

Abstract
A series of 20 novel chromone embedded [1,2,3]-triazoles derivatives were synthesized via an easy and convenient synthetic procedure starting from 2-hydroxy acetophenone. The in vitro anti-mycobacterial evaluation studies carried out in this work reveal that seven compounds exhibit significant inhibition against Mycobacterium tuberculosis H37Rv strain with MIC in the range of 1.56–12.5 µg ml−1. Noticeably, compound 6s was the most potent compound in vitro with a MIC value of 1.56 µg ml−1. Molecular docking and chemoinformatics studies revealed that compound 6s displayed drug-like properties against the enoyl-acyl carrier protein reductase of M. tuberculosis further establishing its potential as a potent inhibitor.
Keywords: chromone, triazole, molecular docking, anti-mycobacterial activity
1. Introduction
Tuberculosis is a major infectious disease caused by Mycobacterium tuberculosis (Mtb) and it is estimated that there were 10.4 million new cases and 1.4 million deaths in 2015 alone, of which developing countries showed a major share [1]. Recent study reveals that the numbers of TB cases in India are two to three times higher than previously estimated suggesting that the global number of TB cases might be largely underestimated [2]. Furthermore, the emergence of a drug-resistant microorganism responsible for TB, especially multidrug-resistant one, along with lethal combination of TB and HIV infection makes this disease even more challenging [3–5]. In the last 50 years, only a few drugs have been approved by the FDA to treat TB. Therefore, the discovery and development of novel anti-TB agents with new chemotypes acting on novel drug targets is an important task for infectious diseases research programmes.
Natural products have a rich history as lead compounds for drug discovery. Further, natural products have contributed significantly to the current portfolio of anti-TB drugs, with one first-line drug (rifampicin) and several second-line agents (kanamycin, viomycin, etc.) being either natural products themselves or being derived from a natural product lead [6,7]. Chromone frameworks are frequently found in a diverse array of natural products, that includes natural flavone/isoflavone products, therapeutically active drugs such as anti-inflammatory, anti-platelet, anti-microbial, anti-obesity, anti-cancer agents, drug candidates for neurodegenerative diseases and adenosine receptors [8–10]. In fact, biological activities of these chromone molecules mainly depend on the conjugated bi- and tricyclic motifs with ketone functionality, but vary depending on the nature, position and variation of substituents. In this context, and in view of our continuing interest in the chemistry of privileged chromone motif [11–13], in particular, the design and synthesis of natural products like small molecules based on the chromone motif for various biological applications, herein, we designed and synthesized a series of novel chromone embedded 1,4 disubstituted [1,2,3]-triazole analogues using chimeric approach [14–16] and evaluated their anti-mycobacterial potential against M. tuberculosis H37Rv (figure 1). The interest in incorporation of [1,2,3]-triazole moiety stems from the advent of click chemistry protocol [17,18], which has been used in various applications including drug discovery process. In addition, triazole embedded heterocyclic frameworks exhibit plethora of biological activities, especially anti-mycobacterial activity (figure 1) [19–22]. Furthermore, these triazole products are considered as aggressive pharmacophores that can actively engage in drug–receptor interactions while maintaining an excellent chemical and metabolic profile [23].
Figure 1.

(a) Representative example of chromones and [1,2,3]-triazole analogues and their anti-tubercular activity; (b) our design of chromone embedded [1,2,3]-triazole framework as chimeric scaffold.
2. Results and discussion
2.1. Synthesis
A four-step synthetic strategy was followed for the preparation of novel chromone embedded [1,2,3]-triazoles 6a–t as outlined in scheme 1. At first, 3-formyl chromone 2 was synthesized by formylation of o-hydroxylacetophenone 1 using Vilsmeier-Haack reagent (POCl3 in DMF) at 55°C for 5 h in 75% yield. Further, 3-formyl chromone 2 was treated for reduction with solid supported basic alumina in isopropanol at 75°C for 4 h to yield 3-hydroxyl methyl chromone 3 in 82% which was subsequently mesylated followed by azidation with sodium azide in DMF at 50°C for 5 h affording the key chromone embedded azide intermediate 4 in 93% yield. Finally, the [1,2,3]-triazole core was incorporated through copper catalysed 1,3 dipolar cycloaddition of 2-azido methyl chromone (4) with commercially available different alkyl/aryl terminal alkynes (5a–t) in the presence of sodium ascorbate in t-BuOH/H2O (1 : 1, v/v) solvent mixture. This resulted in the formation of chromone embedded triazole compounds (6a–t), respectively, in good to excellent yields (scheme 1). The structures of all the newly synthesized compounds 6a–t were confirmed by the 1H NMR, 13C NMR and mass spectral data (electronic supplementary material). In the 1H NMR spectra of compound 6a (representative example), a signal corresponding to the CH2 protons that bridge the chromone with triazole moiety was observed at δ 5.48 ppm (as a singlet). The corresponding 13C resonance signal was delineated at δ 45.5 ppm and the chromone carbonyl was discernible at δ 176.7 ppm. In addition, the appearance of a sharp singlet for 1 proton observed at δ 8.22 ppm in the PMR, suggested the presence of 1,2,3 triazole C–H. The appearance of a sharp singlet (1H) observed at δ 8.15 ppm in the PMR, suggested the presence olefinic C–H of chromone moiety. The HRMS (ESI) for 6a shows the m/z at 304.1086 for C18H13O2N3 [M + H]+.
Scheme 1.

Synthesis of chromone embedded [1,2,3]-triazoles.
2.2. Anti-mycobacterial evaluation
All the new chromone embedded [1,2,3]-triazole derivatives (6a–t) were screened for their in vitro anti-tubercular activity against Mycobacterium tuberculosis H37Rv (ATCC27294) using MABA assay method (see the electronic supplementary material for detailed experimental procedure). The minimum inhibitory concentration (MIC; µg ml−1) was determined for each compound. The MIC is defined as the lowest concentration at which complete inhibition of bacterial growth was observed. Ethambutol and rifampicin were used as reference compounds. The MIC values of the synthesized compounds along with the standard drugs for comparison are reported in table 1.
Table 1.
In vitro anti-tubercular activity of chromone embedded [1,2,3]-triazoles against Mycobacterium tuberculosis H37Rv.
| MIC | MIC | ||||||
|---|---|---|---|---|---|---|---|
| entry | compound | (μg ml−1)a | cytotoxicityb | entry | compound | (μg ml−1)a | cytotoxicityb |
| 6a | 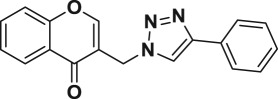 |
25 | 12.86 | 6k | 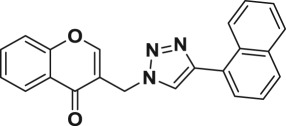 |
50 | 26.12 |
| 6b | 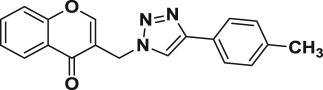 |
25 | 26.82 | 6l | 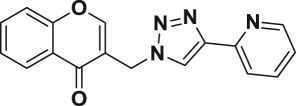 |
50 | 30.60 |
| 6c | 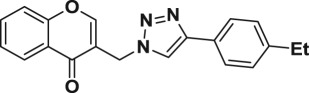 |
25 | 16.12 | 6m | 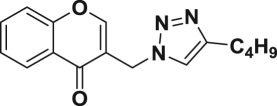 |
6.25 | 36.82 |
| 6d | 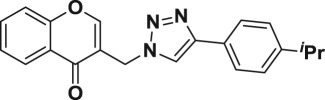 |
50 | 20.60 | 6n | 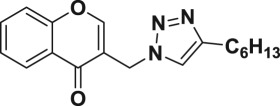 |
50 | 23.74 |
| 6e | 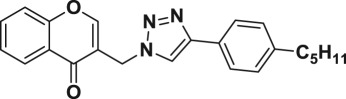 |
25 | 20.12 | 6o | 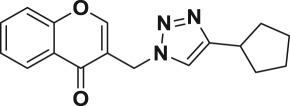 |
3.125 | 18.42 |
| 6f | 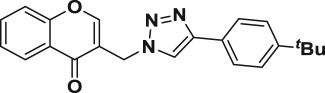 |
3.125 | 20.12 | 6p |  |
12.5 | 20.12 |
| 6g | 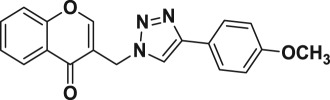 |
6.25 | 28.40 | 6q | 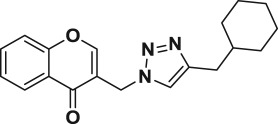 |
25 | 28.62 |
| 6h | 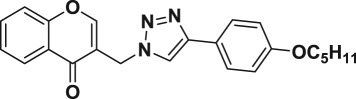 |
3.125 | 18.68 | 6r | 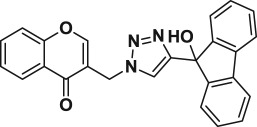 |
50 | 30.60 |
| 6i | 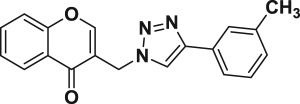 |
25 | 26.82 | 6s | 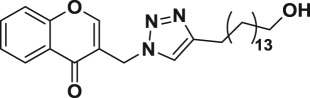 |
1.56 | 24.68 |
| 6j |  |
50 | 30.34 | 6t |  |
50 | 30.12 |
aRifampicin (MIC, 0.24 µg ml−1); ethambutol (MIC, 7.64 µg ml−1).
bCytotoxicity at 50 µg ml−1 (RAW 264.7 cells).
Among the 20 chromone embedded [1,2,3]-triazole derivatives tested, seven compounds (6f–6h, 6m, 6o, 6p and 6s) were found to be active with MIC values in the range of 1.56–12.5 µg ml−1. The compound 6s was found to be highly active among all the compounds tested with a MIC value of 1.56 µg ml−1, which is 4.8 times more active than the standard drug ethambutol (MIC, 7.64 µg ml−1). The preliminary SAR of the chromone embedded triazole analogues reveals that the compounds bearing phenyl group (6a) as well as substituted phenyl such as 4-methyl, 4-ethyl, 4-propyl and 4-pentyl (6b, 6c, 6d and 6e) do not favour better activity, with the exception of 6f possessing 4-t-butyl group (MIC, 3.125 µg ml−1). It was also observed that alkoxy substitution at 4-position of the phenyl ring (6g and 6h) enhances the activity against Mtb. However, addition of another methyl group at 2-position of 6g leads to complete loss of activity, 6i (MIC, 50 µg ml−1). Further, replacement of phenyl group with naphthyl (6k) and pyridyl (6l) does not appear to enhance the activity (MIC, greater than 50 µg ml−1).
Interestingly, modification of the triazole core by changing R group from aromatic to aliphatic group (cyclic or acyclic) enhances the activity against Mtb. For example, the compound 6o possessing cyclopentyl substituent at R position and compound 6m possessing n-butyl substituent exhibit better activity (MIC, 3.125 and 6.25 µg ml−1, respectively), with an exception of 6n possessing n-hexyl group (MIC, greater than 50 µg ml−1). Importantly, the most active compound in the series, 6s possess long aliphatic chain terminated with hydrophilic –OH as a capping group (MIC, 1.56 µg ml−1).
All chromone embedded [1,2,3]-triazole analogues were also tested for in vitro cytotoxicity against RAW 264.7 cells at 50 µg concentration using (4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) assay. All the analogues showed less than 50% inhibition, percentage inhibitions of cells are represented in table 1. The most promising anti-tubercular analogues 6f, 6g, 6h, 6m, 6o and 6s exhibited 20.12%, 28.40%, 18.68%, 36.82%, 18.42% and 24.68% growth inhibition, respectively, at 50 µg ml−1. The results indicated that potent analogues 6f, 6h, 6o and 6s are comparatively less toxic and are suitable for further studies.
3. Computational studies
Mycobacterium tuberculosis inhibitors perform inhibitory action via different mechanistic pathways in the cell. We selected six validated protein targets from each pathway based on their role and importance (table 2) [24]. The biological significance of the selected proteins is discussed in detail herein. Thymidylate kinase (PDB ID: 1G3U) plays a role in the catalysis of the transfer of the phosphoryl moiety from the phosphoryl donor, ATP to TMP which is key intermediate for the DNA-blocking builds [25]. Lumazine synthase (PDB ID: 1W19) catalyses certain steps in riboflavin biosynthesis [26].
Table 2.
List of tuberculosis targets and mechanistic pathway class.
| PDB ID | name of targets | class |
|---|---|---|
| 1G3U | thymidylate kinase | DNA synthesis |
| 1W19 | 6,7-dimethyl-8-ribityllumazine synthase | cofactor biosynthesis |
| 1ZID | enoyl-acyl carrier protein | mycolic acid biosynthesis |
| 2OZ5 | MTB phosphotyrosine phosphatase B | arrest of phagosome maturation |
| 3IUB | pantothenate synthetase | β-alanine metabolism |
| 1DG5 | dihydrofolate reductase | folate metabolism |
Enoyl-acyl carrier protein (PDB ID: 1ZID) is essential for fatty acid synthase system (FAS-II) pathway in mycobacterial cells [27], whereas pantothenate synthase (PDB ID: 3IUB) catalyses the condensation of pantonate with β-alanine to form pantothenate, a precursor coenzyme A biosynthesis [28]. MTB phosphotyosine B [MtbPtpB] (PDB ID: 2OZ5) blocks the signal regulated kinase and p-38 mediated by IL-6 thereby promoting mycobacterial survival in the host [29]. Dihydrofolate reductase (PDB ID: 1DG5) helps in regulating the amount of tetrahydrofolate in the cell. Tetrahydrofolate derivatives are key components in purine and thymidylate synthesis, which is important for cell proliferation and cell growth [30].
4. Methodology
4.1. Preparation of ligands
The two-dimensional structures (.mol) of four compounds, i.e. 6f, 6h, 6o and 6s, were drawn and the structure was analysed by using Marvin view. The compounds were converted to three-dimensional structure (.pdb) using LigPrep tool [31]. LigPrep is a Schrödinger suite tool which is used to generate three-dimensional structures from two-dimensional structures, search tautomers, isomers for compounds and carry out energy minimization by applying the OPLS 2005 force field.
4.2. Preparation of macromolecule
The protein targets retrieved from RCSB Protein Data Bank are proteins associated with metabolic functioning and proliferation of M. tuberculosis. The proteins listed in table 2 served as docking receptors. The proteins were fixed for errors in atomic representations and optimized using Protein Preparation Wizard Maestro v. 10.3 (Maestro, v. 10.3: Schrödinger, LLC, New York, NY, USA). The bond orders were assigned to residues, hydrogen atoms were added at pH 7.0. Minimization was carried out using OPLS 2005 force field with a RMSD cut-off value of 0.3 Ǻ.
4.3. Molecular docking
The molecular docking was performed and analysed via the Glide v. 6.8 docking tool [32]. The receptor grid was centred based on the active site of the protein using receptor grid generation tool. Ligands prepared using LigPrep were flexibly docked in grid box using Monte Carlo-based simulation algorithm. An extra precision (XP) method was employed that generated binding poses based on energy. The favourably docked molecules were ranked according to the Glide Score (tables 3 and 4).
Table 3.
Molecular docking analysis of 6 protein targets with selected compounds. The binding energy was calculated for Glide in kcal mol−1.
| Glide score binding energy (kcal mol−1) |
||||
|---|---|---|---|---|
| PDB target | compound 6f | compound 6h | compound 6o | compound 6s |
| 1G3U | −6.551 | −6.782 | −5.617 | −5.912 |
| 1W19 | −6.852 | −4.880 | −4.165 | −5.600 |
| 1ZID | −7.826 | −9.189 | −7.316 | −11.123 |
| 2OZ5 | −6.899 | −7.344 | −5.572 | −7.967 |
| 3IUB | −6.600 | −6.602 | −5.104 | −5.291 |
| 1DG5 | −4.521 | −4.793 | −4.475 | −6.233 |
Table 4.
Molecular docking analysis of selected compounds.
| protein target | compound name | amino acids involved in intermolecular interactions | binding energy (kcal mol−1) |
|---|---|---|---|
| 1ZID | compound 6f | Thr196 | −7.826 |
| Phe149 | |||
| compound 6h | Met98 | −9.189 | |
| Arg32 | |||
| compound 6s | Asp64 | −11.123 | |
| Trp222 | |||
| Tyr158 | |||
| compound 6o | Thr196 | −7.316 |
4.4. Molecular docking analysis
Automated docking was used to assess the binding modes and conformation of the ligand molecules. Among the 20 chromone embedded [1,2,3]-triazoles, compounds 6f, 6h, 6o and 6s were considered as they showed significant activities (table 1). 1ZID, enoyl-acyl carrier protein yielded better binding scores with four chromone-based triazoles when compared with the rest of the proteins (table 2). Compound 6s gave a better score when compared with other compounds for the target proteins, with binding score ranging from −7.3 to −11.123 kcal mol−1. Enoyl-acyl carrier protein reductase is involved in mycolic acid biosynthesis, the inhibition of which leads to the lysis of Mtb. The key intermolecular protein ligand interactions are depicted in figure 2. Figure 2 represents the intermolecular amino acid interaction with the compounds 6f, 6h, 6s and 6o. Compound 6s showed highest binding energy values of −11.123 kcal mol−1. Asp64, Trp222 and Tyr158 amino acids interacted with compound 6s showing high ligand exposure. Trp222 and Tyr158 had interaction with the compound 6s. Compound 6f and 6o similarly showed interaction with Phe149 and Thr196, respectively. Thus, the above results suggest that interaction improves the docking scores. Compound 6s is bound to the active site amino acid residues in the pocket region as shown in figure 3a,b. The pocket region of 1ZID is present in a loop region flanked by alpha helix chains seen in figure 3a. The location and orientation of the triazole group are complementary to the surrounding InhA side chains, which create a specific binding pocket. These observations indicate that compound 6s may have an important role in anchoring within the active site of the receptor.
Figure 2.

Amino acids involved in intermolecular interactions.
Figure 3.
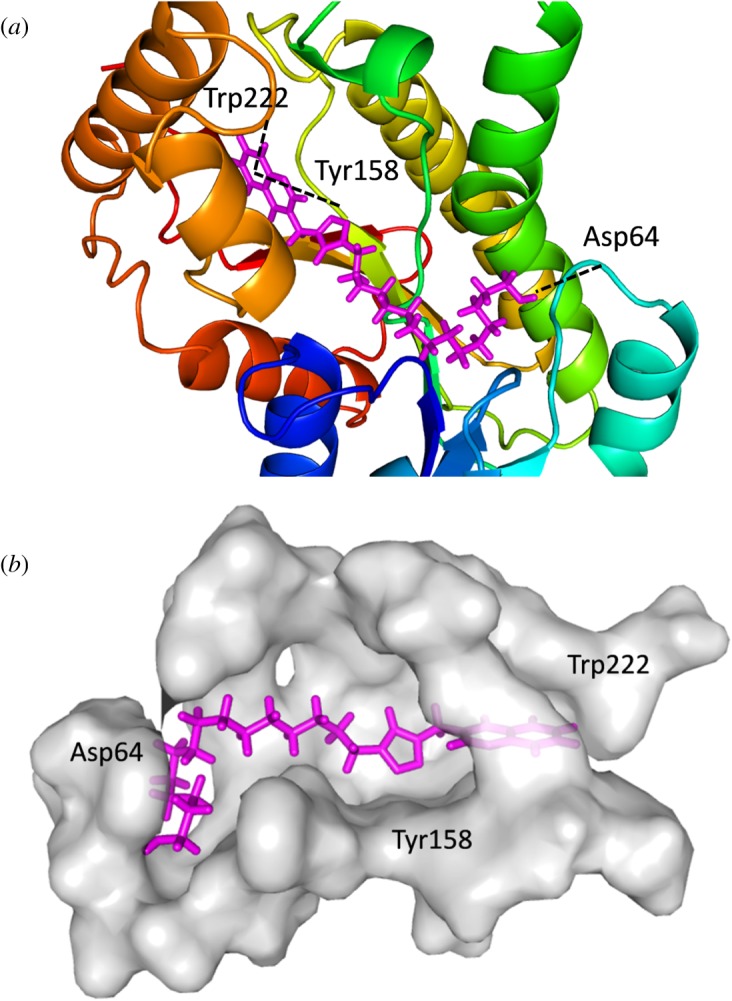
(a) Compound 6s (in magenta) in enoyl-acyl carrier protein (PDB ID:1ZID). (b) Alignment of compound 6s in the binding pocket.
4.5. Chemoinformatics analysis
Six active compounds were analysed for their drug-like properties (table 5). Lipinski rule of five were predicted using Screening Assistant 2 tool [33]. All these compounds including compound 6s displayed good drug-like properties. The drug-like and lead-like property analysis for the compounds generated a score of 0.25 which gave support to the positive results obtained in rule of five. ADME properties were predicted using PreADMET software [34] in order to check their potential as anti-tubercular compounds.
Table 5.
Chemoinformatics analysis.
| compounds | ||||||
|---|---|---|---|---|---|---|
| properties | 6f | 6g | 6h | 6m | 6o | 6s |
| Lipinski rulea | ||||||
| molecular weight | 359.429 | 333.347 | 389.455 | 283.331 | 295.342 | 453.627 |
| HB accept | 3 | 4 | 4 | 3 | 3 | 4 |
| HB donor | 0 | 0 | 0 | 0 | 0 | 1 |
| LogP | 3.335 | 1.792 | 3.631 | 1.533 | 1.409 | 5.188 |
| chemical properties | ||||||
| Weiner patha | 2075 | 1661 | 2737 | 1015 | 1117 | 4787 |
| ring counta | 4 | 4 | 8 | 5 | 3 | 17 |
| PDL/PLLa | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
| ADME properties | ||||||
| BBB (−3.0–1.2)b | 2.88269 | 1.161 | 0.23527 | 0.5112 | 0.36551 | 1.14616 |
| CaCo2 (nms) (<25, poor, >500, best)b | 34.0619 | 23.231 | 32.9739 | 25.026 | 16.0711 | 39.8231 |
| HIA (50–100%)b | 97.41 | 97.16 | 97.72 | 98.34 | 98.27 | 96.48 |
| rotatable bonds (0–15)a | 4 | 4 | 8 | 5 | 3 | 17 |
| TPSA (7.0–200.0)b | 57.01 | 66.24 | 66.24 | 57.01 | 57.01 | 77.24 |
| Toxicity propertiesc | ||||||
| DSSTox carcinogenic potency mutagenecity | Neg. | Neg. | Neg. | Neg. | Neg. | Neg. |
| (p: 0.161) | (p: 0.202) | (p: 0.112) | (p: 0.110) | (p: 0.189) | (p: 0.115) | |
| DSSTox carcinogenic potency mouse | Neg. | Neg. | Neg. | Neg. | Neg. | Neg. |
| (p: 0.083) | (p: 0.093) | (p: 0.099) | (p: 0.107) | (p: 0.141) | (p: 0.208) | |
aComputed using Screening Assistant 2 program. PDL, progressive drug like; PLL, progressive lead like.
bPreADMET software.
cLAZAR wherein Neg., negative and p, probability value.
The blood brain barrier (BBB) model values for compound 6s was 1.14616 which clearly lay in range suggesting the compound can penetrate the BBB on theoretical grounds. Most compounds displayed CaCo2 cell permeability values above 25 nms [35], topological polar surface area (TPSA) above 7.0 and the human intestinal absorption (HIA) quantities in the 50–100% range, indicating that they may be further developed in an oral dosage form [36]. Lazy structure activity relationships (LAZAR) software [37] predicted all the compounds as non-carcinogenic and non-mutagenic, and the probability greater than 0.025 suggesting the predictions to be reliable. The predicted favourable ADME features for compound 6s further indicates that it is a promising anti-tubercular lead candidate.
5. Conclusion
In summary, a series of novel chromone embedded [1,2,3]-triazole derivatives were synthesized via an easy and convenient synthetic protocol starting from 2-hydroxy acetophenone. The novel 20 analogues 6a–t accomplished in four-step synthetic sequences using click chemistry in the key step were fully characterized by their NMR and mass spectral data. The in vitro anti-mycobacterial evaluation study of all the compounds revealed seven compounds found to be active against M. tuberculosis H37Rv. The compound 6s is the most potent compound in vitro with a MIC value of 1.56 µg ml−1. Cross docking studies revealed compound 6s to be more effective against the enoyl-acyl carrier protein reductase of Mtb. Molecular docking and chemoinformatics studies proved that compound 6s possesses drug-like properties. Docking results indicated that Asp64, Trp222 and Tyr158 amino acids in binding pocket as potential ligand binding hot-spot residues. Further, the molecular variation as well as in vivo studies to prove their specificity towards Mtb are underway.
6. Experimental section
6.1. General methods
Solvents were purified and dried by standard procedures prior to use. 1H NMR and 13C NMR spectra were recorded on a Bruker AC-200, 400 & 500 NMR spectrometer. Spectra were obtained in CDCl3. Monitoring of reactions was carried out using TLC plates Merck silica gel 60 F254 and visualization with UV light (254 and 365 nm), I2 and anisaldehyde in ethanol as development reagents. Mass spectra were recorded at ionization energy 70 eV on API Q Star Pulsar spectrometer using electrospray ionization. UV detector: λ 280 nm; elution: mixtures of acetonitrile and water.
6.1.1. 4-oxo-4H-chromene-3-carbaldehyde (2)
To a stirred solution of dry DMF (40 ml), POCl3 (20.6 ml, 220.34 mmol) was added dropwise at 5°C. The mixture was stirred for 15 min and then the solution of 2-hydroxyacetophenone (10 g, 73.44 mmol) in DMF (20 ml) was added dropwise at 5°C. The reaction mixture was stirred at the same temperature for 30 min, then heated and stirred at 55°C for another 4 h. The mixture was cooled to room temperature, poured into ice-water (approx. 400 ml) and stirred for 1.5 h. The precipitate was filtered off, washed with ethanol afforded 2; White solid, yield = 75%; mp = 152–154°C. IR (CHCl3, cm−1) νmax = 3370, 3023, 2921, 2403, 1659, 1612, 1569, 1464, 1423, 1310, 1217, 1027, 930, 766, 671; 1H NMR (400 MHz, CDCl3) δ = 7.50–7.56 (m, 2H), 7.74–7.79 (m, 1H), 8.31 (dd, J = 7.9, 1.6 Hz, 1H), 8.56 (s, 1H), 10.40 (s, 1H); 13C NMR (100 MHz, CDCl3) δ = 188.6 (CO), 175.9 (CO), 160.6 (CH), 156.2 (C), 134.8 (CH), 126.6 (CH), 126.2 (CH), 125.3 (C), 120.3 (C), 118.6 (CH); HRMS(ESI) m/z = Calcd for C10H6O3 [M + H]+ 175.0390, found 175.0392.
6.1.2. 3-(hydroxymethyl)-4H-chromen-4-one (3)
To a stirred solution of 3-formyl chromone 2 (2 g, 5% of alumina weight) in 100 ml of 2-propanol, about 40 g of basic alumina was added. The resulting solution was stirred at 75°C for 4 h. The reaction mixture was filtered through celite bed and the solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel using 7 : 3 pet ether/ethyl acetate to afford compound 3; Viscous liquid, yield = 82%; IR (CHCl3, cm−1) νmax = 3423, 3019, 2925, 2403, 1643, 1469, 1406, 1347, 1217, 1155, 1023, 971, 918, 852, 763, 670; 1H NMR (200 MHz, CDCl3) δ = 2.12 (bs, 1H), 4.60 (s, 2H), 7.40–7.51 (m, 2H), 7.67–7.75 (m, 1H), 7.96 (s, 1H), 8.24 (dd, J = 7.9, 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 178.4 (CO), 156.6 (C), 152.8 (CH), 133.9 (CH), 125.6 (CH), 125.23 (CH), 123.8 (C), 123.3 (C), 118.2 (CH), 58.5 (CH2); HRMS(ESI) m/z = Calcd for C10H8O3 [M + H]+ 177.0546, found 177.0546.
6.1.3. 3-(azidomethyl)-4H-chromen-4-one (4)
To a stirred solution of 3 (2.5 g, 14.2 mmol) and Et3N (5.14 ml, 36.92 mmol), methanesulfonyl chloride (1.49 ml, 18.46 mmol) in CH2Cl2 (30 ml) was added dropwise at 0°C. The resulting reaction mixture was stirred at 0°C for 1 h. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with water (approx. 20 ml) and extracted with CH2Cl2 (3 × 10 ml). The combined organic layers were washed with water and brine. The organic layer was dried over Na2SO4, filtered and concentrated. The crude mesylated product 3a (2.56 g, 71%) was further used for next step without any purification.
To a solution of crude mesylate 3a (2.5 g, 9.84 mol) in anhydrous DMF (20 ml), sodium azide (1.6 g, 24.6 mmol) was added batchwise at room temperature. The resulting solution was heated to 50°C for 5 h. After completion of reaction (monitored by TLC) reaction mixture was poured into ice cold water (approx. 20 ml) and extracted with ethyl acetate (3 × 10 ml). The combined ethyl acetate layers were washed with brine, dried over Na2SO4, and evaporated in vacuo. The residue was purified by flash chromatography to affored azide 4; White solid, yield = 93%; mp = 50–52°C; IR (CHCl3, cm−1) νmax = 3369, 3018, 2922, 2855, 2107, 1648, 1416, 1407, 1349, 1268, 1217, 1106, 1028, 842, 759, 668; 1H NMR (200 MHz, CDCl3) δ = 4.33 (s, 2H), 7.41–7.51 (m, 2H), 7.67–7.76 (m, 1H), 7.97 (s, 1H), 8.26 (dd, J = 7.9, 1.7 Hz, 1H); 13C NMR (50 MHz, CDCl3)δ = 176.9 (CO), 156.5 (C), 153.8 (CH), 134.0 (CH), 125.9 (CH), 125.5 (CH), 123.7 (C), 119.7 (C), 118.2 (CH), 46.4 (CH2); HRMS(ESI) m/z = Calcd for C10H7O2N3Na [M + Na]+ 224.0430, found 224.0432.
6.1.4. General procedure for synthesis of chromone embedded [1,2,3]-triazole derivatives (6a–t)
To a stirred solution of azide 4 (1 equiv) and aliphatic/aromatic alkynes (5a–t) (1.3 equiv) in t-butanol (3 ml) was added sequentially copper sulfate pentahydrate (20 mol %), sodium ascorbate (20 mol %) and distilled water (3 ml). The resulting reaction mixture was stirred for 1–3 h at 60°C. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with EtOAc (1 × 10 ml) and then washed with water (2 × 5 ml), the organic layer was separated, washed with brine solution (2 × 5 ml), dried over anhydrous sodium sulfate and concentrated in vacuo. The crude residue thus obtained was purified over silica gel column chromatography eluted with pet ether/ethyl acetate (1 : 1) to furnish corresponding chromone embedded [1,2,3]-triazole derivatives (6a–t).
6.1.5. 3-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6a)
Yellow solid; yield = 90%; mp = 154–155°C; IR (CHCl3, cm−1) νmax = 3685, 3357, 3022, 2923, 2402, 1649, 1523, 1469, 1423, 1353, 1216, 1030, 927, 765, 671; 1H NMR (400 MHz, CDCl3) δ = 5.48 (s, 2H), 7.30–7.34 (m, 1H), 7.39–7.51 (m, 4H), 7.70–7.74 (m, 1H), 7.83 (d, J = 7.3 Hz, 2H), 8.15 (s, 1H), 8.22 (s, 1H), 8.24 (dd, J = 8.0, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.8 (CH), 134.4 (CH, 2 carbons), 130.1 (C), 128.8 (CH, 2 carbons), 128.3 (CH), 125.8 (CH, 3 carbons), 123.8 (C), 121.3 (C), 119.1 (C), 118.4 (CH, 2 carbons), 45.5 (CH2); HRMS(ESI) m/z = Calcd for C18H13O2N3 [M + H]+ 304.1081, found 304.1086.
6.1.6. 3-((4-(p-tolyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6b)
White solid; yield = 82%; mp = 170–172°C; IR (CHCl3, cm−1) νmax = 3687, 3189, 3022, 2403, 2356, 1645, 1523, 1469, 1422, 1216, 1037, 927, 770, 672; 1H NMR (200 MHz, CDCl3) δ = 2.37 (s, 3H), 5.47 (s, 2H), 7.20 (s, 1H), 7.24 (s, 1H), 7.42–7.51 (m, 2H), 7.68–7.77 (m, 3H), 8.08 (s, 1H), 8.20 (s, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 148.0 (C), 137.9 (C), 134.3 (C), 129.4 (CH, 2 carbons), 127.6 (C), 125.8 (CH), 125.8 (CH), 125.6 (CH, 2 carbons), 123.8 (C), 120.6 (CH), 119.3 (C), 118.4 (CH), 45.2 (CH2), 21.2 (CH3); HRMS(ESI) m/z = Calcd for C19H15O2N3 [M + H]+ 318.1237, found 318.1240.
6.1.7. 3-((4-(4-ethylphenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6c)
Pale yellow solid; yield = 82%; mp = 49–150°C; IR (CHCl3, cm−1) νmax = 3687, 3394, 3022, 2403, 1648, 1529, 1424, 1217, 1030, 927, 769, 672; 1H NMR (200 MHz, CDCl3) δ = 1.25 (t, J = 7.6 Hz, 3H), 2.67 (q, J = 15.3, 7.6 Hz, 2H), 5.47 (s, 2H), 7.22 (s, 1H), 7.26 (s, 1H), 7.42–7.51 (m, 2H), 7.68–7.76 (m, 3H), 8.09 (s, 1H), 8.20 (s, 1H), 8.20 (s, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 148.1 (C), 144.3 (C), 134.3 (CH), 128.2 (CH, 2 carbons), 127.9 (C), 125.8 (CH), 125.8 (CH), 125.7 (CH, 2 carbons), 123.8 (C), 120.7 (CH), 119.3 (C), 118.4 (CH), 45.2 (CH2), 28.6 (CH2), 15.5 (CH3); HRMS(ESI): m/z = Calcd for C20H17O2N3 [M + H]+ 332.1394, found 332.1401.
6.1.8. 3-((4-(4-propylphenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6d)
White solid; yield = 84%; mp = 135–136°C; IR (CHCl3, cm−1) νmax = 3188, 3019, 2596, 2406, 1631, 1433, 1218, 1041, 768, 671; 1H NMR (200 MHz, CDCl3) δ = 0.94 (t, J = 7.3 Hz, 3H), 1.56–1.75 (m, 4H), 2.60 (t, J = 7.6 Hz, 2H), 5.47 (s, 2H), 7.20 (s, 1H), 7.24 (s, 1H), 7.42–7.51 (m, 2H), 7.67–7.76 (m, 3H), 8.08 (s, 1H), 8.19 (s, 1H), 8.23 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 148.1 (C), 142.7 (C), 134.3 (CH), 128.8 (CH, 2 carbons), 127.9 (C), 125.8 (CH), 125.7 (CH), 125.6 (CH, 2 carbons), 123.8 (C), 120.7 (CH), 119.3 (C), 118.4 (CH), 45.2 (CH2), 37.8 (CH2), 24.4 (CH2), 13.8 (CH3); HRMS(ESI): m/z = Calcd for C21H19O2N3 [M + H]+ 346.1550, found 346.1558.
6.1.9. 3-((4-(4-pentylphenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6e)
White solid; yield = 88%; mp = 147–148°C; IR (CHCl3, cm−1) νmax = 3370, 3022, 2926, 2403, 1648, 1524, 1466, 1421, 1353, 1216, 1029, 927, 763, 670; 1H NMR (200 MHz, CDCl3) δ = 0.89 (t, J = 6.7 Hz, 3H), 1.29–1.36 (m, 4H), 1.63–1.70 (m, 2H), 2.58–2.66 (m, 2H), 5.48 (s, 2H), 7.20 (s, 1H), 7.24 (s, 1H), 7.42–7.51 (m, 2H), 7.68–7.76 (m, 3H), 8.08 (s, 1H), 8.19 (s, 1H), 8.25 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 148.1 (C), 143.0 (C), 134.3 (CH), 128.8 (CH, 2 carbons), 127.8 (C), 125.8 (CH), 125.7 (CH), 125.6 (CH, 2 carbons), 123.8 (C), 120.7 (CH), 119.3 (C), 118.4 (CH), 45.2 (CH2), 35.6 (CH2), 31.4 (CH2), 31.0 (CH2), 22.5 (CH2), 14.0 (CH3); HRMS(ESI): m/z = Calcd for C23H23O2N3 [M + H]+ 374.1863, found 374.1868.
6.1.10. 3-((4-(4-(tert-butyl)phenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6f)
White solid; yield = 96%; mp = 218–219°C; IR (CHCl3, cm−1) νmax = 3390, 3021, 2963, 2404, 1648, 1464, 1218, 1032, 927, 769, 673; 1H NMR (200 MHz, CDCl3) δ = 1.34 (s, 9H), 5.48 (s, 2H), 7.41–7.51 (m, 4H), 7.68–7.78 (m, 3H), 8.10 (s, 1H), 8.20 (s, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.6 (CO), 156.5 (C), 155.6 (CH), 151.1 (C), 147.9 (C), 134.3 (CH), 127.7 (C), 125.8 (CH), 125.7 (CH), 125.6 (CH, 2 carbons), 125.4 (CH, 2 carbons), 123.8 (C), 120.7 (CH), 119.3 (C), 118.3 (CH), 45.2 (CH2), 34.6 (C), 31.2 (CH3, 3 carbons); HRMS(ESI): m/z = Calcd for C22H21O2N3 [M + H]+ 360.1707, found 360.1712.
6.1.11. 3-((4-(4-methoxyphenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6g)
White solid; yield = 60%; mp = 170–171°C; IR (CHCl3, cm−1) νmax = 3687, 3022, 2403, 2356, 1648, 1511, 1467, 1424, 1351, 1217, 1030, 927, 770, 672; 1H NMR (200 MHz, CDCl3) δ = 3.84 (s, 3H), 5.47 (s, 2H), 6.92 (s, 1H), 6.97 (s, 1H), 7.42–7.51 (m, 2H), 7.68–7.77 (m, 3H), 8.04 (s, 1H), 8.20 (s, 1H), 8.22–8.27 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.8 (CO), 159.5 (C), 156.5 (C), 155.7 (CH), 147.9 (C), 134.4 (CH), 127.0 (CH, 3 carbons), 125.8 (CH), 123.8 (C), 123.2 (C), 120.2 (CH), 119.3 (C), 118.3 (CH), 114.1 (CH, 2 carbons), 55.3 (CH3), 45.2 (CH2); HRMS(ESI): m/z = Calcd for C19H15O3N3 [M + H]+ 334.1186, found 334.1192.
6.1.12. 3-((4-(4-(pentyloxy)phenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6h)
White solid; yield = 85%; mp = 154–155°C; IR (CHCl3, cm−1) νmax = 3686, 3189, 3021, 2953, 2403, 1647, 1466, 1418, 1352, 1310, 1217, 1039, 926, 768, 671; 1H NMR (200 MHz, CDCl3) δ = 0.94 (t, J = 6.9 Hz, 3H), 1.38–1.50 (m, 4H), 1.77–1.86 (m, 2H), 3.98 (t, J = 6.6 Hz, 2H), 5.47 (s, 2H), 6.91 (s, 1H), 6.95 (s, 1H), 7.42–7.51 (m, 2H), 7.68–7.76 (m, 3H), 8.03 (s, 1H), 8.19 (s, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 159.1 (C), 156.5 (C), 155.6 (CH), 147.9 (C), 134.3 (CH), 127.0 (CH, 2 carbons), 125.8 (CH), 125.7 (CH), 123.8 (C), 123.0 (C), 120.1 (CH), 119.4 (C), 118.4 (CH), 114.7 (CH, 2 carbons), 68.0 (CH2), 45.2 (CH2), 28.9 (CH2), 28.1 (CH2), 22.5 (CH2), 14.0 (CH3); HRMS(ESI): m/z = Calcd for C23H23O3N3 [M + H]+ 390.1812, found 390.1821.
6.1.13. 3-((4-(m-tolyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6i)
Yellow solid; yield = 85%; mp = 124–125°C; IR (CHCl3, cm−1) νmax = 3685, 3190, 3021, 2403, 1646, 1523, 1468, 1418, 1352, 1217, 1043, 926, 767, 671; 1H NMR (200 MHz, CDCl3) δ = 2.37 (s, 3H), 5.45 (s, 2H), 7.09–7.13 (m, 1H), 7.23–7.31 (m, 1H), 7.39–7.49 (m, 2H), 7.57–7.74 (m, 3H), 8.09 (s, 1H), 8.18–8.24 (m, 2H);.13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.4 (C), 155.6 (CH), 148.0 (C), 138.4 (C), 134.3 (CH), 130.3 (C), 128.8 (CH), 128.6 (CH), 126.3 (CH), 125.7 (CH, 2 carbons), 123.7 (C), 122.8 (CH), 120.9 (CH), 119.2 (C), 118.3 (CH), 45.2 (CH2), 21.3 (CH3); HRMS(ESI): m/z = Calcd for C19H15O2N3 [M + H]+ 318.1237, found 318.1245.
6.1.14. 3-((4-(4-methoxy-2-methylphenyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6j)
Yellow solid; yield = 60%; mp = 172–173°C; IR (CHCl3, cm−1) νmax = 3686, 3392, 3022, 2403, 1648, 1473, 1425, 1217, 1033, 927, 769, 672; 1H NMR (200 MHz, CDCl3) δ = 2.45 (s, 3H), 3.82 (s, 3H), 5.49 (s, 2H), 6.79–6.83 (m, 2H), 7.42–7.52 (m, 3H), 7.66–7.77 (m, 2H), 7.99 (s, 1H), 8.21–8.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 159.3 (C), 156.5 (C), 155.6 (CH), 147.1 (C), 137.1 (C), 134.3 (CH), 130.1 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 122.6 (CH), 119.4 (C, 2 carbons), 118.3 (CH), 116.1 (CH), 111.3 (CH), 55.2 (OCH3), 45.1 (CH2), 21.5 (CH3); HRMS(ESI): m/z = Calcd for C20H17O3N3 [M + H]+ 348.1343, found 348.1353.
6.1.15. 3-((4-(naphthalen-1-yl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6k)
Brick red solid; yield = 93%, mp = 154–155°C; IR (CHCl3, cm−1) νmax = 3687, 3189, 3022, 2403, 2355, 1643, 1523, 1472, 1424, 1216, 1038, 928, 770, 672; 1H NMR (200 MHz, CDCl3) δ = 5.56 (s, 2H), 7.42–7.56 (m, 6H), 7.68–7.77 (m, 2H), 7.86–7.91 (m, 2H), 8.22–8.26 (m, 2H), 8.28 (s, 1H), 8.36–8.41 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.7 (CH), 147.0 (C), 134.3 (CH), 133.8 (C), 130.9 (C), 128.8 (CH), 128.3 (CH), 127.9 (C), 127.2 (CH), 126.6 (CH), 125.9 (CH), 125.8 (CH), 125.7 (CH), 125.4 (CH), 125.3 (CH), 123.9 (CH), 123.8 (C), 119.2 (C), 118.3 (CH), 45.3 (CH2); HRMS(ESI): m/z = Calcd for C22H15O2N3 [M + H]+ 354.1237, found 354.1246.
6.1.16. 3-((4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6l)
Green solid; yield = 60%; mp = 175–176°C; IR (CHCl3, cm−1) νmax = 3686, 3189, 3022, 2403, 2355, 1648, 1523, 1469, 1420, 1352, 1217, 1040, 927, 770, 672; 1H NMR (400 MHz, CDCl3) δ = 5.52 (s, 2H), 7.32 (s, 1H), 7.43–7.50 (m, 2H), 7.69–7.73 (m, 1H), 7.84–7.93 (m, 1H), 8.16 (s, 1H), 8.23 (dd, J = 8.0, 1.6 Hz, 2H), 8.60 (s, 1H), 8.71 (s, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.4 (CO), 156.5 (C), 155.4 (CH), 150.1 (C), 149.2 (C), 148.4 (C), 136.9 (CH), 134.3 (CH), 125.9 (CH), 125.8 (CH), 123.8 (CH), 123.3 (CH), 122.8 (CH), 120.3 (C), 119.1 (CH), 118.2 (CH), 45.4 (CH2); HRMS(ESI): m/z = Calcd for C17H12O2N4 [M + H]+ 305.1033, found 305.1038.
6.1.17. 3-((4-butyl-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6 m)
White solid; yield = 93%; mp = 87–88°C; IR (CHCl3, cm−1) νmax = 3686, 2412, 3022, 2963, 2403, 1648, 1529, 1468, 1424, 1350, 1217, 1032, 927, 769, 672; 1H NMR (200 MHz, CDCl3) δ = 0.92 (t, J = 7.1 Hz, 3H), 1.26–1.45 (m, 2H), 1.57–1.74 (m, 2H), 2.75 (t, J = 7.2 Hz, 2H), 5.44 (s, 2H), 7.42–7.52 (m, 2H), 7.69–7.77 (m, 2H), 8.17–8.26 (m, 2H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.5 (CH), 148.7 (C), 134.3 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 121.9 (CH), 119.5 (C), 118.3 (CH), 44.9 (CH2), 31.5 (CH2), 25.3 (CH2), 22.3 (CH2), 13.8 (CH3); HRMS(ESI): m/z = Calcd for C16H17O2N3 [M + H]+ 284.1394, found 284.1396.
6.1.18. 3-((4-hexyl-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6n)
White solid; yield = 82%; mp = 84–85°C; IR (CHCl3, cm−1) νmax = 3414, 3022, 2404, 1647, 1433, 1218, 1030, 928, 769, 673; 1H NMR (400 MHz, CDCl3) δ = 0.86 (t, J = 6.9 Hz, 3H), 1.26–1.37 (m, 6H), 1.61–1.68 (m, 2H), 2.67–2.71 (m, 2H), 5.40 (s, 2H), 7.43–7.49 (m, 2H), 7.64 (s, 1H), 7.69–7.73 (m, 1H), 8.15 (s, 1H), 8.22 (dd, J = 8.0, 1.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 148.4 (C), 134.3 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 122.2 (CH), 119.3 (C), 118.3 (CH), 45.2 (CH2), 31.5 (CH2), 29.3 (CH2), 28.9 (CH2), 25.5 (CH2), 22.5 (CH2), 14.0 (CH3); HRMS(ESI): m/z = Calcd for C18H21O2N3 [M + H]+ 312.1707, found 312.1711.
6.1.19. 3-((4-cyclopentyl-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6o)
Pale yellow solid; yield = 88%; mp = 157–158°C; IR (CHCl3, cm−1) νmax = 3190, 3010, 1630, 1450, 1220, 1040, 770, 670; 1H NMR (200 MHz, CDCl3) δ = 1.64–1.81 (m, 7H), 2.03–2.13 (m, 2H), 3.09–3.22 (m, 1H), 5.39 (s, 2H), 7.41–7.50 (m, 2H), 7.58 (s, 1H), 7.67–7.76 (m, 1H), 8.14 (s, 1H), 8.23 (dd, J = 7.9, 1.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 152.9 (C), 134.3 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 120.9 (CH), 119.4 (C), 118.3 (CH), 44.9 (CH2), 36.7 (CH), 33.1 (CH2, 2 carbons), 25.1 (CH2, 2 carbons); HRMS(ESI): m/z = Calcd for C17H17O2N3 [M + H]+ 296.1394, found 296.1398.
6.1.20. 3-((4-cyclohexyl-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6p)
Yellow solid; yield = 80%; mp = 145–146°C; IR (CHCl3, cm−1) νmax = 3188, 3020, 1632, 1433, 1218, 1042, 768, 670; 1H NMR (200 MHz, CDCl3) δ = 1.28–1.49 (m, 5 H), 1.70–1.85 (m, 3H), 1.96–2.07 (m, 2H), 2.64–2.80 (m, 1H), 5.38 (s, 2H), 7.41–7.50 (m, 2H), 7.56 (s, 1H), 7.67–7.75 (m, 1H), 8.12 (s, 1H), 8.23 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.6 (CH), 153.9 (C), 134.2 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 120.6 (CH), 119.4 (C), 118.3 (CH), 44.9 (CH2), 35.3 (CH), 32.9 (CH2, 2 carbons), 26.1 (CH2, 2 carbons), 25.9 (CH2); HRMS(ESI): m/z = Calcd for C18H19O2N3 [M + H]+ 310.1550, found 310.1557.
6.1.21. 3-((4-(cyclohexylmethyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6q)
White solid; yield = 88%; mp = 133–134°C; IR (CHCl3, cm−1) νmax = 3686, 3189, 3021, 2928, 2853, 2403, 1647, 1524, 1465, 1417, 1350, 1216, 1043, 926, 768, 671; 1H NMR (200 MHz, CDCl3) δ = 0.89–1.03 (m, 2H), 1.14–1.28 (m, 3H), 1.60–1.72 (m, 6H), 2.56 (d, J = 6.7 Hz, 2H), 5.39 (s, 2H), 7.41–7.51 (m, 2H), 7.59 (s, 1H) 7.67–7.76 (m, 1H), 8.13 (s, 1H), 8.23 (dd, J = 8.0, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.5 (CH), 147.2 (C), 134.3 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 122.5 (CH), 119.5 (C), 118.3 (CH), 44.9 (CH2), 38.0 (CH), 33.4 (CH2), 33.0 (CH2, 2 carbons), 26.4 (CH2), 26.1 (CH2, 2 carbons); HRMS(ESI): m/z = Calcd for C19H21O2N3 [M + H]+ 324.1707, found 324.1710.
6.1.22. 3-((4-(9-hydroxy-9H-fluoren-9-yl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6r)
Pale yellow solid; yield = 58%; mp = 239–240°C; IR (CHCl3, cm−1) νmax = 3687, 3188, 3022, 2403, 1645, 1522, 1467, 1421, 1216, 1043, 926, 769, 671; 1H NMR (200 MHz, CDCl3) δ = 1.74 (bs, 1H), 5.35 (s, 2H), 7.30 (s, 1H), 7.33–7.53 (m, 6H), 7.61–7.74 (m, 5H), 8.13 (s, 1H), 8.14–8.19 (m, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.5 (CO), 156.5 (C), 155.7 (CH), 147.6 (C, 2 carbons), 139.6 (C, 2 carbons), 134.3 (CH), 129.7 (C), 129.5 (CH, 2 carbons), 128.5 (C), 128.3 (CH, 2 carbons), 125.9 (CH), 125.7 (CH), 124.9 (CH), 123.7 (C), 120.3 (CH, 2 carbons), 120.2 (CH, 2 carbons), 119.0 (C), 118.3 (CH), 45.0 (CH2); HRMS(ESI): m/z = Calcd for C25H17O3N3 [M + H]+ 408.1343, found 408.1349.
6.1.23. 3-((4-(15-hydroxypentadecyl)-1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6s)
Pale yellow solid; yield = 84%; mp = 123–124°C; IR (CHCl3, cm−1) νmax = 3688, 3391, 3023, 2930, 2403, 2354, 1648, 1524, 1427, 1216, 1026, 928, 768, 671; 1H NMR (200 MHz, CDCl3) δ = 1.25–1.37 (m, 22H), 1.57–1.72 (m, 4H), 2.68 (t, J = 6.57 Hz, 2H), 3.65 (t, J = 6.6 Hz, 2H), 5.39 (s, 2H), 7.41–7.51 (m, 2H), 7.61 (s, 1H), 7.68–7.76 (m, 1H), 8.13 (s, 1H), 8.23 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.5 (CH), 148.7 (C), 134.3 (CH), 125.8 (CH), 125.7 (CH), 123.8 (C), 121.9 (CH), 119.5 (C), 118.4 (CH), 63.0 (CH2), 44.9 (CH2), 32.8 (CH2), 29.6 (CH2, 7 carbons), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 25.7 (CH2), 25.6 (CH2); HRMS(ESI): m/z = Calcd for C27H39O3N3 [M + H]+ 454.3064, found 454.3074.
6.1.24. 2-((1H-1,2,3-triazol-1-yl)methyl)-4H-chromen-4-one (6t)
Pale yellow solid; yield = 38%; mp = 117–118°C; IR (CHCl3, cm−1) νmax = 3687, 3412, 3022, 2403, 2356, 1649, 1523, 1470, 1421, 1216, 1069, 1025, 927, 770, 672; 1H NMR (200 MHz, CDCl3) δ = 5.47 (s, 2H), 7.42–7.51 (m, 2H), 7.69–7.76 (m, 2H), 7.92 (s, 1H), 8.16 (s, 1H), 8.23 (dd, J = 7.9, 1.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 176.7 (CO), 156.5 (C), 155.5 (CH), 134.3 (CH), 133.9 (CH), 125.8 (CH, 2 carbons), 124.8 (CH), 123.8 (C), 119.3 (C), 118.4 (CH), 45.0 (CH2); HRMS(ESI): m/z = Calcd for C12H9O2N3 [M + H]+ 228.0768, found 228.0771.
Supplementary Material
Acknowledgements
V.N. and A.S. thanks CSIR-New Delhi for the award of senior research fellowships.
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
M.M. and R.V. conceived, designed the study and wrote the manuscript. V.N. and A.S. performed synthetic experiments. P.Y. and D.S. performed anti-tubercular assay. S.B., R.V. and M.K. performed computational analysis.
Competing interests
The authors declare no competing interests.
Funding
Financial support from the CSIR Network projects (BSC0121 and CSC0130) is gratefully acknowledged.
References
- 1.World Health Organization Global Tuberculosis Report. 2016. (http://www.who.int/tb/publications/global_report/en/ )
- 2.Kalia NP, et al. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl Acad. Sci. USA 114, 7426–7431. (doi:10.1073/pnas.1706139114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, Dye C. 2003. The growing burden of tuberculosis. Arch. Intern. Med. 163, 1009–1021. (doi:10.1001/archinte.163.9.1009) [DOI] [PubMed] [Google Scholar]
- 4.Long R. 2000. Drug-resistant tuberculosis. CMAJ 163, 425–428. [PMC free article] [PubMed] [Google Scholar]
- 5.Pozniak A. 2000. HIV-associated tuberculosis in the era of HAART. Int. J. Tuberc. Lung Dis. 4, 993–994. [PubMed] [Google Scholar]
- 6.Dong M, Pfeiffer B, Altmann K-H. 2017. Recent developments in natural product-based drug discovery for tuberculosis. Drug Discov. Today 22, 585–591. (doi:10.1016/j.drudis.2016.11.015) [DOI] [PubMed] [Google Scholar]
- 7.Newman DJ, Cragg GM. 2016. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. (doi:10.1021/acs.jnatprod.5b01055) [DOI] [PubMed] [Google Scholar]
- 8.Reis J, Gaspar A, Milhazes N, Borges F. 2017. Chromone as a privileged scaffold in drug discovery: recent advances. J. Med. Chem. 60, 7941–7957. (doi:10.1021/acs.jmedchem.6b01720) [DOI] [PubMed] [Google Scholar]
- 9.Gaspar A, Matos MJ, Garrido J, Uriarte E, Borges F. 2014. Chromone: a valid scaffold in medicinal chemistry. Chem. Rev. 114, 4960–4992. (doi:10.1021/cr400265z) [DOI] [PubMed] [Google Scholar]
- 10.Keri RS, Budagumpi S, Pai RK, Balakrishna RG. 2014. Chromones as a privileged scaffold in drug discovery: a review. Eur. J. Med. Chem. 78, 340–374. (doi:10.1016/j.ejmech.2014.03.047) [DOI] [PubMed] [Google Scholar]
- 11.Mujahid M, et al. 2015. Spirochromone-chalcone conjugates as antitubercular agents: synthesis, bio evaluation and molecular modeling studies. RSC Adv. 5, 106 448–106 460. (doi:10.1039/C5RA21737G) [Google Scholar]
- 12.Mujahid M, Gonnade RG, Yogeeswari P, Sriram D, Muthukrishnan M. 2013. Synthesis and antitubercular activity of amino alcohol fused spirochromone conjugates. Bioorg. Med. Chem. Lett. 23, 1416–1419. (doi:10.1016/j.bmcl.2012.12.073) [DOI] [PubMed] [Google Scholar]
- 13.Muthukrishnan M, Mujahid M, Yogeeswari P, Sriram D. 2011. Syntheses and biological evaluation of new triazole-spirochromone conjugates as inhibitors of Mycobacterium tuberculosis. Tetrahedron Lett. 52, 2387–2389. (doi:10.1016/j.tetlet.2011.02.099) [Google Scholar]
- 14.Kumar K, Singh P, Kremer L, Guérardel Y, Biot C, Kumar V. 2012. Synthesis and in vitro anti-tubercular evaluation of 1,2,3-triazole tethered β-lactam–ferrocene and β-lactam–ferrocenylchalcone chimeric scaffolds. Dalton Trans. 41, 5778–5781. (doi:10.1039/c2dt30514c) [DOI] [PubMed] [Google Scholar]
- 15.Araújo AC, Nicotra F, Airoldi C, Costa B, Giagnoni G, Fumagalli P, Cipolla L. 2008. Synthesis and biological evaluation of novel rigid 1,4-benzodiazepine-2,5-dione chimeric scaffolds. Eur. J. Org. Chem. 2008, 635–639. (doi:10.1002/ejoc.200700952) [Google Scholar]
- 16.Abrous L, Jokiel PA, Friedrich SR, Hynes J Jr, Smith AB III, Hirschmann R. 2004. Novel chimeric scaffolds to extend the exploration of receptor space: hybrid β-d-glucose−benzoheterodiazepine structures for broad screening. Effect of amide alkylation on the course of cyclization reactions. J. Org. Chem. 69, 280–302. (doi:10.1021/jo0352068) [DOI] [PubMed] [Google Scholar]
- 17.Kolb HC, Finn MG, Sharpless KB. 2001. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 40, 2004–2021. (doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5) [DOI] [PubMed] [Google Scholar]
- 18.Thirumurugan P, Matosiuk D, Jozwiak K. 2013. Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev. 113, 4905–4979. (doi:10.1021/cr200409f) [DOI] [PubMed] [Google Scholar]
- 19.De Carvalho da Silva F, do Carmo Cardoso MF, Ferreira PG, Ferreira VF. 2015. Biological properties of 1H-1,2,3- and 2H-1,2,3-triazoles. In Topics in heterocyclic chemistry, vol. 40. (eds Dehaen W, Bakulev VA), pp. 117 Berlin, Germany: Springer-Verlag. [Google Scholar]
- 20.Xu Z, Song X-F, Hu Y-Q, Qiang M, Lv Z-S. 2017. Azide-alkyne cycloaddition towards 1H-1,2,3-triazole-tethered gatifloxacin and isatin conjugates: design, synthesis and in vitro anti-mycobacterial evaluation. Eur. J. Med. Chem. 138, 66–71. (doi:10.1016/j.ejmech.2017.05.057) [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Xu Z, Gao C, Ren Q-C, Chang L, Lv Z-S, Feng L-S. 2017. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 138, 501–513. (doi:10.1016/j.ejmech.2017.06.051) [DOI] [PubMed] [Google Scholar]
- 22.Zhou B, et al. 2010. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl Acad. Sci. USA 107, 4573–4578. (doi:10.1073/pnas.0909133107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massarotti A, Brunco A, Sorba G, Tron GC. 2014. ZINClick: a database of 16 million novel, patentable, and readily synthesizable 1, 4-disubstituted triazoles. J. Chem. Inf. Model. 54, 396–406. [DOI] [PubMed] [Google Scholar]
- 24.Mdluli K, Spigelman M. 2006. Novel targets for tuberculosis drug discovery. Curr. Opin. Pharmacol. 6, 459–467. (doi:10.1016/j.coph.2006.06.004) [DOI] [PubMed] [Google Scholar]
- 25.Munier-Lehmann H, Chaffotte A, Pochet S, Labesse G. 2001. Thymidylate kinase of Mycobacterium tuberculosis: a chimera sharing properties common to eukaryotic and bacterial enzymes. Protein Sci. 10, 1195–1205. (doi:10.1110/ps.45701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgunova E, Meining W, Illarionov B, Haase I, Jin G, Bacher A, Cushman M, Fischer M, Ladenstein R. 2005. Crystal structure of lumazine synthase from Mycobacterium tuberculosis as a target for rational drug design: binding mode of a new class of purinetrione inhibitors. Biochemistry 44, 2746–2758. (doi:10.1021/bi047848a) [DOI] [PubMed] [Google Scholar]
- 27.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR Jr, Sacchettini JC. 1998. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102. (doi:10.1126/science.279.5347.98) [DOI] [PubMed] [Google Scholar]
- 28.Tizon L, et al. 2011. A prodrug approach for improving antituberculosis activity of potent Mycobacterium tuberculosis type II dehydroquinase inhibitors. J. Med. Chem. 54, 6063–6084. (doi:10.1021/jm2006063) [DOI] [PubMed] [Google Scholar]
- 29.Grundner C, Perrin D, Hooft van Huijsduijnen R, Swinnen D, Gonzalez J, Gee CL, Wells TN, Alber T. 2007. Structural basis for selective inhibition of Mycobacterium tuberculosis protein tyrosine phosphatase PtpB. Structure 15, 499–509. (doi:10.1016/j.str.2007.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y-S, Sacchettini JC. 2016. Structural insights into Mycobacterium tuberculosis Rv2671 protein as a dihydrofolate reductase functional analogue contributing to para-aminosalicylic acid resistance. Biochemistry 55, 1107–1119. (doi:10.1021/acs.biochem.5b00993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rucci N, et al. 2006. Inhibition of protein kinase c-Src reduces the incidence of breast cancer metastases and increases survival in mice: implications for therapy. J. Pharmacol. Exp. Ther. 318, 161–172. (doi:10.1124/jpet.106.102004) [DOI] [PubMed] [Google Scholar]
- 32.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. 2004. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. (doi:10.1021/jm030644s) [DOI] [PubMed] [Google Scholar]
- 33.Le Guilloux V, Arrault A, Colliandre L, Bourg S, Vayer P, Morin-Allory L. 2012. Mining collections of compounds with screening assistant 2. J. Cheminf. 4, 20 (doi:10.1186/1758-2946-4-20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SK, Park SH, Lee IH, No KT. 2007. PreAD-MET Ver. v2.0. Seoul, Korea: BMDRC.
- 35.Tripathi M, Khan SI, Thakur A, Ponnan P, Rawat DS. 2015. 4-Aminoquinoline-pyrimidine-aminoalkanols: synthesis, in vitro antimalarial activity, docking studies and ADME predictions. New J. Chem. 39, 3474–3483. (doi:10.1039/C5NJ00094G) [Google Scholar]
- 36.Ren S, Lien EJ. 2000. Caco-2 cell permeability vs human gastro-intestinal absorption: QSPR analysis. In Progress in drug research (ed. Jucker E.), pp. 1–23, Basel, Switzerland: Birkhauser. [DOI] [PubMed] [Google Scholar]
- 37.Maunz A, Gutlein M, Rautenberg M, Vorgrimmler D, Gebele D, Helma C. 2013. Lazar: a modular predictive toxicology framework. Front. Pharmacol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.