepared using general procedure 1 and 3-methoxyphenol (0.248 g, 2 mmol). The product was obtained as a clear oil (223 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 7.18 (t, J = 8.1 Hz, 1H), 6.59–6.44 (m, 3H), 4.20 (dd, J = 11.0, 3.2 Hz, 1H), 3.95 (dd, J = 11.0, 5.6 Hz, 1H), 3.93 (s, 3H), 3.38–3.33 (m, 1H), 2.95–2.86 (m, 1H), 2.76 (dd, J = 4.9, 2.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 160.5, 159.5, 130.0, 106.6 (2C), 100.8, 69.0, 55.1, 49.7, 43.8; IR υmax/cm−1: 2833 (O–CH3), 1490 (arom.), 1198 (epox.) 1148 (C–O–C), 761 (arom.), 839 (epox.); LRMS (EI) m/z: 180 (M+, 90%), 124 (100) [52].
4.15. 2-((2-Methoxyphenoxy)methyl)oxirane (5l)
Compound 5l was prepared using general procedure 1 and 2-methoxyphenol (0.248 g, 2 mmol). The product was obtained as a white solid (252 mg, 70%), m.p.: less than 40°C. 1H NMR (400 MHz, acetone-d6) δ 6.97 (dt, J = 8.0, 1.5 Hz, 2H), 6.92 (td, J = 7.7 Hz, 1H), 6.86 (td, J = 7.6, 1.8, 1H), 4.28 (dd, J = 11.3, 2.9 Hz, 1H), 3.88 (dd, J = 11.3, 6.2 Hz, 1H), 3.81 (s, 3H), 3.31 (ddd, J = 9.1, 4.2, 2.7 Hz, 1H), 2.82 (dd, J = 4.9, 4.1 Hz, 1H), 2.69 (dd, J = 5.2, 2.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 149.1, 147.7, 121.4, 120.7, 113.6, 112.2, 69.8, 55.4, 49.8, 43.8; IR υmax/cm−1: 2840 (O–CH3), 1508 (arom.), 1229 (epox.) 1023 (C–O–C), 860 (epox.), 745 (arom.); LRMS (EI) m/z: 180 (M+, 80%), 124 (100%) [42].
4.16. Methyl 4-(oxirane-2-ylmethoxy)benzoate (5m)
Compound 5m was prepared using general procedure 1 and methyl 4-hydroxybenzoate (0.304 g, 2 mmol). The product was obtained as a white solid (228 mg, 67%), m.p.: 57–60°C. 1H NMR (400 MHz, acetone-d6) δ 8.06–7.85 (m, 2H), 7.17–6.84 (m, 2H), 4.45 (dd, J = 11.3, 2.6 Hz, 1H), 3.97 (dd, J = 11.3, 6.4 Hz, 1H), 3.84 (s, 1H), 3.34 (ddt, J = 6.7, 4.2, 2.6 Hz, 1H), 2.85 (dd, J = 5.0, 4.3 Hz, 2H), 2.74 (dd, J = 5.1, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 166.8, 163.5, 132.2 (2C), 123.8, 115.2 (2C), 70.4, 52.0, 50.4, 44.4; IR υmax/cm−1: 2882 (O–CH3), 1708 (C = O), 1508 (arom.), 1254 (epox.) 1025 (C–O–C), 847 (epox.); LRMS (EI) m/z: 208 (M+, 50%), 121 (100%) [53].
4.17. General procedure 2
Using a Vapourtec RS-200 equipped with collection valve kit and synthesis auto-sampler, a 10 ml reaction loop was charged with a 0.05 M solution of alkene (1 mmol) in acetone. The solution was pumped at 0.333 µl min−1 through PFA tubing and mixed with a stream of 0.315 M sodium hydrogen carbonate(aq) (6.30 mmol) at 0.333 µl min−1. The outgoing solution then passed through an 8 cm PFA tube and mixed with a stream of 0.394 M Oxone(aq) (7.88 mmol, 0.333 µl min−1). The resulting stream was subsequently passed through two 10 ml PFA coil reactors in series at 60°C, 5 bar and 1 ml min−1 (residence time 20 min). The resulting reaction stream was then quenched in-line (immediately after the back pressure regulator) using a stream 0.4 M sodium sulfite(aq). The solution was then collected, concentrated in vacuo to remove acetone and diluted up to 50 ml with water (pH 7). The pH of the solution was adjusted to pH ∼7 using saturated ammonium chloride(aq). The aqueous solution was extracted with ethyl acetate (3 × 50 ml), the organic layers were combined and washed with brine (50 ml). The organic layer was separated, dried over magnesium sulfate and concentrated in vacuo to afford the desired product; no further purification was required unless stated.
4.18. 2-(4-Methoxybenzyl)oxirane (8a)
Compound 8a was prepared using general procedure 2 and 4-allylanisole (0.153 ml, 1 mmol). The product was obtained as a colourless oil (153 mg, 93%). 1H NMR (400 MHz, acetone-d6) δ 7.20 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 3.77 (s, 3H), 3.07–3.00 (m, 1H), 2.75 (dd, J = 5.5, 1.9 Hz, 2H), 2.68 (dd, J = 5.1, 4.0 Hz, 1H), 2.49 (dd, J = 5.2, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 159.5, 130.8 (2C), 130.5, 114.6 (2C), 55.5, 52.9, 46.7, 38.6; IR υmax/cm−1: 2836 (O–CH3), 1511 (arom.), 1243 (epox.) 1032 (C–O–C), 830 (arom.), 815 (epox.); LRMS (EI) m/z: 164 (M+, 30%), 121 (100%) [54].
4.19. 2-Phenyloxirane (8b)
Compound 8b was prepared using general procedure 2 and styrene (0.114 ml, 1 mmol). The product was obtained as a clear oil (115 mg, 98%). 1H NMR (400 MHz, acetone-d6) δ 6.95–6.81 (m, 5H), 3.47 (dd, J = 4.0, 2.7 Hz, 1H), 2.66 (dd, J = 5.4, 4.2 Hz, 1H), 2.39 (dd, J = 5.4, 2.6 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 128.0, 118.7 (2C), 118.4, 116.0 (2C), 41.8, 40.7; IR υmax/cm−1: 3046, 2990, 2910 (arom.), 873 (epox.) 755 (arom.); LRMS (EI) m/z: 120 (M+, 30%), 91 (100%) [55].
4.20. 4-(Oxiran-2-yl)benzoic acid (8c)
Compound 8c was prepared using general procedure 2 and styrene (0.114 ml, 1 mmol). The product was obtained as an off-white solid (107 mg, 65%), m.p.: 132–133°C. 1H NMR (400 MHz, acetone-d6) δ 8.02 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.3 Hz, 2H), 3.98 (dd, J = 3.9, 2.5 Hz 1H), 3.16 (dd, J = 5.6, 4.2 Hz, 1H), 2.81 (dd, J = 5.6, 2.5 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 167.4, 144.4, 131.3, 130.6 (2C), 126.4 (2C), 52.1, 51.6; IR υmax/cm−1: 3057 (COOH) 2853 (arom.), 1681 (C=O), 1294 (C–O), 949 (C–O–C) 857 (epox.), 769 (arom.); LRMS (ESI+) m/z: 165 (M + H, 100%); HRMS (ESI) calcd for C9H9O3 (M + H), 165.0546; found 165.0547.
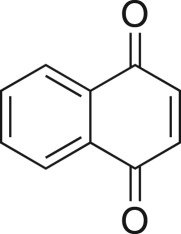
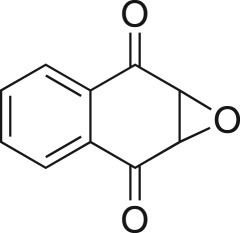
4.21. 1α,7α-dihydronaphtho[2,3-β]oxirene-2,7-dione (8d)
Compound 8d was prepared using general procedure 2 and 1,4-naphthoquinone (0.158 g, 1 mmol). The product was obtained as a white solid (129 mg, 74%), m.p.: 132–134°C. 1H NMR (400 MHz, acetone-d6) δ 7.99 (dd, J = 5.7, 3.4 Hz, 2H), 7.89 (dd, J = 5.8, 3.3 Hz, 2H), 4.12 (s, 2H); 13C NMR (101 MHz, acetone-d6) δ 191.5 (2C), 135.5 (2C), 132.9 (2C), 127.6 (2C), 56.4 (2C); IR υmax/cm−1: 2878 (C–O), 1709 (C=O), 1683 (C=O), 1211 (epox.) 1027 (C–O–C), 880 (arom.), 818 (epox.); LRMS (EI) m/z: 174 (M+, 40%), 146 (40%) 105 (100%). [56]
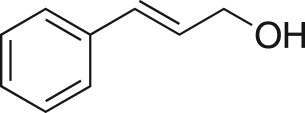
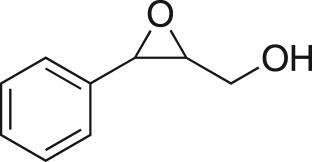
4.22. (3-Phenyloxiran-2-yl)methanol (8e)
Compound 8e was prepared using general procedure 2 and cinnamyl alcohol (0.130 ml, 1 mmol). The product was obtained as a clear oil (96 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 4.05 (d, J = 12.7 Hz, 1H), 3.93 (d, J = 2.1 Hz, 1H), 3.85–3.77 (m, 1H), 3.26–3.21 (m, 1H), 1.89 (bs, 1H); 13C NMR (101 MHz, CDCl3) δ 136.8, 128.7 (2C), 128.5 (2C), 125.9, 62.5, 61.4, 55.7; IR υmax/cm−1: 3420 (OH), 1244 (epox.) 1025 (C–O–C), 750 (epox.); LRMS (EI) m/z: 150 (M+, 20%), 91 (100%) [55].
4.23. Epibromohydrin (8j)
Compound 8j was prepared using general procedure 2 and allybromide (0.086 ml, 1 mmol). The product was obtained as a colourless oil (122 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 3.43 (dd, J = 10.5, 5.9 Hz, 1H), 3.32 (dd, J = 10.5, 5.6 Hz, 1H), 3.29–3.24 (m, 1H), 2.96–2.92 (m, 1H), 2.67 (dd, J = 4.8, 2.4 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 51.9, 48.8, 34.6; IR υmax/cm−1: 2921, 1464, (CH2), 1252, 1071 (epox.), 659 (C–Br); LRMS (EI) m/z: 120 (M+, 30%), 91 (100) [57].
4.24. 1-(3,3-Dimethyloxiran-2-yl)ethan-1-one (8k)
Compound 8k was prepared using general procedure 2 and mesityl oxide (0.171 ml, 1.5 mmol). The product was obtained as a yellow oil (108 mg, 63%). 1H NMR (400 MHz, CD3OD) δ 3.60 (s, 1H), 2.23 (s, 3H), 1.42 (s, 3H), 1.22 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 204.0, 65.9, 60.8, 28.2, 24.7, 18.4; IR υmax/cm−1: 2971 (CH3), 1737 (C = O), 1217 (epox.) 1062 (C–O–C); LRMS (EI) m/z: 114 (M+, 5%), 99 (40), 43 (100) [58].
4.25. General procedure 3
A suspension of epoxide (1.00 mmol), amine (1.10 mmol) and bismuth (III) trifluoromethane sulfonate (15 mol%) in CH3CN (3 ml) was subjected to microwave irradiation at the below stated temperature and time (typical graph of the temperature/power/pressure in the electronic supplementary material, figure S1). The reaction mixture was concentrated in vacuo and crude material subjected to column chromatography.
4.26. General procedure 4
Using a Vapourtec RS-400 equipped with fraction collection kit and auto-sampler, either a 0.5 ml (loop A), 1.0 ml (loop B) or 2.0 ml (loop C) sample loop was charged with a 0.4 M solution of epoxide in toluene. An additional sample loop (either 0.5 ml (loop A), 1.0 ml (loop B) or 2.0 ml (loop C)) was charged with a 2.0 M amine solution in ethanol. The solutions were flowed together and the resulting stream was then passed through two 10 ml PFA coil reactors in series at 120°C, 6 bar back pressure and 1 ml min−1 (residence time 20 min). The resulting reaction mixture was collected and purified as described.
4.27. General procedure 5
Using a Vapourtec RS-400 equipped with fraction collection kit and auto-sampler, either a 0.5 ml (loop A), 1.0 ml (loop B) or 2.0 ml (loop C) sample loop was charged with a 0.4 M solution of epoxide in toluene. An additional sample loop (either 0.5 ml (loop A), 1.0 ml (loop B) or 2.0 ml (loop C)) was charged with either a 2.0 M or 2.8 M amine solution in ethanol (as stated). The solutions were flowed together and the resulting stream was then passed through two PFA coil reactors in series at 150°C, 10 bar back pressure and 1 ml min−1 (residence time 20 min). The resulting reaction mixture was collected and purified as described.
4.28. 1-(tert-Butylamino)-3-(4-nitrophenoxy)propan-2-ol (10a)
Compound 10a was prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.078 g, 0.4 mmol, loop B) and tert-butylamine (9a) (0.22 ml, 2.0 mmol, loop B). The resulting reaction mixture was concentrated using a stream of compressed air to remove solvent and volatile reagents to afford the desired product as a yellow solid (71 mg, 66%), m.p.: 80–83°C. 1H NMR (400 MHz, CD3OD) δ 8.27–8.17 (m, 2H), 7.17–7.09 (m, 2H), 4.18–4.01 (m, 3H), 2.84–2.70 (m, 2H), 1.16 (s, 9H); 13C NMR (151 MHz, CD3OD) δ 165.5, 143.0, 126.8 (2C), 115.9 (2C), 72.7, 70.0, 49.2, 46.0, 28.5 (3C); IR υmax/cm−1: 3302 (OH), 1648 (C–N), 1589 (C–NO2), 1329 (Ar–NO2), 843 (arom.); LRMS (ESI+) m/z 269 (100%, M + H); HRMS (ESI) calcd for C13H21N2O4 (M + H), 269.1496; found 269.1493.
4.29. 1-(Cyclohexylamino)-3-(4-nitrophenoxy)propan-2-ol (10c)
Compound 10c was prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.156 g, 0.8 mmol, loop C) and cyclohexylamine (9c) (0.46 ml, 4.0 mmol, loop C). The resulting reaction mixture was concentrated in vacuo and recrystallized from 1 : 1 v/v hexane : diethyl ether to afford the desired compound as a creamy white solid (168 mg, 71%), m.p.: 124–126°C. 1H NMR (400 MHz, CD3OD) δ 8.22 (d, J = 7.9 Hz, 2H), 7.12 (d, J = 7.9 Hz, 2H), 4.16–4.03 (m, 3H), 2.87 (d, J = 11.9 Hz, 1H), 2.77–2.66 (m, 1H), 2.53–2.41 (m, 1H), 1.94 (d, J = 11.5 Hz, 2H), 1.77 (d, J = 12.9 Hz, 2H), 1.66 (d, J = 11.5 Hz, 1H), 1.40–1.04 (m, 5H); 13C NMR (151 MHz, CD3OD) δ 165.5, 142.9, 126.8 (2C), 115.9 (2C), 72.8, 69.7, 58.1, 50.0, 33.94, 33.90, 27.2, 26.1; IR υmax/cm−1: 3247 (OH), 1593 (C–NO2), 1328 (Ar–NO2), 847 (arom.); LRMS (ESI+) m/z 295 (100%, M + H); HRMS (ESI) calcd for C15H23N2O4 (M + H), 295.1652; found 295.1649.
4.30. 1-Morpholino-3-(4-nitrophenoxy)propan-2-ol (10d)
Compound 10d was prepared using general procedure 3; 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.194 g, 1.00 mmol), morpholine (9d) (0.095 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% CH3OH in CH2Cl2) to afford the product as a cream solid (113 mg, 40%).
Compound 10d was also prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.039 g, 0.2 mmol, loop A) and morpholine (0.09 ml, 1.0 mmol, loop A). The resulting reaction mixture was concentrated in vacuo to afford the desired product as a brown solid (20 mg, 36%), m.p.: 80–88°C. 1H NMR (400 MHz, acetone-d6) δ 8.22 (d, J = 9.3 Hz, 2H), 7.17 (d, J = 9.3 Hz, 2H), 4.27 (q, J = 6.2 Hz, 1H), 4.20–4.04 (m, 3H), 3.62 (t, J = 4.7 Hz, 4H), 2.60–2.44 (m, 6H); 13C NMR (101 MHz, acetone-d6) δ 165.4, 142.3, 126.6 (2C), 115.8 (2C), 72.8, 67.5 (2C), 67.3, 62.1, 55.1 (2C); IR υmax/cm−1: 3302 (OH),, 1589 (C–NO2), 1329 (Ar–NO2), 843 (arom.); LRMS (ESI+) m/z 283 (100%, M + H) [59].
4.31. 1-(4-Nitrophenoxy)-3-(phenylamino)propan-2-ol (10e)
Compound 10e was prepared using general procedure 3; 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.194 g, 1.00 mmol), aniline (9e) (0.100 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (1.5% CH3OH in CH2Cl2) to afford the product as cream solid (238 mg, 82%).
Compound 10e was also prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.039 g, 0.2 mmol, loop A) and aniline (9e) (0.09 ml, 1.0 mmol, loop A). The resulting reaction mixture was concentrated using a stream of compressed air to remove solvent and volatile reagents to afford the desired product as a yellow solid (32 mg, 56%), m.p.: 126–130°C. 1H NMR (400 MHz, acetone-d6) δ 8.23 (d, J = 9.3 Hz, 2H), 7.18 (d, J = 9.3 Hz, 2H), 7.10 (dd, J = 8.3, 7.5 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 6.59 (t, J = 7.3 Hz, 1H), 5.02–4.91 (m, 1H), 4.49 (d, J = 4.3 Hz, 1H), 4.35–4.18 (m, 3H), 3.45 (ddd, J = 12.5, 6.4, 4.9 Hz, 1H), 3.34–3.23 (m, 1H); 13C NMR (101 MHz, acetone-d6) δ 165.2, 149.8, 142.4, 129.8 (2C), 126.6 (2C), 117.5, 115.8 (2C), 113.5 (2C), 72.3, 69.9, 47.21; IR υmax/cm−1: 3274 (OH), 1596 (C–NO2), 1330 (Ar–NO2), 843 (arom.); LRMS (ESI+) m/z 289 (100%, M + H) [60].
4.32. 1-(Benzylamino)-3-(4-nitrophenoxy)propan-2-ol (10f)
Compound 10f was prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.156 g, 0.8 mmol, loop C) and benzylamine (9f) (0.44 ml, 4.0 mmol, loop C). The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% CH3OH in CH2Cl2) to afford the desired product as a yellow solid (138 mg, 57%), m.p.: 117–119°C. 1H NMR (400 MHz, DMSO-d6) δ 8.23–8.16 (m, 2H), 7.34–7.27 (m, 4H), 7.24–7.19 (m, 1H), 7.17–7.11 (m, 2H), 5.07 (d, J = 4.8 Hz, 1H), 4.16 (dd, J = 10.0, 4.1 Hz, 1H), 4.03 (dd, J = 10.0, 6.3 Hz, 1H), 3.97–3.90 (m, 1H), 3.76–3.68 (m, 2H), 2.61 (qd, J = 11.9, 5.9 Hz, 2H), 2.19 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.2, 140.9, 140.7, 128.1 (2C), 127.9 (2C), 126.5, 125.9 (2C), 115.1 (2C), 71.7, 68.0, 53.1, 51.5; IR υmax/cm−1: 3284 (OH), 1594 (C–NO2), 1339 (Ar–NO2), 846 (arom.); LRMS (ESI+) m/z 303 (100%, M + H); HRMS (ESI) calcd for C16H19N2O4 (M + H), 303.1339; found 303.1336.
4.33. 1-(Benzyl(methyl)amino)-3-(4-nitrophenoxy)propan-2-ol (10g)
Compound 10g was prepared using general procedure 3; 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.194 g, 1.00 mmol), N-benzylmethylamine (9 g) (0.169 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (45% EtOAc in hexanes) to afford the product as a yellow oil (176 mg, 53%).
Compound 10g was also prepared using general procedure 4 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.039 g, 0.2 mmol, loop A) and N-benzylmethylamine (9g) (0.13 ml, 1.0 mmol, loop A). The resulting reaction mixture was concentrated in vacuo adsorbed to silica and subjected to column chromatography (45% EtOAc in hexanes) to afford the desired product as a brown oil (51 mg, 80%). 1H NMR (400 MHz, acetone-d6) δ 8.22 (d, J = 9.3 Hz, 2H), 7.42–7.19 (m, 5H), 7.13 (d, J = 9.3 Hz, 2H), 4.25 (dd, J = 9.4, 3.0 Hz, 1H), 4.21–4.04 (m, 3H), 3.60 (s, 2H), 2.61 (ddd, J = 27.6, 12.7, 6.3 Hz, 2H), 2.28 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 165.4, 142.3, 140.2, 129.8 (2C), 129.0 (2C), 127.8, 126.6 (2C), 115.8 (2C), 72.75, 68.0, 63.5, 60.6, 43.2; IR υmax/cm−1: 3431 (OH), 2842 (CH2), 1593 (C–NO2), 1511 (arom.), 1330 (Ar–NO2), 1112 (C–O–C), 1023 (C–O), 843 (arom.); LRMS (ESI+) m/z 331 (100%, M + H); HRMS (ESI) calcd for C17H21N2O4 (M + H), 317.1496; found 317.1494.
4.34. 1-(4-Nitrophenoxy)-3-[{2-(4-(trifluoromethyl)-2-pyrimidinyl)}amino]-2-propanol (1)
Compound 1 was prepared using general procedure 3; 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.194 g, 1.00 mmol), 2 (0.101 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 5 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (1 : 10 : 89 NH4OH : CH3OH : CH2Cl2) to afford the desired product as a white solid (183 mg, 45%).
Compound 1 was also prepared using general procedure 5 with 2-((4-nitrophenoxy)methyl)oxirane (5a) (0.078 g, 0.4 mmol, loop B) and 2 (0.577 g, 2.8 mmol, loop B, 2.8 M), with a residence time of 40 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (1 : 10 : 89 NH4OH : CH3OH : CH2Cl2) to afford the desired product as a white solid (100 mg, 63%), m.p.: 130–131°C. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (brs, 1H), 8.19 (d, J = 9.2 Hz, 2H), 7.78 (brd, J = 26.1 Hz, 1H), 7.14 (d, J = 9.3 Hz, 2H), 6.93 (d, J = 4.9 Hz, 1H), 5.06 (d, J = 3.9 Hz, 1H), 4.07 (ddd, J = 16.2, 10.0, 5.2 Hz, 2H), 3.89 (d, J = 3.9 Hz, 1H), 3.40–3.35 (m, 2H), 2.79–2.57 (m, 4H), 1.89 (brs, 1H); 13C NMR (151 MHz, CDCl3) δ 163.7, 162.7, 160.6, 156.6 (dd, J = 36.2, 36.8 Hz), 141.9, 126.1 (2C), 120.6 (dd, J = 550.4, 274.9 Hz), 114.7 (2C), 105.9 (d, J = 2.3 Hz), 71.2, 68.4, 51.4, 48.9, 41.4; IR υmax/cm−1: 3039 (OH), 1588 (C–NO2), 1515 (NH), 1341 (C–NO2), 1254 (C–F), 1130 (C–O–C), 848 (arom.); LRMS (ESI+) m/z 402 (M+, 100%); HRMS (ESI) calcd for C16H18F3N5O4 (M + H), 402.1384; found 402.1380.
4.35. 1-(tert-Butylamino)-3-(4-methoxyphenyl)propan-2-ol (11a)
Compound 11a was prepared using general procedure 5 with compound 8a (0.131 g, 0.8 mmol, loop C) and tert-butylamine (9a) (0.44 ml, 4.0 mmol, loop C, 2.0 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography to afford the desired product as a brown solid (90 mg, 21%), m.p.: 44–46°C. 1H NMR (400 MHz, acetone-d6) δ 7.16 (d, J = 8.6 Hz, 2H), 6.86–6.79 (m, 2H), 3.75 (s, 3H), 3.69–3.61 (m, 1H), 2.69–2.61 (m, 3H), 2.40 (dd, J = 11.3, 7.9 Hz, 1H), 1.05 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 178.2, 151.4, 150.3 (2C), 133.4 (2C), 91.6, 74.5, 69.5, 67.5, 60.8, 48.5 (3C); IR υmax/cm−1: 3297 (OH), 2928 (CH2, CH3), 1463 (arom.), 1244 (C–O), 1033 (C–O–C); LRMS (ESI+) m/z 238 (100%, M + H); HRMS (ESI) calcd for C14H23N2NaO2 (M + Na), 260.1626; found 260.1633.
4.36. 1-(Cyclohexylamino)-3-(4-methoxyphenyl)propan-2-ol (11c)
Compound 11c was prepared using general procedure 5 with compound 8a (0.131 g, 0.8 mmol, loop C) and cyclohexylamine (9c) (0.46 ml, 4.0 mmol, loop C, 2.0 M). The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (10% CH3OH in CH2Cl2) to afford the product as a pale brown solid (53 mg, 40%), m.p.: 61–64°C. 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 3.79–3.74 (m, 4H), 2.83–2.61 (m, 3H), 2.47 (dd, J = 12.0, 9.1 Hz, 1H), 2.39 (tt, J = 10.4, 3.7 Hz, 1H), 1.91–1.79 (m, 2H), 1.70 (d, J = 12.9 Hz, 2H), 1.65–1.54 (m, 1H), 1.28–1.11 (m, 3H), 1.09–0.97 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 158.3, 130.5, 130.4 (2C), 114.0 (2C), 71.0, 56.8, 55.4, 51.8, 40.9, 34.0, 33.6, 26.2, 25.2, 25.1; IR υmax/cm−1: 3281 (OH), 2921 (CH2, CH3), 1461 (arom.), 1244 (C–O), 1038 (C–O–C); LRMS (ESI+) m/z 264 (100%, M + H); HRMS (ESI) calcd for C16H25NO2 (M + H), 264.1958; found 264.1956.
4.37. 1-(4-Methoxyphenyl)-3-morpholinopropan-2-ol (11d)
Compound 11d was prepared using general procedure 3; compound 8a (0.164 g, 1.0 mmol), morpholine (9d) (0.096 ml, 1.1 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN (2 ml) were subjected to microwave irradiation at 140°C for 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (50% EtOAc in hexanes) to afford the product as a pale brown oil (56 mg, 23%).
Compound 11d was also prepared using general procedure 5 with compound 8a (0.033 g, 0.2 mmol, loop A) and morpholine (9d) (0.09 ml, 1.0 mmol, loop A, 2.0 M). The resulting reaction mixture was concentrated in vacuo to afford the desired product as a brown oil (20 mg, 36%). 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 3.93–3.88 (m, 1H), 3.79 (s, 3H), 3.70 (dt, J = 5.6, 3.8 Hz, 4H), 3.27 (bs, 1H), 2.77 (dd, J = 13.8, 6.9 Hz, 1H), 2.63 (dd, J = 13.6, 5.5 Hz, 3H), 2.41–2.30 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 158.4, 130.4 (2C), 130.1, 114.0 (2C), 67.3, 66.9 (2C), 64.1 (2C), 55.4, 53.8, 40.5; IR υmax/cm−1: 3423 (OH), 2928, 2839 (CH2, CH3), 1511 (arom.), 1244 (C–O), 1115 (C–O–C); LRMS (ESI+) m/z 252 (100%, M + H); HRMS (ESI) calcd for C14H22NO3 (M + H), 252.1592; found 252.1595.
4.38. 1-(4-Methoxyphenyl)-3-(phenylamino)propan-2-ol 3 (11e)
A mixture 14e/11e was prepared using general procedure 3; compound 8a (0.164 g, 1.0 mmol), aniline (9e) (0.100 ml, 1.1 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN (2 ml) were subject to microwave irradiation at 140°C for 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (50% EtOAc in hexanes) to afford the product as a colourless oil (57 mg, 16%; ratio 14e : 11e, 3 : 2).
Compound 11e was also prepared using general procedure 5 with compound 8a (0.131 g, 0.8 mmol, loop C) and aniline (9e) (0.36 ml, 4.0 mmol, loop C, 2.0 M). The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% CH3OH in CH2Cl2) to afford the desired product as a brown solid (121 mg, 59%), m.p.: 92–94°C. 1H NMR (400 MHz, CDCl3) δ 7.19–7.15 (m, 4H), 6.89–6.86 (m, 2H), 6.72 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 7.6 Hz, 2H), 4.07–4.01 (m, 1H), 3.80 (d, J = 4.3 Hz, 3H), 3.31 (dd, J = 12.8, 3.4 Hz, 1H), 3.08 (dd, J = 12.8, 8.0 Hz, 1H), 2.84 (dd, J = 13.8, 5.2 Hz, 1H), 2.76 (dd, J = 13.8, 7.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 158.6, 148.4, 130.5 (2C), 129.7, 129.4 (2C), 118.0, 114.3 (2C), 113.4 (2C), 71.4, 55.4, 49.6, 40.8; IR υmax/cm−1: 3384 (OH), 2928 (CH2, CH3), 1493 (arom.), 1243 (C–O), 1031 (C–O–C); LRMS (ESI+) m/z 258 (100%, M + H); HRMS (ESI) calcd for C16H20NO2 (M + H), 258.1489; found 258.1489.
4.39. 1-(Benzylamino)-3-(4-methoxyphenyl)propan-2-ol (11f)
Compound 11f was prepared using general procedure 5 with compound 8a (0.131 g, 0.8 mmol, loop C) and benzylamine (9c) (0.46 ml, 4.0 mmol, loop C, 2.0 M). The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (10% CH3OH in CH2Cl2) to afford the product as a pale brown solid (109 mg, 50%), m.p.: 63–66°C. 1H NMR (400 MHz, CD3OD) δ 7.33–7.22 (m, 5H), 7.11 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 3.93–3.87 (m, 1H), 3.79–3.68 (m, 5H), 2.72–2.61 (m, 3H), 2.56–2.50 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 158.3, 140.1, 130.40 (2C), 130.36, 128.6 (2C), 128.2 (2C), 127.2 (2C), 114.0, 70.9, 55.4, 54.3, 53.8, 40.8; IR υmax/cm−1: 3200 (OH), 2936 (CH2), 2835 (O-CH3), 1510 (arom.), 1242 (C–O), 1033 (C–O–C); LRMS (ESI+) m/z 272 (100%, M + H); HRMS (ESI) calcd for C17H22NO2 (M + H), 272.1645; found 272.1642.
4.40. 1-(Benzyl(methyl)amino)-3-(4-methoxyphenyl)propan-2-ol (11g)
Compound 11g was prepared using general procedure 3; compound 8a (0.164 g, 1.0 mmol), N-benzylmethylamine (9g) (0.169 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN (2 ml) were subject to microwave irradiation at 140°C and 10 min. The resulting reaction was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (10% CH3OH in CH2Cl2) to afford the product as a yellow oil (58 mg, 20%).
Compound 11g was also prepared using general procedure 5 with compound 8a (0.131 g, 0.8 mmol, loop C) and N-benzylmethylamine (9g) (0.103 ml, 4.0 mmol, loop C, 2.0 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography (50% EtOAc in hexanes) to afford the desired product as a brown oil (150 mg, 66%). 1H NMR (400 MHz, acetone-d6) δ 7.39–7.22 (m, 5H), 7.17 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 3.97–3.88 (m, 1H), 3.77 (s, 3H), 3.63 (d, J = 13.2 Hz, 1H), 3.51 (d, J = 13.2 Hz, 1H), 2.67 (ddd, J = 20.9, 13.8, 6.1 Hz, 2H), 2.48–2.36 (m, 2H), 2.21 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 159.1, 140.0, 132.0, 131.2 (2C), 129.8 (2C), 129.0 (2C), 127.8, 114.3 (2C), 69.7, 64.0, 63.2, 55.4, 42.6, 41.4; IR υmax/cm−1: 3442 (OH), 2930 (CH2), 2837 (–O–CH3), 1512 (arom.), 1244 (C–O), 1032 (C–O–C); LRMS (ESI+) m/z 286 (100%, M + H); HRMS (ESI) calcd for C18H24NO2 (M + H), 286.1802; found 286.1800.
4.41. 2-(tert-Butylamino)-1-phenylethanol (12a)
Compound 12a was prepared using general procedure 5 with styrene oxide (8b) (0.092 ml, 0.8 mmol, loop C) and tert-butylamine (9a) (0.59 ml, 5.6 mmol, loop C, 2.8 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography to afford the desired product as a white solid (121 mg, 78%), m.p.: 85–87°C. 1H NMR (400 MHz, CD3OD) δ 7.41–7.31 (m, 4H), 7.30–7.23 (m, 1H), 4.70 (dd, J = 9.1, 4.0 Hz, 1H), 2.76 (dd, J = 11.4, 9.1 Hz, 1H), 2.69 (dd, J = 11.5, 4.0 Hz, 1H), 1.13 (s, 9H); 13C NMR (151 MHz, CD3OD) δ 165.5, 142.7, 126.8 (2C), 115.9 (2C), 72.7, 70.0, 46.0, 28.5 (3C); IR υmax/cm−1: 3288 (OH), 2973 (CH), 1605 (N–H), 1224 (C–O); LRMS (ESI+) m/z 194 (100%, M + H) [30].
4.42. 2-(Cyclohexylamino)-1-phenylethanol (12c)
Compound 12c was prepared using general procedure 5 with styrene oxide (8b) (0.092, 0.8 mmol, loop C) and cyclohexylamine (9c) (0.46 ml, 4.0 mmol, loop C, 2.8 M). The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (10% CH3OH in CH2Cl2) to afford the product as a crystalline tan solid (0.98 g, 58%), m.p.: 86–88°C. 1H NMR (400 MHz, acetone-d6) δ 7.39 (d, J = 7.2 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 4.69 (dd, J = 9.1, 3.3 Hz, 1H), 2.93 (dd, J = 11.9, 3.6 Hz, 1H), 2.65 (dd, J = 11.9, 9.2 Hz, 1H), 2.52 (tt, J = 10.1, 3.7 Hz, 1H), 1.97–1.85 (m, 2H), 1.76–1.67 (m, 2H), 1.63–1.54 (m, 1H), 1.33–1.06 (m, 5H); 13C NMR (101 MHz, CDCl3) δ 144.9, 128.9 (2C), 127.8, 126.7 (2C), 72.6, 57.2, 55.6, 34.1, 33.8, 26.9, 25.6 (2C); IR υmax/cm−1: 3284 (OH), 2931 (CH), (N–H), 1492 (arom.), 1263 (C–O); LRMS (ESI+) m/z 220 (100%, M + H); HRMS (ESI) calcd for C14H22NO (M + H), 220.1696; found 220.1695.
4.43. 2-Morpholino-1-phenylethan-1-ol (12d)
Compound 12d was prepared using general procedure 3; styrene oxide (8b) (0.114 ml, 1.0 mmol), morpholine (9d) (0.096 ml, 1.1 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% CH3OH in CH2Cl2) to afford the product as a white solid (133 mg, 64%).
Compound 12d was also prepared using general procedure 5 with styrene oxide (8b) (0.092 ml, 0.8 mmol, loop C) and morpholine (9d) (0.48 ml, 5.6 mmol, loop C, 2.8 M). The resulting reaction mixture concentrated in vacuo to afford the desired product as a brown solid (0.032 g, 78%), m.p.: 81–82°C. 1H NMR (400 MHz, acetone-d6) δ 7.40 (d, J = 7.6 Hz, 2H), 7.32 (t, J = 7.6 Hz, 2H), 7.23 (t, J = 7.2 Hz, 1H), 4.79 (dd, J = 9.0, 3.4 Hz, 1H), 4.10 (s, 1H), 3.72–3.58 (m, 4H), 2.69–2.60 (m, 2H), 2.52–2.41 (m, 4H); 13C NMR (101 MHz, acetone-d6) δ 144.5, 128.9 (2C), 128.0, 127.0 (2C), 69.9, 68.0 (2C), 67.5, 54.6 (2C); IR υmax/cm−1: 3122 (OH), 2926, 2817 (CH2, CH3), 1510 (arom.), 1134 (C–O), 1111 (C–O–C); LRMS (ESI+) m/z 208 (100%, M + H); HRMS (ESI) calcd for C12H18NO2 (M + H), 208.1332; found 208.1332.
4.44. 1-Phenyl-2-(phenylamino)ethan-1-ol (12e)
Compound 12e was prepared using general procedure 3; styrene oxide (8b) (0.114 ml, 1.0 mmol), aniline (9e) (0.100 ml, 1.1 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction mixture was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% EtOAc in hexanes) to afford the product as a brown oil (68 mg, 32%).
Compound 12e was also prepared using general procedure 5 with styrene oxide (8b) (0.092 ml, 0.8 mmol, loop C) and aniline (9e) (0.51 ml, 5.6 mmol, loop C, 2.8 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography to afford the desired product as a brown oil (56 mg, 33%). 1H NMR (400 MHz, acetone-d6) δ 7.44 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 7.25–7.17 (m, 1H), 7.00 (dd, J = 8.5, 7.4 Hz, 2H), 6.61–6.49 (m, 3H), 5.33 (d, J = 3.0 Hz, 1H), 4.46 (dt, J = 8.0, 4.9 Hz, 1H), 4.06 (t, J = 5.9 Hz, 1H), 3.82 (ddd, J = 10.4, 5.7, 4.5 Hz, 1H), 3.66 (ddd, J = 11.0, 7.8, 6.0 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 149.3, 142.8, 129.6 (2C), 129.2 (2C), 127.8 (3C), 117.5, 114.3 (2C), 67.8, 61.2; IR υmax/cm−1: 3391 (OH), 2928 (CH), 1601 (N–H), 1502 (arom.), 1263 (C–O); LRMS (ESI+) m/z 214 (100%, M + H) [30,61].
4.45. 2-(Benzylamino)-1-phenylethanol (12f)
Compound 12f was prepared using general procedure 5 with styrene oxide (8b) (0.092 ml, 0.8 mmol, loop C) and benzylamine (9f) (0.61 ml, 5.6 mmol, loop C, 2.8 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography to afford the desired product as a brown solid (138 mg, 76%), m.p.: 83–87°C. 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 10H), 4.74 (dd, J = 8.9, 3.6 Hz, 1H), 3.89–3.81 (m, 2H), 2.95 (dd, J = 12.2, 3.6 Hz, 1H), 2.76 (dd, J = 12.2, 8.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 128.7, 128.5, 128.3, 127.7, 127.4, 126.0, 77.4, 71.9, 56.6, 53.6, 31.1; IR υmax/cm−1: 3289 (OH), 2933 (CH), 1626 (N–H), 1502 (arom.), 1299 (C–O).); LRMS (ESI+) m/z 228 (100%, M + H) [62].
4.46. 2-(Benzyl(methyl)amino)-2-phenylethanol (12g) :2-(benzyl(methyl)amino)-1-phenylethanol (15g)
A mixture 12g/15g was prepared using general procedure 3; styrene oxide (8b) (0.114 ml, 1.00 mmol), N-benzylmethylamine (9g) (0.169 ml, 1.10 mmol) and bismuth (III) trifluoromethane sulfonate (0.100 g, 15 mol%) in 3 ml CH3CN were subject to microwave irradiation at 140°C and 10 min. The resulting reaction was concentrated in vacuo, adsorbed to silica and subjected to column chromatography (5% CH3OH in CH2Cl2) to afford the product as a yellow oil (0.126 g, 52%; ratio of 12 g : 15 g, 1 : 5).
Mixture 12g/15g was also prepared using general procedure 5 with styrene oxide (8b) (0.092 ml, 0.8 mmol, loop C) and N-benzylmethylamine (9g) (0.72 ml, 5.6 mmol, loop C, 2.8 M). The resulting reaction mixture was adsorbed to silica and subjected to column chromatography to afford the desired product as a yellow oil (126 mg, 60%). Major regioisomer: 1H NMR (400 MHz, acetone-d6) δ 7.51–7.13 (m, 10H), 4.80 (dd, J = 8.9, 4.4 Hz, 1H), 4.11 (s, 1H), 3.72 (d, J = 13.4 Hz, 1H), 3.56 (d, J = 13.4 Hz, 1H), 2.64–2.47 (m, 2H), 2.30 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 144.6, 140.0, 129.8 (2C), 129.1 (2C), 128.8 (2C), 127.8, 127.8, 126.8 (2C), 70.7, 66.6, 63.0, 42.4; minor regioisomer: 1H NMR (400 MHz, acetone-d6) δ 7.57–7.08 (m, 10H), 4.07–4.02 (m, 1H), 3.79–3.73 (m, 2H), 3.62 (d, J = 13.4 Hz, 1H), 3.42 (d, J = 13.4 Hz, 1H), 2.13 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 129.8, 129.6, 128.9, 128.1, 127.7, 70.3, 62.9, 59.6, 38.2; IR υmax/cm−1: 3431 (OH), 2941 (CH2), 1442 (CH2), 1019 (C–O–C); LRMS (ESI+) m/z 242 (100%, M + H); HRMS (ESI) calcd for C16H20NO (M + H) 242.1539; found 242.1539.
Supplementary Material
Acknowledgements
P.J.C. acknowledges the receipt of a University of Newcastle Postgraduate Research Scholarship. J.R.B. acknowledges the receipt of an Australian Postgraduate Award.
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
This work was conceived by P.J.C. and N.C. The chemical synthesis reported herein was conducted by P.J.C., J.R.B. and N.C. A.M., P.J.C. and J.R.B. co-wrote the manuscript. A.M. is responsible for obtaining the funding to support this project.
Competing interests
The authors declare we have no competing interests.
Funding
A.M. is the recipient of funding from the Australian Cancer Research Foundation, the Ramaciotti Foundation and the Australian Research Council (grant no. DP14101565). P.J.C. is the recipient of a University of Newcastle Postgraduate Research Scholarship. J.R.B. is the recipient of an Australian Postgraduate Award.
References
- 1.Ager DJ, Prakash I, Schaad R. 1996. 1,2-Amino alcohols and their heterocyclic derivatives as chiral auxiliaries in asymmetric synthesis. Chem. Rev. 96, 835–876. (doi:10.1021/cr9500038) [DOI] [PubMed] [Google Scholar]
- 2.Bergmeier SC. 2000. The synthesis of vicinal amino Alcohols. Tetrahedron, 56, 2561–2576. (doi:10.1016/S0040-4020(00)00149-6) [Google Scholar]
- 3.Karjalainen OK, Koskinen AMP. 2012. Diastereoselective synthesis of vicinal amino alcohols. Org. Biomol. Chem. 10, 4311–4326. (doi:10.1039/c2ob25357g) [DOI] [PubMed] [Google Scholar]
- 4.Lin GQ, Xu MH, Zhong YW, Sun XW. 2008. An advance on exploring N-tert-butanesulfinyl imines in asymmetric synthesis of chiral amines. Acc. Chem. Res. 41, 831–840. (doi:10.1021/ar7002623) [DOI] [PubMed] [Google Scholar]
- 5.Burchak ON, Py S. 2009. Reductive cross-coupling reactions (RCCR) between C═N and C═O for β-amino alcohol synthesis. Tetrahedron 65, 7333–7356. (doi:10.1016/j.tet.2009.06.003) [Google Scholar]
- 6.Curini M, Epifano F, Marcotullio MC, Rosati O. 2001. Zirconium sulfophenyl phosphonate as a heterogeneous catalyst in the preparation of β-amino alcohols from epoxides. Eur. J. Org. Chem. 2001, 4149–4152. (doi:10.1002/1099-0690(200111)2001:21<4149::AID-EJOC4149>3.0.CO;2-R) [Google Scholar]
- 7.Kureshy RI, Agrawal S, Kumar M, Khan NUH, Abdi SHR, Bajaj HC. 2010. Hβ zeolite: An efficient and reusable catalyst for ring-opening of epoxides with amines under microwave irradiation. Catal. Lett. 134, 318–323. (doi:10.1007/s10562-009-0237-z) [Google Scholar]
- 8.McCluskey A, Leitch SK, Garner J, Caden CE, Hill TA, Odell LR, Stewart SG. 2005. BiCl3-mediated opening of epoxides, a facile route to chlorohydrins or amino alcohols: one reagent, two paths. Tetrahedron Lett. 46, 8229–8232. (doi:10.1016/j.tetlet.2005.09.088) [Google Scholar]
- 9.Cossar PJ, Ma C, Gordon CP, Ambrus JI, Lewis PJ, McCluskey A. 2010. Identification and validation of small molecule modulators of the NusB-NusE interaction. Bioorg. Med. Chem. Lett. 27, 162–167. (doi:10.1016/j.bmcl.2016.11.091) [DOI] [PubMed] [Google Scholar]
- 10.Wegner J, Ceylan S, Kirschning A. 2012. Flow chemistry: a key enabling technology for (multistep) organic synthesis. Adv. Syn. Cat. 354, 17–57. (doi:10.1002/adsc.201100584) [Google Scholar]
- 11.Petersen TP, Mirsharghi S, Rummel PC, Thiele S, Rosenkilde MM, Ritzen A, Ulven T. 2013. Multistep continuous-flow synthesis in medicinal chemistry: discovery and preliminary structure–activity relationships of CCR8 ligands. Chem., Eur. J. 19, 9343–9350. (doi:10.1002/chem.201204350) [DOI] [PubMed] [Google Scholar]
- 12.Sharma S, Maurya RA, Min K-I, Jeong G-Y, Kim D-P. 2013. Odorless isocyanide chemistry: an integrated microfluidic system for a multistep reaction sequence. Angew. Chem. Int. Ed. Engl. 52, 7564–7568. (doi:10.1002/anie.201303213) [DOI] [PubMed] [Google Scholar]
- 13.Brzozowski M, O'Brien M, Ley SV, Polyzos A. 2015. Flow chemistry: intelligent processing of gas-liquid transformations using a tube-in-tube reactor. Acc. Chem. Res. 48, 349–362. (doi:10.1021/ar500359m) [DOI] [PubMed] [Google Scholar]
- 14.Salvador CEM, Pieber B, Neu PM, Torvisco A, Kleber C, Andrade Z, Kappe CO. 2015. A sequential Ugi multicomponent/Cu-catalyzed azide-alkyne cycloaddition approach for continuous flow generation of cyclic peptoids. J. Org. Chem. 80, 4590–4602. (doi:10.1021/acs.joc.5b00445) [DOI] [PubMed] [Google Scholar]
- 15.Kim H, Lee H-J, Kim D-P. 2016. Flow-assisted synthesis of [10]Cycloparaphenylene through serial microreactions under mild conditions. Angew. Chem. Int. Ed. Engl. 55, 1422–1426. (doi:10.1002/anie.201509748) [DOI] [PubMed] [Google Scholar]
- 16.McQuade DT, Seeberger PH. 2013. Applying flow chemistry: methods, materials, and multistep synthesis. J. Org. Chem. 78, 6384–6389. (doi:10.1021/jo400583m) [DOI] [PubMed] [Google Scholar]
- 17.Gilmore K, Kopetzki D, Lee JW, Horváth Z, McQuade DT, Seidel-Morgenstern A, Seeberger PH. 2014. Continuous synthesis of artemisinin-derived medicines. Chem. Commun. 50, 12 652–12 655. (doi:10.1039/C4CC05098C) [DOI] [PubMed] [Google Scholar]
- 18.Wiles C, Watts P. 2012. Continuous flow reactors: a perspective. Green Chem. 14, 38–54. (doi:10.1039/C1GC16022B) [Google Scholar]
- 19.Noël T, Buchwald SL. 2011. Cross-coupling in flow. Chem. Soc. Rev. 40, 5010–5029. (doi:10.1039/c1cs15075h) [DOI] [PubMed] [Google Scholar]
- 20.Battilocchio C, Feist F, Hafner A, Simon M, Tran DN, Allwood DM, Blakemore DC, Ley SV. 2016. Iterative reactions of transient boronic acids enable sequential C–C bond formation. Nature Chem. 8, 360–367. (doi:10.1038/nchem.2439) [DOI] [PubMed] [Google Scholar]
- 21.Trinh TN, Hizartzidis L, Lin AJS, McCluskey A, Gordon CP. 2014. An efficient continuous flow approach to furnish furan-based biaryls. Org. Biomol. Chem. 12, 9562–9571. (doi:10.1039/C4OB01641F) [DOI] [PubMed] [Google Scholar]
- 22.Hopkin MD, Baxendale IR, Ley SV. 2013. An expeditious synthesis of imatinib and analogues utilising flow chemistry methods. Org. Biomol. Chem. 11, 1822–1839. (doi:10.1039/C2OB27002A) [DOI] [PubMed] [Google Scholar]
- 23.Ley SV, Fitzpatrick DE, Ingham RJ, Myers RM. 2015. Organic synthesis: march of the machines. Angew. Chem. Int. Ed. Engl. 54, 3449–3464. (doi:10.1002/anie.201410744) [DOI] [PubMed] [Google Scholar]
- 24.Ingham RJ, Riva E, Nikbin N, Baxendale IR, Ley SV. 2012. A ‘Catch–react–release’ method for the flow synthesis of 2-aminopyrimidines and preparation of the Imatinib base. Org. Lett. 14, 3920–3923. (doi:10.1021/ol301673q) [DOI] [PubMed] [Google Scholar]
- 25.Porta R, Benaglia M, Puglisi A. 2015. Flow chemistry: recent developments in the synthesis of pharmaceutical products. Org. Proc. Res. Develop. 20, 2–25. (doi:10.1021/acs.oprd.5b00325) [Google Scholar]
- 26.Hizartzidis L, Tarleton M, Gordon CP, McCluskey A. 2014. Chemoselective flow hydrogenation approaches to isoindole-7-carboxylic acids and 7-oxa-bicyclio[2.2.1]heptanes. RSC Adv. 4, 9709–9722. (doi:10.1039/c3ra47657j) [Google Scholar]
- 27.Mastronardi F, Gutmann B, Kappe CIO. 2013. Continuous flow generation and reactions of anhydrous diazomethane using a teflon AF-2400 tube-in-tube reactor. Org. Lett. 15, 5590–5593. (doi:10.1021/ol4027914) [DOI] [PubMed] [Google Scholar]
- 28.Müller STR, Wirth T. 2015. Diazo compounds in continuous-flow technology. ChemSusChem 8, 245–250. (doi:10.1002/cssc.201402874) [DOI] [PubMed] [Google Scholar]
- 29.Nobuta T, Xiao G, Ghislieri D, Gilmore K, Seeberger PH. 2015. Continuous and convergent access to vicinyl amino alcohols. Chem. Commun. 51, 15 133–15 136. (doi:10.1039/C5CC06093A) [DOI] [PubMed] [Google Scholar]
- 30.Bedore MW, Zaborenko N, Jensen KF, Jamison TF. 2010. Aminolysis of epoxides in a microreactor system: a continuous flow approach to β-amino alcohols. Org. Process Res. Dev. 14, 432–440. (doi:10.1021/op9003136) [Google Scholar]
- 31.Gutmann B, Cantillo D, Kappe CO. 2015. Continuous-flow technology: a tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 54, 6688–6728. (doi:10.1002/anie.201409318) [DOI] [PubMed] [Google Scholar]
- 32.Hizartzidis L, Cossar PJ, Robertson MJ, Simone MI, Young KA, McCluskey A, Gordon CP. 2014. Expanding the utility of flow hydrogenation: a robust protocol restricting hydrodehalogenation. RSC Adv. 4, 56 743–56 748. (doi:10.1039/C4RA09605C) [Google Scholar]
- 33.Pastre JC, Browne DL, Ley SV. 2013. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 42, 8849–8869. (doi:10.1039/c3cs60246j) [DOI] [PubMed] [Google Scholar]
- 34.Baxendale IR, Hornung C, Ley SV, Molina J de MM, Wikström A. 2013. Flow microwave technology and microreactors in synthesis. Aust. J. Chem. 66, 131–144. (doi:10.1071/CH12365) [Google Scholar]
- 35.Kappe CO, Dallinger D. 2006. The impact of microwaves on drug discovery. Nat. Rev. Drug Disc. 5, 51–63. (doi:10.1038/nrd1926) [DOI] [PubMed] [Google Scholar]
- 36.Glasnov TN, Kappe CO. 2010. Toward a continuous-flow synthesis of Boscalid. Adv. Synth. Catal. 352, 3089–3097. (doi:10.1002/adsc.201000646) [Google Scholar]
- 37.Baumann M, Baxendale IR. 2013. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 9, 2265–2319. (doi:10.3762/bjoc.9.265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molette J, et al. 2013. Identification and optimization of an aminoalcohol-carbazole series with antimalarial properties. ACS Med. Chem. Lett. 4, 1037–1041. (doi:10.1021/ml400015f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherfaoui B, Guo TK, Sun HP, Cheng WL, Liu F, Jiang F, Xu XL, You QD. 2016. Synthesis and evaluation of 4-(2-hydroxypropyl)piperazin-1-yl) derivatives as Hsp90 inhibitors. Bioorganic Med. Chem. 24, 2423–2432. (doi:10.1016/j.bmc.2016.03.049) [DOI] [PubMed] [Google Scholar]
- 40.Kong X-D, Ma Q, Zhou J, Zeng B-B, Xu J-H. 2014. A smart library of epoxide hydrolase variants and the top hits for synthesis of (S)-β-blocker precursors. Angew. Chem. Int. Ed. 53, 6641–6644. (doi:10.1002/anie.201402653) [DOI] [PubMed] [Google Scholar]
- 41.Jana NK, Verkade JG. 2003. Phase-vanishing methodology for efficient bromination, alkylation, epoxidation, and oxidation reactions of organic substrates. Org. Lett. 5, 3787–3790. (doi:10.1021/ol035391b) [DOI] [PubMed] [Google Scholar]
- 42.Lifchits O, Mahlau M, Reisinger CM, Lee A, Fare C, Polyak I. 2013. The cinchona primary amine-catalyzed asymmetric epoxidation and hydroperoxidation of α,β-unsaturated carbonyl compounds with hydrogen peroxide. J. Am. Chem. Soc. 135, 6677–6693. (doi:10.1021/ja402058v) [DOI] [PubMed] [Google Scholar]
- 43.Kakei H, Tsuji R, Ohshima T, Shibasaki M. 2005. Catalytic asymmetric epoxidation of α,β-unsaturated esters using an yttrium-piphenyldiol complex. J. Am. Chem. Soc. 127, 8962–8963. (doi:10.1021/ja052466t) [DOI] [PubMed] [Google Scholar]
- 44.Lin AJS, Russell CC, Baker JR, Frailey SL, Sakoff JA, McCluskey A. 2016. A Facile hybrid ‘flow and batch’ access to substituted 3,4-dihydro-2H-benzo[b][1,4]oxazine-6-carboxamides. Org. Biomol. Chem. 14, 8732–8742. (doi:10.1039/C6OB01153E) [DOI] [PubMed] [Google Scholar]
- 45.Bach RD, Dmitrenko O, Adam W, Schambony S. 2003. Relative reactivity of peracids versus dioxiranes (DMDO and TFDO) in the epoxidation of alkenes. A combined experimental and theoretical analysis. J. Am. Chem. Soc. 125, 924–934. (doi:10.1021/ja026882e) [DOI] [PubMed] [Google Scholar]
- 46.Baumstark AL, Vasquez PC. 1988. Epoxidation by dimethyldioxirane. Electronic and steric effects. J. Org. Chem. 53, 3437–3439. (doi:10.1021/jo00250a007) [Google Scholar]
- 47.Lidström P, Tierney J, Wathey B, Westman J. 2001. Microwave assisted organic synthesis: a review. Tetrahedron 57, 9225–9283. (doi:10.1016/S0040-4020(01)00906-1) [Google Scholar]
- 48.Kappe CO. 2004. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 43, 6250–6280. (doi:10.1002/anie.200400655) [DOI] [PubMed] [Google Scholar]
- 49.Watts P, Wiles C. 2012. Micro reactors, flow reactors and continuous flow synthesis. J. Chem. Res. 36, 181–193. (doi:10.3184/174751912X13311365798808) [Google Scholar]
- 50.Zhao J, Chu Y-Y, Li A-T, Ju X, Kong X-D, Pan J, Tang Y, Xu J-H. 2011. An unusual (R)-selective epoxide hydrolase with high activity for facile preparation of enantiopure glycidyl ethers. Adv. Synth. Catal. 353, 1510–1518. (doi:10.1002/adsc.201100031) [Google Scholar]
- 51.Toda Y, Komiyama Y, Kikuchi A, Suga H. 2016. Tetraarylphosphonium salt-catalyzed carbon dioxide fixation at atmospheric pressure for the synthesis of cyclic carbonates. ACS Catal. 6, 6906–6910. (doi:10.1021/acscatal.6b02265) [Google Scholar]
- 52.Pchelka BK, Loupy A, Petit A. 2006. Improvement and simplification of synthesis of 3-aryloxy-1,2-epoxypropanes using solvent-free conditions and microwave irradiations. Relation with medium effects and reaction mechanism. Tetrahedron 62, 10 968–10 979. (doi:10.1016/j.tet.2006.08.067) [Google Scholar]
- 53.Schmit ML, et al. 2013. Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J. Med. Chem. 56, 7334–7342. (doi:10.1021/jm400792m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jensen KL, Standley EA, Jamison TF. 2014. Highly regioselective nickel-catalyzed cross-coupling of N-tosylaziridines and alkylzinc reagents. J. Am. Chem. Soc. 136, 11 145–11 152. (doi:10.1021/ja505823s) [DOI] [PubMed] [Google Scholar]
- 55.Geng X, Wang Z, Li X, Zhang C. 2005. A simple method for epoxidation of olefins using sodium chlorite as an oxidant without a catalyst. J. Org. Chem. 70, 9610–9613. (doi:10.1021/jo0511400) [DOI] [PubMed] [Google Scholar]
- 56.Yang SG, Hwang JP, Park MY, Lee K, Kim YH. 2007. Highly efficient epoxidation of electron-deficient olefins with tetrabutylammonium peroxydisulfate. Tetrahedron 63, 5184–5188. (doi:10.1016/j.tet.2007.03.167) [Google Scholar]
- 57.Brunetto G, Gori S, Fraschi R, Napolitano E. 2002. Crystallization-induced asymmetric transformations. Enantiomerically pure (−)-(R)- and (+)-(S)-2,3-dibromopropan-1-ol and epibromohydrins. A study of dynamic resolution via the formation of diastereoisomeric esters. Helv. Chim. Acta 85, 3785–3791. (doi:10.1002/1522-2675(200211)85:11<3785::AID-HLCA3785>3.0.CO;2-A) [Google Scholar]
- 58.Borude VS, Shah RV, Shukla SR. 2013. Synthesis of β-amino alcohol derivatives from phenols in presence of phase transfer catalyst and lipase biocatalyst. Curr. Chem. Lett. 2, 1–12. (doi:10.5267/j.ccl.2012.10.002) [Google Scholar]
- 59.Forkel NV, Henderson DA, Fuchter MJ. 2014. Calcium-mediated stereoselective reduction of α,β-epoxy ketones. Tetrahedron Lett. 55, 5511–5514. (doi:10.1016/j.tetlet.2014.08.050) [Google Scholar]
- 60.Tan W, Zhao B, Sha L, Jiao P, Wan M, Su L, Shin D. 2006. Microwave-assisted ring opening of epoxides in solvent-free conditions. Synth. Commun. 36, 1353–1359. (doi:10.1080/00397910500522025) [Google Scholar]
- 61.Negrón-Silva G, Hernández-Reyes CX, Angeles-Beltrán D, Lomas-Romero L, González-Zamora E, Méndez-Vivar J. 2007. Comparative study of regioselective synthesis of β-aminoalcohols under solventless conditions catalyzed by sulfated zirconia and SZ/MCM-41. Molecules 12, 2515–2532. (doi:10.3390/12112515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Padwa A, Chen Y-Y, Chiacchio U, Dent W. 1985. Diastereofacial selectivity in azomethine ylide cycloaddition reactions derived from chiral α-cyanoaminosilanes. Tetrahedron 41, 3529–3535. (doi:10.1016/S0040-4020(01)96706-7) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.