

Abstract
The unicellular protozoan parasite Leishmania causes the neglected tropical disease leishmaniasis, affecting 12 million people in 98 countries. In South America, where the Viannia subgenus predominates, so far only L. (Viannia) braziliensis and L. (V.) panamensis have been sequenced, assembled and annotated as reference genomes. Addressing this deficit in molecular information can inform species typing, epidemiological monitoring and clinical treatment. Here, L. (V.) naiffi and L. (V.) guyanensis genomic DNA was sequenced to assemble these two genomes as draft references from short sequence reads. The methods used were tested using short sequence reads for L. braziliensis M2904 against its published reference as a comparison. This assembly and annotation pipeline identified 70 additional genes not annotated on the original M2904 reference. Phylogenetic and evolutionary comparisons of L. guyanensis and L. naiffi with 10 other Viannia genomes revealed four traits common to all Viannia: aneuploidy, 22 orthologous groups of genes absent in other Leishmania subgenera, elevated TATE transposon copies and a high NADH-dependent fumarate reductase gene copy number. Within the Viannia, there were limited structural changes in genome architecture specific to individual species: a 45 Kb amplification on chromosome 34 was present in all bar L. lainsoni, L. naiffi had a higher copy number of the virulence factor leishmanolysin, and laboratory isolate L. shawi M8408 had a possible minichromosome derived from the 3’ end of chromosome 34. This combination of genome assembly, phylogenetics and comparative analysis across an extended panel of diverse Viannia has uncovered new insights into the origin and evolution of this subgenus and can help improve diagnostics for leishmaniasis surveillance.
Keywords: Leishmania, leishmaniasis, genome, assembly, aneuploidy
1. Introduction
Most cutaneous leishmaniasis (CL) and mucocutaneous leishmaniasis (MCL) cases in the Americas are the result of infection by Leishmania parasites belonging to the Viannia subgenus. The complexity of the molecular, epidemiological and ecological challenges associated with Leishmania in South America remains opaque due to our limited understanding of the biology of Viannia parasites. Nine Viannia (sub)species have been described so far: L. (V.) braziliensis, L. (V.) peruviana, L. (V.) guyanensis, L. (V.) panamensis, L. (V.) shawi, L. (V.) lainsoni, L. (V.) naiffi, L. (V.) lindenbergi and L. (V.) utingensis. CL and MCL are endemic in 18 out 20 countries in the Americas [1] and are mainly associated with L. braziliensis, L. guyanensis and L. panamensis, whose frequency varies geographically. Other species are less frequently associated with human disease, and some are restricted to certain areas [2].
Human CL is partially driven by transmission from sylvatic and peridomestic mammalian reservoirs [3], via sand flies of the genus Lutzomyia (sensu Young and Duncan, 1994) in the Americas, distinct from Phlebotomus sand flies in the Old World [4]. Although CL has spread to domestic and peridomestic niches due to migration, new settlements and deforestation [5–7], there is still a high incidence of some Leishmania in sylvatic environments, such that human infection is accidentally acquired due to sand fly bites when handling livestock [8]. Leishmania naiffi and L. guyanensis are among the Viannia species that show variable responses to treatment, and diversity in the types of clinical manifestations presented, and are adapting to environmental niche and transmission changes driven by humans.
Leishmania naiffi was formally described from a parasite isolated in 1989 from its primary reservoir, the nine-banded armadillo (Dasypus novemcinctus), in Pará state of northern Brazil [9–11]. Leishmania naiffi was initially placed in the Viannia subgenus based on its molecular and immunological characteristics [9]. Many phlebotomine species are likely to participate in the transmission of L. naiffi in Amazonia [12], including Lu. (Psathyromyia) ayrozai and Lu. (Psychodopygus) paraensis in Brazil [13], Lu. (Psathyromyia) squamiventris and Lu. tortura in Ecuador [14], and Lu. trapidoi and Lu. gomezi in Panama [15]. Leishmania naiffi has been isolated from humans and armadillos [9,10] and detected in Thrichomys pachyurus rodents found in the same habitat as D. novemcinctus in Brazil [16]. The nine-banded armadillo is hunted, handled and consumed in the Americas and is regarded as a pest [11,17,18]. People in the same vector range as these armadillos could be exposed to infective sand flies: three L. naiffi CL cases followed contact with armadillos in Suriname [19]. Leishmania naiffi causes localized CL in humans with small discrete lesions on the hands, arms or legs [10,20,21], which has been observed in Brazil, French Guiana, Ecuador, Peru and Suriname [19,22]. CL due to L. naiffi usually responds to treatment [10,22] and can be self-limiting [23], though poor response to antimonial or pentamidine therapy was reported in two patients in Manaus, Brazil [20].
L. guyanensis was first described in 1954 [24] and its primary hosts are the forest dwelling two-toed sloth (Choloepus didactylus) and the lesser anteater Tamandua tetradactyl [25]. Potential secondary reservoirs of L. guyanensis are Didelphis marsupialis (the common opossum) [26,27], rodents from the genus Proechimys [25], Marmosops incanus (the grey slender opposum) [28] in Brazil and D. novemcinctus [29]. Lu. umbratilis, Lu. anduzei and Lu. whitmani are prevalent in forests [30] and act as vectors of L. guyanensis [31–33]. Leishmania guyanensis has been found in French Guiana, Bolivia, Brazil, Colombia, Guyana, Venezuela, Ecuador, Peru, Argentina and Suriname [34–39].
More precise genetic screening of Viannia isolates is necessary to trace hybridization between species. Infection of humans, dogs and Lu. ovallesi with L. guyanensis/L. braziliensis hybrids was reported in Venezuela [40,41]. A L. shawi/L. guyanensis hybrid causing CL was detected in Amazonian Brazil [42], and L. naiffi has produced viable progeny with L. lainsoni [43] and L. braziliensis (Elisa Cupolillo 2018, unpublished data). There is extensive evidence of interbreeding among L. braziliensis complex isolates, including more virulent L. braziliensis/L. peruviana hybrids with higher survival rates within hosts in vitro [44].
Leishmania genomes are characterized by several key features. Genes are organized as polycistronic transcription units that have a high degree of synteny across Leishmania species [45]. These polycistronic transcription units are co-transcribed by RNA polymerase II as polycistronic pre-mRNAs that are 5′-transpliced and 3′-polyadenylated [46,47]. This means translation and stability of these mature mRNAs determine gene expression rather than transcription rates. In addition, Leishmania display extensive aneuploidy, frequently possess extrachromosomal amplifications driven by homologous recombination at repetitive sequences, and have variable gene copy numbers [48]. The Leishmania subgenus genomes of L. infantum, L. donovani and L. major have 36 chromosomes [49], whereas Viannia genomes have 35 chromosomes due to a fusion of chromosomes 20 and 34 [45,50]. In contrast to the species of the Leishmania subgenus, Viannia parasites possess genes encoding functioning RNA interference (RNAi) machinery that may mediate infective viruses and transposable elements [51].
Fully annotated genomes have been described in detail for only two Viannia species: L. panamensis [51] and L. braziliensis [45,48], limiting our comprehension of their evolutionary origin, genetic diversity and functional adaptations. Consequently, we present reference genomes for L. guyanensis LgCL085 and L. naiffi LnCL223 to address these critical gaps. These new annotated reference genomes were compared with other Viannia species genomes to examine structural variation, sequence divergence, gene synteny and chromosome copy number changes. We contrasted the genomic configuration of L. guyanensis LgCL085 and L. naiffi LnCL223 with the L. braziliensis MHOM/BR/1975/M2903 assembly, two unannotated L. peruviana chromosome-level scaffold assemblies [52], the L. panamensis MHOM/PA/1994/PSC-1 reference and the L. braziliensis MHOM/BR/1975/M2904 reference. Furthermore, we assessed aneuploidy in five unassembled Viannia datasets originally isolated from humans, armadillos and primates, which are commonly used in studies on Viannia parasites [53–56]: L. shawi reference isolate MCEB/BR/1984/M8408 also known as IOC_L1545, L. guyanensis MHOM/BR/1975/M4147 (iz34), L. naiffi MDAS/BR/1979/M5533 (IOC_L1365), L. lainsoni MHOM/BR/1981/M6426 (IOC_L1023), L. panamensis MHOM/PA/1974/WR120 [53] (IOC stands for Instituto Oswaldo Cruz).
2. Results
2.1. Genome assembly from short reads
The genomes of L. (Viannia) guyanensis LgCL085 and L. (V.) naiffi LnCL223 were assembled from short reads, along with an assembly of L. braziliensis M2904 generated in the same way as a positive control [48] (table 1). This facilitated comparison with the published M2904 genome, which was assembled by capillary sequencing of a plasmid clone library together with extensive finishing work and with fosmid end sequencing [45], so that the ability of short reads to correctly and comprehensively resolve Leishmania genome architecture could be quantified.
Table 1.
Data used in this study. The World Health Organization (WHO) numbers are structured such that M is mammal, R is reptile, HOM is Homo, CAN is canine, DAS is Dasypus (an armadillo), CEB is Cebus (a primate), ARV is Arvicanthis (a rodent), TAR is Tarentolae and LAT is Latastia (a long-tailed lizard). The top two rows indicate the isolates for L. guyanensis and L. naiffi genomes published here.
| species | source | data type | name or WHO number | SRAa | number and length of reads | reference |
|---|---|---|---|---|---|---|
| L. guyanensis | SRA | reads | LgCL085 | ERX180458 | 15 272 969 (100 bp paired-end) | this study |
| L. naiffi | SRA | reads | LnCL223 | ERX180449 | 8 131 246 (100 bp paired-end) | this study |
| L. braziliensis | Sanger FTP site | genome and reads | MHOM/BR/1975/M2904 | ERX005631 (LbrM2904 v3) | 26 007 384 (76 bp paired-end) | Rogers et al. [48] |
| L. guyanensis | SRA | reads | MHOM/BR/1975/M4147 | SRX767379 | 6 225 035 (100 bp paired-end) | Harkins et al. [53] |
| L. lainsoni | SRA | reads | MHOM/BR/1981/M6426 | SRX764333 | 4 630 952 (100 bp paired-end) | Harkins et al. [53] |
| L. naiffi | SRA | reads | MDAS/BR/1979/M5533 | SRX764332 | 9 646 461 (100 bp paired-end) | Harkins et al. [53] |
| L. panamensis | Genbank and SRA | genome and reads | MHOM/PA/1994/PSC-1 | SRX681913; (CP009370: CP009404) | 5 875 837 (100 bp paired-end) | Llanes et al. [51] |
| L. panamensis | SRA | reads | MHOM/PA/1974/WR120 | SRX767384 | 4 536 341 (100 bp paired-end) | Harkins et al. [53] |
| L. shawi | SRA | reads | MCEB/BR/1984/M8408 | SRX764331 | 5 110 479 (100 bp paired-end) | Harkins et al. [53] |
| L. peruviana | Genbank and SRA | genome and reads | PAB-4377 | ERX556165 (Bioproject ID: PRJEB7263) | 16 117 316 (100 bp paired end) | Valdivia et al. [52] |
| L. peruviana | Genbank and SRA | genome and reads | LEM1537 (MHOM/PE/1984/LC39) | ERX556164 (Bioproject ID: PRJEB7263) | 9 378 317 (100 bp paired end) | Valdivia et al. [52] |
aSRA stands for SRA or TriTrypDB accession ID.
Firstly, the L. guyanensis LgCL085, L. naiffi LnCL223 and the L. braziliensis M2904 control reads were filtered to remove putative contaminant sequences identified by aberrant GC content, trimmed at the 3′ ends to remove low-quality bases, and polymerase chain reaction (PCR) primer sequences were removed (see Methods for details) resulting in 26 067 692 properly paired reads for L. guyanensis, 13 979 628 for L. naiffi, 34 592 618 for the L. (V.) braziliensis control (electronic supplementary material, table S1). These filtered reads for L. guyanensis, L. naiffi and L. braziliensis were de novo assembled into contigs using Velvet [57] with k-mers of 61 for L. guyanensis, 43 for L. naiffi and 43 for the L. braziliensis control optimized for eachlibrary.
The initial contigs were scaffolded using read pair information with SSPACE [58] to yield 2800 L. guyanensis scaffolds with an N50 of 95.4 Kb, 6530 L. naiffi scaffolds with an N50 of 24.3 Kb, and 3782 L. braziliensis scaffolds with an N50 of 20.6 Kb (table 2). The corrected scaffolds for L. guyanensis, L. naiffi and the L. braziliensis control were contiguated (aligned, ordered and oriented) using the extensively finished L. braziliensis M2904 reference with ABACAS [59]. The output was split into 35 pseudo-chromosomes and REAPR [60] broke scaffolds at possible misassemblies to assess contiguation accuracy. The pseudo-chromosome lengths of each sample approximated the length of each corresponding L. braziliensis M2904 reference chromosome with the exceptions of shorter L. guyanensis chromosomes 2, 4, 12 and 21, and a longer L. naiffi chromosome 1 (electronic supplementary material, figure S1). Post-assembly alignment of all bin contigs using BLASTn identified 44 L. guyanensis sequences spanning 4 566 791 bp as putative contaminants that were removed: half had high similarity to bacterium Niastella koreensis (electronic supplementary material, table S2).
Table 2.
Summary of L. braziliensis reference M2904, L. braziliensis control, L. guyanensis LgCL085 and L. naiffi LnCL223 genome assembly contigs, scaffolds, gaps, read coverage, assembled chromosomal and contig sequence and levels of gene annotation.
|
L. braziliensis |
||||
|---|---|---|---|---|
| M2904 | control | L. guyanensis LgCL085 | L. naiffi LnCL223 | |
| initial number of contigs | 13 601 | 10 308 | 14 682 | |
| initial contig N50 (Kb) | 5.1 | 9.6 | 5.7 | |
| number of scaffolds | 3782 | 2800 | 6530 | |
| scaffold N50 (Kb) | 20.6 | 95.4 | 24.3 | |
| number of gaps | 919 | 3352 | 1557 | 3853 |
| median read coverage | 75 | 74 | 56 | 36 |
| N content (%) | 0.29 | 0.99 | 0.45 | 1.07 |
| chromosomes total length (bp) | 31 238 104 | 28 985 156 | 28 274 008 | 29 179 723 |
| bin sequence total length (bp) | 850 747 | 1 024 497 | 2 740 314 | 1 161 372 |
| total genome length (bp) | 32 088 851 | 30 009 653 | 31 014 322 | 30 341 095 |
| protein-coding genes | 8357 | 8001 | 8230 | 8104 |
| genes on chromosomes | 8432 | 7873 | 7757 | 7952 |
| genes on bin contigs | 188 | 288 | 619 | 310 |
| total number of genes | 8620 | 8161 | 8376 | 8262 |
When the reads for each were mapped to its own assembled genome, the median read coverage was 56 for L. guyanensis, 36 for L. naiffi and 75 for the L. braziliensis control. The latter was on par with the 74-fold median coverage observed when M2904 short reads were mapped to the L. braziliensis reference [45,48] (electronic supplementary material, table S3). The differing coverage levels correlated with the numbers of gaps in the final genome assembly of L. guyanensis (1557, table 2) and L. naiffi (3853).
2.2. Multi-locus sequencing analysis of L. guyanensis LgCL085 and L. naiffi LnCL223 with the Viannia subgenus
As a first step in investigating the genetic origins of these isolates, we examined their species identity using MLSA (multi-locus sequencing analysis). Four housekeeping gene sequences published for 95 Viannia isolates including L. braziliensis, L. lainsoni, L. lindenbergi, L. utingensis, L. guyanensis, L. shawi and L. naiffi [56] were compared with orthologues of each gene extracted from assemblies of L. naiffi LnCL223, L. guyanensis LgCL085, the L. braziliensis reference, L. panamensis PSC-1 and L. peruviana PAB-4377. Among the 95 were four samples with reads available [53]: L. shawi MCEB/BR/1984/M8408 (IOC_L1545), L. guyanensis MHOM/BR/1975/M4147 (iz34), L. naiffi MDAS/BR/1979/M5533 (IOC_L1365) and L. lainsoni MHOM/BR/1981/M6426 (IOC_L1023). The genes were aligned using Clustal Omega v1.1 [61] to create a network for the 102 isolates with SplitsTree v4.13.1 [62]. This replicated the expected highly reticulated structure [56], where L. braziliensis M2904 and L. peruviana PAB-4377 were in the L. braziliensis cluster (figure 1).
Figure 1.

Middle: a neighbour-Net network of the uncorrected p-distances from concatenated 2902-base sequences from four housekeeping genes for 102 Viannia samples. The genes were glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (6PGD), mannose phosphate isomerase (MPI) and isocitrate dehydrogenase (ICD). Leishmania naiffi LnCL223 (cyan) is ‘New_L_naiffi_Reference’ and is related to M5533 (IOC_L1365). Leishmania guyanensis LgCL085 (blue) is ‘New_L_guyanensis_Reference’ and is related to the L. shawi M8408 (IOC_L1545) assembly and the L. panamensis PSC-1 genome, but less so to L. guyanensis M4147 (iz34). The L. braziliensis M2904 reference and control are ‘M2904_Reference’ and ‘M2904_Control’, proximal to L. peruviana PAB-4377. L. lainsoni M6426 (IOC_L1023) (green), L. utingensis (orange) and L. lindenbergi (pink) are shown. The isolate names and detail for each species complex are shown by insets in red (L. braziliensis), dark blue (L. guyanensis) and light blue (L. naiffi). For detailed viewing, the nexus file can be downloaded at https://figshare.com/s/eecf1c6b42ac4deb6acc and high-resolution PDF at https://doi.org/10.6084/m9.figshare.5687329.
Previous work suggests that the L. guyanensis species complex includes L. panamensis and L. shawi because they show little genetic differentiation from one another [56,63–65]. The MLSA here showed that the new L. guyanensis LgCL085 reference clustered phylogenetically in the L. guyanensis species complex, had no sequence differences compared with L. panamensis PSC-1, and seven differences versus L. shawi M8408 across the 2902 sites aligned (figure 1). Leishmania guyanensis LgCL085 grouped with isolates classified as zymodeme Z26 by multi-locus enzyme electrophoresis (MLEE) associated with L. shawi [54]. This was supported by the number and the alleles of genome-wide single-nucleotide polymorphisms (SNPs) called using reads mapped to the L. braziliensis M2904 reference for L. guyanensis (355 267 SNPs), L. guyanensis M4147 (326 491), L. panamensis WR120 (294 459) and L. shawi M8408 (296 095) (electronic supplementary material, table S4).
The L. naiffi LnCL223 was closest to L. naiffi ISQU/BR/1994/IM3936, with two differences. It clustered with MLEE zymodeme Z49 based on the correspondence between the MLSA network and previously typed zymodemes, though L. naiffi is associated with more zymodemes than other Viannia. The number and the alleles of genome-wide SNPs called using reads mapped to the L. braziliensis reference were similar for L. naiffi (548 256) and M5533 (633 560) (electronic supplementary material, table S4) and consistent with the MLSA genetic distances.
There was no evidence of recent gene flow between these three species at any genome-wide 10 Kb segment and L. naiffi LnCL223 had fewer SNPs compared with L. braziliensis M2904 than L. guyanensis LgCL085 (electronic supplementary material, figure S2). Linking the MLSA network topology with previous work [56,63–65], four genetically distinct species complexes are represented by the genome-sequenced Viannia at present: (i) braziliensis including L. peruviana, (ii) guyanensis including L. panamensis and L. shawi, (iii) naiffi and (iv) lainsoni (electronic supplementary material, table S4), and the less explored (v) lindenbergi and (vi) utingensis complexes (figure 1).
2.3. Ancestral diploidy and constitutive aneuploidy in Viannia
The normalized chromosomal coverage of the L. guyanensis LgCL085 and L. naiffi LnCL223 reads mapped to L. braziliensis M2904 showed aneuploidy on a background of a diploid nuclear genome (figure 2). The coverage levels of reads for L. peruviana LEM1537, L. peruviana PAB-4377, L. panamensis PSC-1 and the triploid L. braziliensis control mapped to the M2904 reference, confirmed previous work (electronic supplementary material, figure S3), including the L. braziliensis control (electronic supplementary material, figure S4), and demonstrated that assemblies from short read data were sufficient to estimate chromosome copy number differences. Repeating this for L. shawi M8408, L. naiffi M5533, L. guyanensis M4147, L. panamensis WR120 and L. lainsoni M6426 showed that all these Viannia were predominantly disomic and thus diploidy was the likely ancestral state of this subgenus (figure 2).
Figure 2.

Normalized chromosome copy numbers of L. naiffi LnCL223 reads mapped to its assembly, L. guyanensis LgCL085 reads mapped to its assembly, and L. guyanensis M4147, L. lainsoni M6426, L. naiffi M5533, L. panamensis WR120 and L. shawi M8408 reads mapped to L. braziliensis M2904. Dashed lines indicate disomic, trisomic and tetrasomic states. Results for L. panamensis PSC-1 and L. peruviana PAB-4377 were previously published and are in electronic supplementary material, figure S3.
The somy patterns were supported by the results of mapping the reads of each sample to their own assembled genome or to the M2904 reference to produce the read depth allele frequency (RDAF) distributions from heterozygous SNPs. The majority of L. braziliensis M2904 control chromosomes had peaks with modes at approximately 33% and approximately 67% indicating trisomy, rather than a single peak at approximately 50% consistent with disomy (electronic supplementary material, figure S5). The RDAF distributions from reads mapped to its own assembly for L. guyanensis LgCL085 and L. naiffi LnCL223 had a mode of approximately 50% (electronic supplementary material, figure S6), including peaks indicating trisomy for LgCL085 chromosomes 13, 26 and 35 (electronic supplementary material, figure S7).
2.4. 8262 L. naiffi and 8376 L. guyanensis genes annotated
A total of 8262 genes were annotated on L. naiffi LnCL223: of these 8104 were protein-coding genes, 78 were tRNAs, 15 rRNA genes, four snoRNA genes, two snRNA genes and 59 pseudogenes. In total, 310 genes were on unassigned contigs (electronic supplementary material, table S3) and 8376 genes were annotated on L. guyanensis LgCL085: of these, 8230 were protein-coding genes, 75 tRNAs, 14 rRNA genes, four snoRNA genes, two snRNA genes and 51 pseudogenes. Six hundred and nineteen genes were on unassigned contigs.
There were 8161 genes (8001 protein coding) transferred to the control L. braziliensis genome, along with 76 tRNAs, two snRNA genes, four snoRNA genes, 13 rRNA genes and 65 pseudogenes (table 2). There were 7719 of the protein-coding genes (96.5%) clustered into 7244 orthologous groups (OGs), whereas 8137 of the 8375 (97.2%) protein-coding genes on the L. braziliensis reference grouped into 7383 OGs. This indicated that 97% of protein-coding genes in OGs were recovered, and only 2.8% (235) across 201 OGs were absent in the M2904 control, mainly hypothetical or encoded ribosomal proteins (electronic supplementary material, table S5). In the same way, we found 70 protein-coding genes (electronic supplementary material, table S6) in 62 OGs on the M2904 control absent in the published L. braziliensis annotation.
Few genes were present in L. braziliensis but absent in L. guyanensis LgCL085 and L. naiffi LnCL223. Coverage depth was used to predict each gene's haploid copy number, such that genes with haploid copy numbers at least twice the assembled copy number indicated partially assembled genes in the reference assembly. Thus, we investigated all OGs with haploid copy numbers at least twice the assembled copy number to quantity completeness of the assembly. Only 145 genes in 92 OGs on L. guyanensis LgCL085 (electronic supplementary material, table S7), 142 genes in 90 OGs on L. naiffi LnCL223 (electronic supplementary material, table S8) and 102 genes in 71 OGs (electronic supplementary material, tableS9) on the L. braziliensis control met this criterion, indicating few unassembled genes in each assembly. One hypothetical gene (LnCL223_272760) in L. naiffi LnCL223 with no retrievable information had a haploid copy number of 15 (OG5_173495), whereas all other genomes examined here had zero to two copies.
2.5. A 245 Kb rearrangement akin to a minichromosome in L. shawi M8408
We discovered a putative minichromosome or amplification at the 3′ end of L. shawi M8408 chromosome 34 based on elevated coverage across a pair of inverted repeats spanning 245 Kb (figure 3). This locus spanned at least bases 1 840 001 to 1 936 232 (the end) of L. braziliensis M2904 chromosome 34 (electronic supplementary material, figure S8 and table S10). It was orthologous to a known 100 Kb amplification on L. panamensis PSC-1 chromosome 34 that was predicted to produce a minichromosome when amplified, and contained the frequently amplified LD1 (Leishmania DNA 1) region [66]. In contrast to the L. panamensis PSC-1 minichromosome, the L. shawi M8408 amplification was approximately 30 Kb longer and closer in length to the L. braziliensis M2903 245 Kb minichromosome [67].
Figure 3.

Read depth coverage (blue, y-axis) in 10 Kb blocks for reads mapped to L. braziliensis M2904 chromosome 34 (x-axis) for nine Viannia isolates. The black horizontal line is the median chromosome 34 coverage. L. panamensis PSC-1 (a) and L. shawi M8408 (d) showed a 3′ jump in coverage (green) consistent with an amplification of inverted repeats that could form a linear minichromosome. In addition, this pair shared a 45 Kb amplification (pink) also found in the L. braziliensis M2904 control (g), L. naiffi M5533 (b), L. panamensis WR120 (e), L. peruviana LEM1537 (h), L. peruviana PAB-4377 (c) and L. guyanensis M4147 (f). This was absent in L. lainsoni M6426 (i).
2.6. A 45 Kb locus was amplified in most Viannia genomes
A 45 Kb amplification on chromosome 34 spanning a gene encoding a structural maintenance of chromosome family protein and ten hypothetical genes had between two and four copies in all samples except L. lainsoni M6426 (figure 3; electronic supplementary material, table S10). Using the L. guyanensis gene annotation, putative functions were assigned to five of the ten hypothetical genes. This duplication spanned chromosomal location 1.32–1.35 Mb in the L. braziliensis M2904 reference and had two additional hypothetical genes in L. naiffi LnCL223 (LnCL223_343280 and LnCL223_343290; figure 4).
Figure 4.
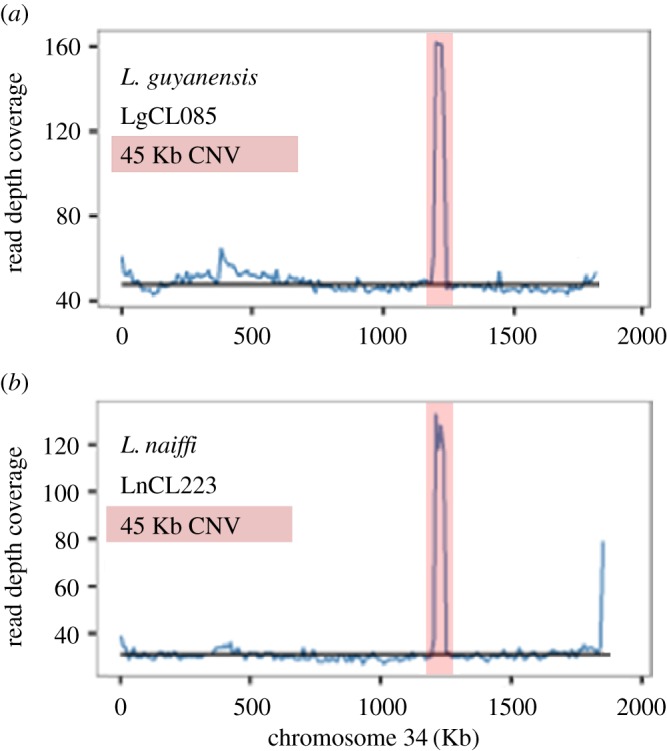
Median coverage (blue) in 10 Kb blocks for L. guyanensis LgCL085 reads mapped to its own assembled chromosome 34 (a) and L. naiffi LnCL223 reads mapped to its own assembled chromosome 34 (b). The black horizontal line is the median chromosome 34 read coverage. There was a 45 Kb amplification to three copies (pink) in L. guyanensis LgCL085 (at chromosome 34 bases 1 195 232–1 239 355, 44 123 bases in length). Similarly, there was a 45 Kb fourfold amplification (pink) in L. naiffi LnCL223 (at chromosome 34 bases 1 206 328–1 251 119, 44 791 bases in length). The latter encompassed two additional hypothetical genes relative to L. guyanensis LgCL085. Neither had evidence of a 3′ minichromosome.
2.7. Genes exclusive to Viannia genomes
A total of 7961 (96.7%) of the 8230 genes annotated for L. guyanensis LgCL085 were assigned to 7381 OGs, 7893 (97.4%) of the 8104 L. naiffi LnCL223 genes to 7324 OGs, and 7692 (99.3%) of the L. panamensis PSC-1 7748 to 7245 OGs. A total of 6835 of these OGs were shared with nine species from the Leishmania, Sauroleishmania and Viannia subgenera: L. (L.) major, L. (L.) mexicana, L. (L.) donovani (infantum), L. (V.) guyanensis, L. (V.) naiffi, L. (V.) braziliensis, L. (V.) panamensis, L. (S.) adleri, L. (S.) tarentolae (electronic supplementary material, table S11).
We identified 22 OGs exclusive to Viannia (electronic supplementary material, table S12): three OGs contained the RNAi pathway genes (DCL1, DCL2, RIF4). Another OG was the telomere-associated mobile elements (TATE) DNA transposons (OG5_132061), a dynamic feature of Viannia genomes [51] (electronic supplementary material, Results). Four OGs encoded a diacylglycerol kinase-like protein (OG5_133291), a nucleoside transporter (OG5_134097), a beta-tubulin/amastin (OG5_183241) and a zinc transporter (OG5_214682). The remaining 14 OGs contained hypothetical genes.
An NADH-dependent fumarate reductase gene (OG5_128620) was amplified in the Viannia examined here: L. guyanensis LgCL085 had 14 copies, L. naiffi LnCL223 had 16, L. panamensis PSC-1 had 16, L. peruviana PAB4377 had 23, L. peruviana LEM1537 had 14 and braziliensis M2904 had 12. This contrasted with the Leishmania and Sauroleishmania subgenera for which three to four copies had been reported for L. infantum, L. mexicana, L. major, L. adleri and L. tarentolae [68,69]. This gene has been implicated in enabling parasites to resist oxidative stress and potentially aiding persistence, drug resistance and metastasis [70,71].
2.8. Few species-specific genes in L. guyanensis LgCL085 and L. naiffi LnCL223
Four genes from four OGs unique to L. naiffi LnCL223 were identified compared with other Leishmania (electronic supplementary material, table S13). Of these four, hypothetical genes LnCL223_312570 and LnCL223_292920 had orthologues in T. brucei and T. vivax, respectively. The LnCL223_341350 protein product had 44–45% sequence identity with a Leptomonas transferase family protein, and LnCL223_352070 was a methylenetetrahydrofolate reductase (OG5_128744), but had no orthologues in the other eight Leishmania or five Trypanosoma species investigated here. Leishmania guyanensis LgCL085 had 31 unique genes in 30 OGs, 25 of which were on unplaced contigs. Four of the six chromosomal genes were also in Trypanosoma genomes, encoding two hypothetical proteins (a tuzin and a poly ADP-ribose glycohydrolase). Twenty eight of the 31 had orthologues in eukaryotes, of which three had orthologues in the free-living freshwater ciliate protozoan Tetrahymena thermophile (electronic supplementary material, table S14) [72].
2.9. Leishmania guyanensis LgCL085 and L. naiffi LnCL223 had over 300 gene arrays
Gene arrays are genes in the same OG with more than two haploid gene copies: they can be cis or trans. There were 327 gene arrays on L. naiffi LnCL223 (electronic supplementary material, table S15), 334 on L. guyanensis LgCL085 (electronic supplementary material, table S16) and 255 on the control L. braziliensis M2904 (electronic supplementary material, table S17)—half the arrays on each genome had two copies of each gene. Twenty-two of the L. guyanensis, 18 of the L. naiffi LnCL223 and 15 of the control L. braziliensis gene arrays contained 10+ haploid gene copies (table 3). The L. panamensis PSC-1 genome had approximately 400 tandem arrays, of which 71% had more than two copies. The L. braziliensis M2904 genome had 615 arrays corresponding to 763 OGs in OrthoMCL v5. Thus, the control genome underestimated the number of gene arrays due to either gene absence or incomplete assembly, indicating that the number of arrays on L. naiffi LnCL223 and L. guyanensis LgCL085 was underestimated.
Table 3.
Arrays with 10 or more gene copies predicted by read depth for each species. OG stands for orthologous group. Genes in OG show the number of genes associated with that OG functional category. OG haploid copy number indicates the numbers of haploid gene copies found in each genome: B stands for the L. braziliensis M2904 control, G for L. guyanensis LgCL085 and N for L. naiffi LnCL223. The grey shading highlights the genes with elevated OG haploid copy numbers.
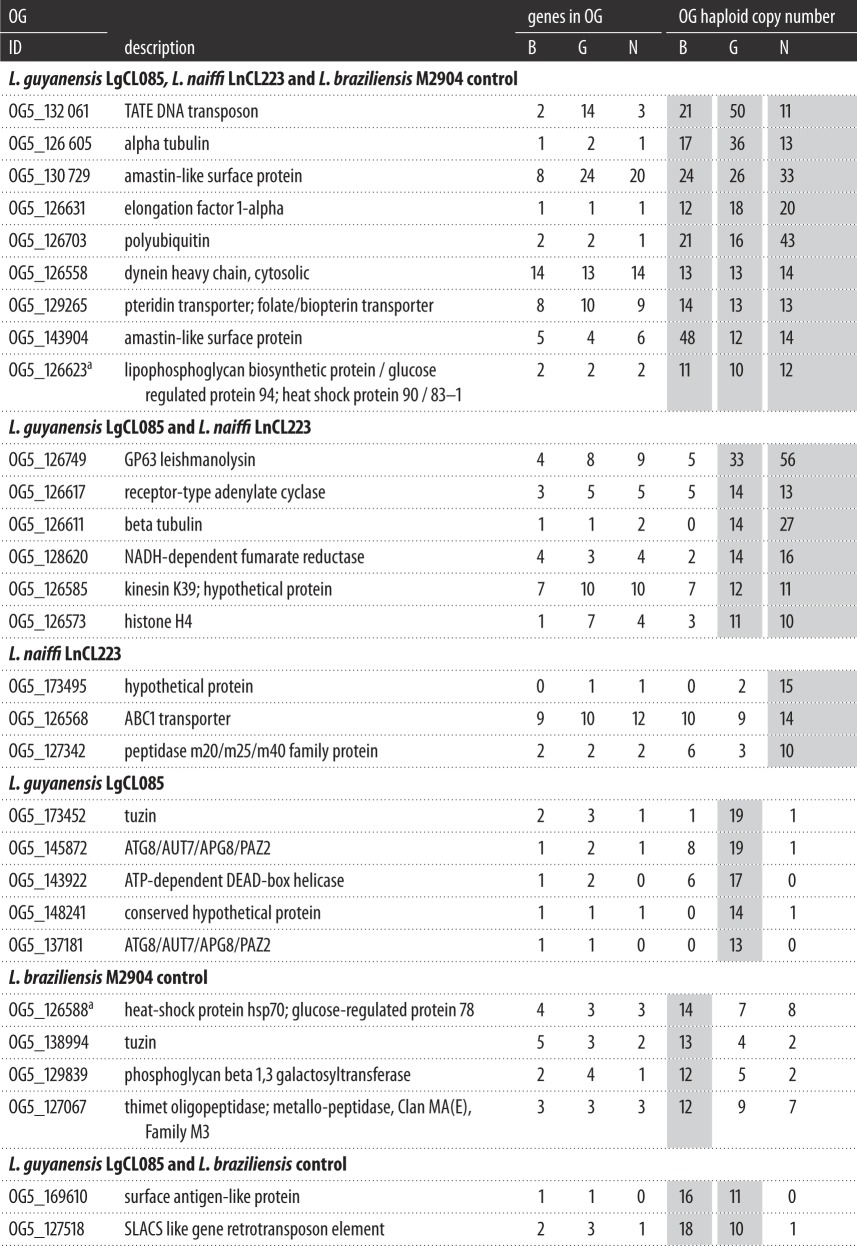 |
aFor OG5_126623 and OG5_126588, the elevated copy numbers were due to amplificated heat shock protein (hsp) genes rather than the glucose-regulated protein (grp) loci, a potential limitation of OG analyses.
The most expanded array on L. guyanensis LgCL085 contained TATE DNA transposons (OG5_132061) with 50 haploid gene copies (table 3) compared with 11 on L. naiffi LnCL223, 21 on the L. braziliensis control and 16 on L. panamensis PSC-1. The L. braziliensis M2904 assembly had 40 TATE DNA transposons, but only two were annotated on the control here, illustrating that more accurate estimates of copy number may be possible.
Leishmania naiffi LnCL223 had the highest haploid gene copy number of the M8 family metalloprotease leishmanolysin (GP63) array (OG5_126749) with 56 haploid gene copies, compared with 33 in L. guyanensis LgCL085, 28 in L. panamensis PSC-1 and 31 in L. braziliensis M2904. This was the sole protease-related OG amplified in all three species (electronic supplementary material, table S23). This family was not expanded in L. peruviana LEM1537 or PAB4377. This was consistent with previous work on L. guyanensis leishmanolysin [73] indicating it is a highly expressed virulence factor in promastigotes [74] affecting the survival during the initial stages of infection [74–77]. Sauroleishmania genomes also had high array copy numbers: 37 for L. adleri [69] and 84 for L. tarentolae (electronic supplementary material, table S12). Leishmania subgenus genomes had lower copy numbers, with 13 for L. mexicana, 15 for L. infantum and five for L. major (OG4_10176 for L. braziliensis M2904, L. mexicana, L. infantum and L. major).
A tuzin gene array (OG5_173452) had higher haploid copy numbers on L. guyanensis LgCL085 (19) and L. panamensis PSC-1 (22) compared with the two copies in L. naiffi, L. mexicana, L. infantum, L. major, L. braziliensis, L. adleri and L. tarentolae. Tuzins are conserved transmembrane proteins in Trypanosoma and Leishmania associated with surface glycoprotein expression [78]. They are often contiguous with δ-amastin genes, whose products are abundant cell surface transmembrane glycoproteins potentially involved in the infection or survival within macrophages. They are absent in Crithidia and Leptomonas species, who lack a vertebrate host stage [78]. Tuzins may play a role in pathogenesis [79], which may be related to leishmaniasis caused by L. guyanensis.
3. Discussion
3.1. Leishmania (Viannia) guyanensis and L. (V.) naiffi draft reference genomes
We assembled high-quality reference genomes for two isolates, L. (Viannia) guyanensis LgCL085 and L. (V.) naiffi LnCL223, from short read sequence libraries to illuminate genomic diversity in the Viannia subgenus and extend previous work [52]. This process combined the de novo assembly with a reference-guided approach using the published genome of L. braziliensis M2904 to assemble the L. guyanensis LgCL085 and L. naiffi LnCL223 into 35 chromosomes each (table 2). An essential feature of this process was to identify and remove contamination in the L. guyanensis and L. braziliensis M2904 libraries and to trim low-quality bases in L. naiffi LnCL223 to ensure that the reads used were informative and free of exogenous impurities. A second screen for contamination in unassigned contigs also removed several L. guyanensis LgCL085 contigs, which improved subsequent annotation and gene copy number estimates.
3.2. Genomes assembled from short reads capture aneuploidy and nearly all genes
Our strategy was tested by applying the same protocol to the L. braziliensis M2904 short read library, which acted as a positive control and quantified the precision of the final output. This facilitated the detection of structural variation or annotation problems, chiefly underestimated copy numbers at certain genes and the incorrect assembly of some loci that were fixed manually. The resulting genomes were largely complete: for comparison, the control L. braziliensis M2904 genome had only four homozygous SNPs, 97.2% of the protein-coding genes of the reference (231 were missing) and 70 additional genes missed in the reference sequence. These findings highlight scope to resolve Leishmania chromosomal architecture more accurately, particularly at repetitive regions and gene arrays, using longer sequencing reads and hybrid assembly approaches.
We showed that the majority of Viannia were diploid and had 35 chromosomes. Aneuploidy was evident for L. guyanensis LgCL085, L. guyanensis M4147, L. naiffi LnCL223, L. naiffi M5533, L. lainsoni M6426, L. panamensis WR120 and L. shawi M8408 as anticipated [80]. This was verified using read depth allele frequency distributions of reads mapped to L. braziliensis M2904 and to their own assemblies.
The L. guyanensis LgCL085 genome had more protein-coding genes (8230) than L. naiffi LnCL223 (8104). These numbers were similar to those for L. panamensis PSC-1 (7748) [51] and L. braziliensis M2904 (8357) [48]. The vast majority of protein coding gene models were computationally transferred [81] from the L. braziliensis M2904 reference with perfect matching, and were verified and improved manually. Both the L. guyanensis and L. naiffi reference genomes contained unassigned bin contigs, and chromosomal regions homologous to multiple chromosomal loci or containing partially collapsed gene arrays. Ninety (L. naiffi) and 92 (L. guyanensis) collapsed gene arrays were identified where haploid gene copy numbers were at least twice the assembled copy number when the reads were mapped to the assembled genomes.
3.3. A better resolution of the Viannia species complexes
This study illustrated that high-throughput sequencing approaches, alignment methods and annotation tools can improve the accuracy of Leishmania gene copy number estimates, gene organization and genome structure resolution. This yielded insights into features differentiating the isolates examined here, including a 45 Kb duplication on chromosome 34 of most Viannia, variable gene repertoires across Viannia species, and a potential minichromosome derived from the 3′ end of L. shawi M8408 chromosome 34. Further work is required to investigate L. utingensis and L. lindenbergi and other potential distinct lineages [82].
Both single-gene and large-scale copy number variations (CNVs) were tolerated by all Leishmania genomes. Leishmania genomes have extensive conservation of gene content with few species-specific genes [45,48]: here, only 31 L. guyanensis LgCL085 and four L. naiffi LnCL223 species-specific genes were found. These four genes unique to L. naiffi LnCL223, its leishmanolysin hyper-amplification, the 31 genes only in L. guyanensis LgCL085 and its tuzin arrays all represent potential targets for improving species-specific typing and better disease surveillance. This is important because infections by the Viannia are spread by many hosts and all sources of infections need to be addressed. Immunological screening of anti-Leishmania antibodies could be enhanced by genetic testing to identify infections from non-endemic or rarer sources like L. naiffi, which has longer parasite survival rates in macrophages in vitro[83].
MLSA of 100 Viannia isolates across four genes and genome-wide diversity inferred from mapped reads indicated that L. guyanensis LgCL085 was closest to L. panamensis PSC-1 within the L. guyanensis species complex, but was assigned the L. guyanensis classification because L. guyanensis, L. panamensis and L. shawi were a monophyletic species complex as shown by MLSA [56], multi-locus microsatellite typing [64], hsp70 [65], internal transcribed spacer [84,85], MLEE [86] and random amplified polymorphic DNA data [87]. Further typing of a more extensive L. guyanensis, L. panamensis and L. shawi isolate set might clarify if these are distinct species or a single genetic group.
More precise genetic screening of Viannia isolates is necessary to trace hybridization between species. Infection of humans, dogs and Lu. ovallesi with L. guyanensis/L. braziliensis hybrids was reported in Venezuela [40,41]. A L. shawi/L. guyanensis hybrid causing CL was detected in Amazonian Brazil [42], and L. naiffi has produced viable progeny with L. lainsoni [43] and L. braziliensis (Elisa Cupolillo 2018, unpublished data). There is extensive evidence of interbreeding among L. braziliensis complex isolates, including more virulent L. braziliensis/L. peruviana hybrids with higher survival rates within hosts in vitro[44].
4. Conclusion
This study highlighted the utility of genome sequencing for the identification, characterization and comparison of Leishmania species. We demonstrated that short reads were sufficient for assembly of most Leishmania genomes so that SNP, chromosome copy number, structural and somy changes can be investigated comprehensively. The L. (V.) guyanensis and L. (V.) naiffi genomes represent a further advance in refining the taxonomical complexity of the Viannia by illustrating their genomic characteristics and the extent to which these are shared across Viannia species, which will assist examining the extent to which they can hybridize. This improved understanding of Leishmania genomes should be used to explore the complex epidemiology of CL and MCL pathologies in the Americas and the roles of non-human reservoirs and sand flies in these processes. Future work could tackle transmission, drug resistance and pathogenesis in the Viannia by applying long-read high-throughput sequencing to examine broader sets of isolates, their genetic diversity, contributions to microbiome variation, and control of transcriptional dosage at gene amplifications.
5. Methods
5.1. Leishmania guyanensis and L. naiffi whole genome sequencing
Extracted DNA for L. guyanensis LgCL085 and L. naiffi LnCL223 was received from Charité University Medicine (Berlin) at the Wellcome Trust Sanger Institute on 6 February 2012. Paired-end 100 bp read Illumina HiSeq 2000 libraries were prepared for both during which L. guyanensis required 12 cycles of PCR. The DNA was sequenced (run 7841_5#12) on 15 (L. guyanensis, run 7841_5#12) and 23 (L. naiffi, run 7909_7#9) March 2012. The library preparation, sequencing and read quality verification were conducted as outlined previously [69]. The resulting L. guyanensis library contained 15 272 969 reads with a median insert size of 327.0 (NCBI accession ERX180458) and the L. naiffi one had 8 131 246 reads with a medianinsert size of 335.4 (ERX180449).
5.2. Viannia comparative genome, annotation and proteome files
The L. braziliensis reference genome (MHOM/BR/1975/M2904) was a positive control whose short reads were examined using the same methods. It was originally sequenced using an Illumina Genome Analyzer II [48] yielding 26 007 384 76 bp paired-end reads with a median insert size of 244.1 bp (ERX005631). Protein sequences were retrieved from the EMBL files using Artemis [88]. Two L. panamensis genomes, two L. peruviana genome assemblies and five 100 bp paired-end Illumina HiSeq 2000 read libraries of other Viannia isolates [53] were used for comparison (table 1). We included the genomes of L. panamensis MHOM/PA/1994/PSC-1, L. peruviana PAB-4377 and LEM1537 (MHOM/PE/1984/LC39), and the 100 bp Illumina HiSeq 2000 paired-end reads for each L. peruviana PAB-4377 (16 117 316 reads) and L. peruviana LEM1537 (9 378 317 reads).
5.3. Library quality control, contaminant removal and screening
Electronic supplementary material, figure S9, presents an overview of the bioinformatic steps used in this paper. Quality control of the L. guyanensis LgCL085, L. naiffi LnCL223, L. braziliensis M2904, the five Viannia libraries from [53], two L. peruviana libraries and L. panamensis PSC-1 read library was carried out using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). No corrections were required for the other libraries. An abnormal distribution of GC content per read observed as an extra GC content peak outside the normal peak for the L. braziliensis M2904 and L. guyanensis reads indicated sequence contamination that was removed (electronic supplementary material, figure S10). Two Illumina PCR primers in the L. braziliensis M2904 reads were removed (electronic supplementary material, table S1). Further evaluation using GC content filtering and the non-redundant nucleotide database with BLASTn [89] to remove contaminant sequences (electronic supplementary material, figure S10) with subsequent correction of read pairing arrangements reduced the initial 52 014 768 reads to 34 592 618 properly paired reads for assembly.
The M2904 reads used to assemble a control genome were used for read mapping, error correction and SNP calling, so the contamination did not affect the published reference. However, it did reduce the number of reads mapped as shown in [48] where only 84% of the L. braziliensis M2904 short reads mapped to the L. braziliensis assembly, compared with 92% of reads for L. infantum reads mapped to its own assembly, 93% of L. major reads mapped to its own assembly and 97% of L. mexicana reads mapped to its own assembly.
The 8 131 246 100 bp paired-end L. naiffi LnCL223 reads and 15 272 969 100 bp paired-end L. guyanensis LgCL085 reads were filtered (electronic supplementary material, table S1) in the same manner using BLASTn and the smoothness of the GC content distribution to remove putative contaminants. Low-quality bases were trimmed at the 3′ end of L. naiffi LnCL223 reads to remove bases with a phred base quality less than 30 using Trimmomatic [90] (electronic supplementary material, table S1 and figure S11). This resulted in 13 033 846 paired-end L. guyanensis LgCL085 sequences and 6 989 814 paired-end L. naiffi LnCL223 sequences—85% and 86% of the initial reads, respectively (electronic supplementary material, table S1).
5.4. Genome evaluation, assembly and optimization
Processed reads were assembled into contigs using Velvet v1.2.09 and assemblies for all odd-numbered k-mer lengths from 21 to 75 were evaluated. The expected k-mer coverage was determined for each assembly using the mode of a k-mer coverage histogram from the velvet-estimate-exp_cov.pl script in Velvet to maximize resolution of repetitive and unique regions [57]. This suggested optimal k-mers of 61 for L. guyanensis LgCL085 and 43 for both L. naiffi LnCL223 and L. braziliensis, which produced assemblies with the highest N50 lengths. Each assembly was assembled with this expected coverage, and contigs were removed if their average k-mer coverage was less than half the expected coverage levels. An expected coverage of 16 and a coverage cut-off of 8 was applied to L. naiffi reads, an expected coverage of 19 and coverage cut-off of 8.5 to L. guyanensis LgCL085, and an expected coverage of 28 and coverage cut-off of 14 to L. braziliensis.
The assembly with the highest N50 for each was scaffolded using SSPACE [58]. In the initial assemblies, 76% of gaps in scaffolds (3592/4754) were closed in for L. guyanensis LgCL085, 63% (4096/6530) for L. naiffi LnCL223 and 67% (4834/8786) for L. braziliensis using Gapfiller [58]. Erroneous bases were corrected by mapping reads to the references with iCORN [91] (electronic supplementary material, figure S12). Misassemblies detected and broken using REAPR [60] were aligned to the L. braziliensis M2904 reference (excluding the bin chromosome 00). Scaffolds were evaluated and broken at putative misassemblies detected from the fragment coverage distribution (FCD) error and regions with low coverage when the reads were mapped to both broken and unbroken options. Additionally, the L. braziliensis broken and unbroken scaffolds were used to verify that removing misassemblies prior to (but not after) the contiguation of scaffolds resulted in more accurate assembled chromosomes. Mis-assembled regions without a gap were replaced with N bases. REAPR corrected 444 errors in L. naiffi LnCL223, of which 59 were caused by low fragment coverage, 206 in L. guyanensis LgCL085 (eight due to low fragment coverage) and 232 in the L. braziliensis control (57 caused by low fragment coverage). Each assembly step improved the corrected N50 and percentage of error-free bases (EFB%) assessed using REAPR (electronic supplementary material, table S18), with the sole exception of L. braziliensis control at the error-correction stage, likely due to its higher heterozygosity. The EFB% was the fraction of the total bases whose reads had no mismatches, matched the expected insert length, had a small FCD error and at least five read pairs oriented in the expected direction.
Gaps > 100 bp were reduced to 100 bp and 200 bp at the edge of each unplaced scaffold was aligned with the 200 bp flanking all pseudo-chromosome gaps using BLASTn to verify that no further gaps could be closed using unplaced scaffolds. Unplaced bin scaffolds less than 1 Kb were discarded, and the resulting assemblies were visualized and compared to L. braziliensis using the Artemis Comparison Tool [92]. Leishmania guyanensis LgCL085 bin sequences with BLASTn E-values less than 1 × 10−5 and percentage identities greater than 40% to non-Leishmania species in non-redundant nucleotide database were removed as possible contaminants. The final scaffolds were contiguated using the L. braziliensis reference with ABACAS [59], unincorporated segments were labelled as unassigned ‘bin’ contigs, and kDNA contigs were annotated (electronic supplementary material).
5.5. Phylogenomic multi-locus sequencing analysis characterization
An MLSA approach was adopted to verify the Leishmania species identity using four housekeeping genes: glucose-6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (6PGD), mannose phosphate isomerase (MPI) and isocitrate dehydrogenase (ICD). Orthologues from other genomes and assemblies were obtained using BLASTn alignment with thresholds of E-value less than 0.05 and percentage identity greater than 70%. Leishmania peruviana LEM-1537 genome had gaps at the MPI and 6PGD genes and was excluded. The four housekeeping genes spanning 2902 sites were concatenated in the order G6PD, 6PGD, MPI and ICD, and aligned using Clustal Omega v1.1 to create a Neighbour-Net network of uncorrected p-distances using SplitsTree v4.13.1.
5.6. Genome annotation and manual curation
Annotation of the L. guyanensis LgCL085, L. naiffi LnCL223 and L. braziliensis control genomes was completed using Companion [80] using L. braziliensis M2904 as the reference as outlined previously [69], including manual checking and correction of gene models. A control run with the L. braziliensis M2904 reference genome using itself as a reference was performed. In L. naiffi LnCL223, 13 genes and one pseudogene were removed because they overlapped existing superior gene models that had improved sequence identity with L. braziliensis M2904 orthologues. Forty-six protein-coding genes were also manually added. Thirty-four protein-coding genes on L. guyanensis LgCL085 were manually added and one protein coding gene was removed. Two hundred and sixty-nine gene models on L. naiffi LnCL223 and 198 on L. guyanensis with multiple joins mainly caused by the presence of short gaps were corrected by extending the gene model across the gap where the gap length was known (less than 100 bp). If the gap length was unknown (greater than 100 bp), the gene was extended to the nearest start or stop codon.
5.7. Measuring ploidy, chromosome copy numbers and copy number variations
By mapping the reads with SMALT v5.7 (www.sanger.ac.uk/resources/software/smalt/) to L. braziliensis M2904, the coverage at each site was determined to quantify the chromosome copy numbers and RDAF distributions at heterozygous SNPs as per previous work [69]. The RDAF distribution was based on the coverage level of each allele at heterozygous SNPs and this feature differed across chromosomes for each isolate (electronic supplementary material, Results). The median coverage per chromosome was obtained, and the median of the 35 values combined with the RDAF distribution mode approximating 50% indicated that all isolates examined here were mostly diploid (except the triploid L. braziliensis M2904). These were visualized with R packages ggplot2 and gridExtra.
After PCR duplicate removal, the mapped reads were used to detect CNVs across genes or within non-overlapping 10 Kb blocks for all chromosomes and bin contigs using the median depth values normalized by the median of the chromosome (or bin contig). Loci with a copy number ≥ 2 were analysed for L. naiffi LnCL223, L. guyanensis LgCL085 and the L. braziliensis control using their reads mapped to their own assembly. This was also repeated for reads mapped to the L. braziliensis M2904 reference for L. guyanensis M4147, L. naiffi M5533, L. shawi M8408, L. lainsoni M6426, L. panamensis WR120, L. panamensis PSC-1, L. peruviana LEM1537 and L. peruviana PAB-4377. Leishmania panamensis PSC-1 reads were mapped to its own reference genome to verify that we could find previously identified amplified loci, and we mapped L. panamensis WR120 to it so that CNVs shared by both L. panamensis could be obtained. The BAM files of L. naiffi LnCL223, L. guyanensis LgCL085 and L. braziliensis M2904 reads mapped to its own assembly were visualized in Artemis to confirm and refine the boundaries of amplified loci.
5.8. Identification of orthologous groups and gene arrays
Protein-coding genes from L. guyanensis LgCL085, L. naiffi LnCL223 and the L. braziliensis M2904 control genome were produced from the EMBL files for each genome and these were submitted to the ORTHOMCLdb v5 webserver [93] to identify OGs. 11 825 OGs with associated gene IDs in at least one of four Leishmania species (L. major strain Friedlin, L. infantum, L. braziliensis and L. mexicana) or five Trypanosoma species (T. vivax, T. brucei, T. brucei gambiense, T. cruzi strain CL Brener and T. congolense) were retrieved from the OrthoMCL database and compared with OGs for each genome. The copy number of each OG was estimated by summing the haploid copy number of each gene in the OG. Gene arrays in each genome were identified by finding all OGs with haploid copy number ≥ 2. Large arrays (greater than or equal to 10 gene copies) were examined and arrays with unassembled gene copies were identified by finding those with haploid gene copy number at least twice the assembled gene number.
5.9. Single-nucleotide polymorphism screening and detection
The filtered reads with Smalt as mapped above were used for calling SNPs using Samtools Pileup v0.1.11 and Mpileup v0.1.18 and quality-filtered with Vcftools v0.1.12b and Bcftools v0.1.17-dev as previously [69] such that SNPs called by both Pileup and Mpileup post-screening were considered valid. These SNPs all had: base quality greater than 25; mapping quality greater than 30; SNP quality greater than 30; a non-reference RDAF greater than 0.1; forward–reverse read coverage ratios greater than 0.1 and less than 0.9; five or more reads; 2+ forward reads; and 2+ reverse reads. Low-quality and repetitive regions of the assemblies were identified and variants in these regions were masked as outlined elsewhere [69]. SNPs were classed as homozygous for an alternative allele to the reference if their RDAF ≥ 0.85 and heterozygous if it was greater than 0.1 and less than 0.85.
The high level of nucleotide accuracy of the assembled genomes was indicated by the low rate of homozygous SNPs when the reads mapped to its own assembly (50 for L. naiffi LnCL223, 12 for L. guyanensis LgCL085, 68 for the L. braziliensis reference and four for the L. braziliensis control). Likewise, the numbers and alleles of heterozygous SNPs for the L. braziliensis control (25 474) matched that for the reference (25 975), suggesting that the 705 (L. naiffi LnCL223) and 14 739 (L. guyanensis LgCL085) heterozygous SNPs were accurate. The difference in homozygous and heterozygous SNP rates for L. braziliensis here versus the original 2011 study [48] was likely due to differing methodology. The genetic divergence of L. naiffi LnCL223 and L. guyanensis LgCL085 compared with L. braziliensis was quantified using the density of heterozygous and homozygous SNPs per 10 Kb non-overlapping window on each chromosome, visualized using Bedtools.
Supplementary Material
Supplementary Material
Acknowledgements
The authors thank Matthew Berriman and members of the WTSI DNA pipelines team for generating the two sequence libraries; Elisa Cupolillo (Instituto Oswaldo Cruz, Brazil) for discussions and comments on the manuscript; Katrin Kuhls (Technical University of Applied Sciences Wildau), Cathal Seoighe (NUI Galway), Hideo Imamura and Jean-Claude Dujardin (both Institute of Tropical Medicine Antwerp) for help; Anne Stone and Kelly Harkins (both Arizona State University) for releasing valuable sequence read data; and the DJEI/DES/ SFI/HEA Irish Centre for High-End Computing (ICHEC) for computational facilities.
Data accessibility
The BioProject ID is PRJEB20208 for L. guyanensis LgCL085 and PRJEB20209 for L. naiffi LnCL223. The DNA reads are available at the NCBI Short Read Archive (SRA) and European Nucleotide Archive at ERX180458 for L. guyanensis LgCL085 and ERX180449 for L. naiffi LnCL223 (these are associated with BioProject PRJEB2600). The consensus genome sequence FASTA files are on figshare at https://doi.org/10.6084/m9.figshare.5693290 for L. guyanensis LgCL085 and https://doi.org/10.6084/m9.figshare.5693272 for L. naiffi LnCL223. The chromosome and bin contig annotation EMBL files are at https://doi.org/10.6084/m9.figshare.5693284 for L. guyanensis LgCL085 and https://doi.org/10.6084/m9.figshare.5693278 for L. naiffi LnCL223. The electronic supplementary material tables are on figshare at https://doi.org/10.6084/m9.figshare.5697064. For ease of reader access, the above genome sequence and annotation files, electronic supplementary material, tables and supplementary data are also available on the Dryad Digital Repository at: https://doi.org/10.5061/dryad.4bm23 [94].
Authors' contributions
S.C. completed the genome assembly, comparative genomics, phylogenetic analysis, mutation investigation, helped design the study and wrote the main manuscript text. S.C., A.S.T. and E.F. completed the genome annotation. M.S. completed genome sequencing. G.S. helped design the study and wrote the main manuscript text. J.A.C. helped design the study and wrote the main manuscript text. T.D. coordinated and designed the study and wrote the main manuscript text. All authors gave approval for publication.
Competing interests
We declare we have no competing interests.
Funding
The authors acknowledge financial support from the NUI Galway PhD fellowship scheme (S.C.) and the Wellcome Trust core funding of the Wellcome Trust Sanger Institute (WTSI, grant 098051) (J.A.C. and M.S.).
References
- 1.Pan American Health Organization. 2017. Leishmaniases: epidemiological report in the Americas See http://www2.paho.org/hq/index.php?option=com_docman&task=doc_view&Itemid=270&gid=39646&lang=en.
- 2.World Health Organization. 2010. Control of the leishmaniases. World Health Organization Technical Report Series 949.
- 3.Gramiccia M, Gradoni L. 2005. The current status of zoonotic leishmaniases and approaches to disease control. Int. J. Parasitol. 35, 1169–1180. (doi:10.1016/j.ijpara.2005.07.001) [DOI] [PubMed] [Google Scholar]
- 4.Killick-Kendrick R. 1999. The biology and control of phlebotomine sand flies. Clin. Dermatol. 17, 279–289. (doi:10.1016/S0738-081X(99)00046-2) [DOI] [PubMed] [Google Scholar]
- 5.Maroli M, Feliciangeli MD, Bichaud L, Charrel RN, Gradoni L. 2013. Phlebotomine sandflies and the spreading of leishmaniases and other diseases of public health concern. Med. Vet. Entomol. 27, 123–147. (doi:10.1111/j.1365-2915.2012.01034.x) [DOI] [PubMed] [Google Scholar]
- 6.Walsh JF, Molyneux DH, Birley MH. 1993. Deforestation: effects on vector-borne disease. Parasitology 106, S55–S75. (doi:10.1017/S0031182000086121) [DOI] [PubMed] [Google Scholar]
- 7.Davies CR, Reithinger R, Campbell-lendrum D, Feliciangeli D, Borges R, Rodriguez N. 2000. The epidemiology and control of leishmaniasis in Andean countries. Cad. Saude Pública 16, 925–950. (doi:10.1590/S0102-311X2000000400013) [DOI] [PubMed] [Google Scholar]
- 8.Rotureau B. 2006. Are New World leishmaniases becoming anthroponoses? Med. Hypotheses 67, 1235–1241. (doi:10.1016/j.mehy.2006.02.056) [DOI] [PubMed] [Google Scholar]
- 9.Lainson R, Shaw JJ. 1989. Leishmania (Viannia) naiffi sp. n., a parasite of the armadillo, Dasypus novemcinctus (L.) in Amazonian Brazil. Ann. Parasitol. Hum. Comp. 64, 3–9. (doi:10.1051/parasite/19896413) [DOI] [PubMed] [Google Scholar]
- 10.Naiff RD, Freitas RA, Naiff MF, Arias JR, Barrett TV, Momen H, Grimaldi Júnior G. 1991. Epidemiological and nosological aspects of Leishmania naiffi Lainson & Shaw, 1989. Mem. Inst. Oswaldo Cruz 86, 317–321. (doi:10.1590/S0074-02761991000300006) [DOI] [PubMed] [Google Scholar]
- 11.Roque AL, Jansen AM. 2014. Wild and synanthropic reservoirs of Leishmania species in the Americas. Int. J. Parasitol. Parasites Wildl. 3, 251–262. (doi:10.1016/j.ijppaw.2014.08.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Souza AAA, et al. 2017. Natural Leishmania (Viannia) infections of phlebotomines (Diptera: Psychodidae) indicate classical and alternative transmission cycles of American cutaneous leishmaniasis in the Guiana Shield, Brazil. Parasite 24, 13 (doi:10.1051/parasite/2017016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arias JR, Miles MA, Naiff RD, Povoa MM, de Freitas RA, Biancardi CB, Castellon EG. 1985. Flagellate infections of Brazilian sand flies (Diptera: Psychodidae): isolation in vitro and biochemical identification of Endotrypanum and Leishmania. Am. J. Trop. Med. Hyg. 34, 1098–1108. (doi:10.4269/ajtmh.1985.34.1098) [DOI] [PubMed] [Google Scholar]
- 14.Kato H, Gomez EA, Yamamoto Y, Calvopiña M, Guevara AG, Marco JD, Barroso PA, Iwata H, Hashiguchi Y. 2008. Natural infection of Lutzomyia tortura with Leishmania (Viannia) naiffi in an Amazonian area of Ecuador. Am. J. Trop. Med. Hyg. 79, 438–440. [PubMed] [Google Scholar]
- 15.Azpurua J, De La Cruz D, Valderama A, Windsor D. 2010. Lutzomyia sand fly diversity and rates of infection by Wolbachia and an exotic Leishmania species on Barro Colorado Island, Panama. PLoS Negl. Trop. Dis. 4, e627 (doi:10.1371/journal.pntd.0000627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cássia-Pires R, Boité MC, D'Andrea PS, Herrera HM, Cupolillo E, Jansen AM, Roque AL. 2014. Distinct Leishmania species infecting wild caviomorph rodents (Rodentia: Hystricognathi) from Brazil. PLoS Negl. Trop. Dis. 8, e3389 (doi:10.1371/journal.pntd.0003389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abba AM, Superina M. 2010. The 2009/2010 armadillo red list assessment. BioOne 11, 135–184. [Google Scholar]
- 18.Ober HK, Degroote LW, McDonough CM, Mizell RF, Mankin RW. 2011. Identification of an attractant for the nine-banded armadillo, Dasypus novemcinctus. Wildl. Soc. Bull. 35, 421–429. [Google Scholar]
- 19.van Thiel PP, Gool TV, Kager PA, Bart A. 2010. First cases of cutaneous leishmaniasis caused by Leishmania (Viannia) naiffi infection in Surinam. Am. J. Trop. Med. Hyg. 82, 588–590. (doi:10.4269/ajtmh.2010.09-0360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fagundes-Silva GA, Sierra Romero GA, Cupolillo E, Yamashita EP, Gomes-Silva A, De Oliveira Guerra JA, Da-Cruz AM. 2015. Leishmania (Viannia) naiffi: rare enough to be neglected? Mem. Inst. Oswaldo Cruz 110, 797–800. (doi:10.1590/0074-02760150128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lainson R, Shaw JJ, Silveira FT, Braga RR, Ishikawa EA. 1990. Cutaneous leishmaniasis of man due to Leishmania (Viannia) naiffi Lainson and Shaw, 1989. Ann. Parasitol. Hum. Comp. 65, 282–284. [PubMed] [Google Scholar]
- 22.Pratlong F, Deniau M, Darie H, Eichenlaub S, Pröll S, Garrabe E, Le Guyadec T, Dedet JP. 2002. Human cutaneous leishmaniasis caused by Leishmania naiffi is wide-spread in South America. Ann. Trop. Med. Parasitol. 96, 781–785. (doi:10.1179/000349802125002293) [DOI] [PubMed] [Google Scholar]
- 23.Van der Snoek EM, Lammers AM, Kortbeek LM, Roelfsema JH, Bart A, Jaspers CA. 2009. Spontaneous cure of American cutaneous leishmaniasis due to Leishmania naiffi in two Dutch infantry soldiers. Clin. Exp. Dermatol. 34, e889–e891. (doi:10.1111/j.1365-2230.2009.03658.x) [DOI] [PubMed] [Google Scholar]
- 24.Floch H. 1954. Leishmania tropica guyanensis n.sp. agent de la leishmaniose tegumentarie de Guyanes et de l'Amerique Centralele Arch Inst Pasteur La Guyane Française du Teritoire L'Inni 15, 328.
- 25.Lainson R, Shaw JJ, Povoa M. 1981. The importance of edentates (sloths and anteaters) as primary reservoirs of Leishmania braziliensis guyanensis, causative agent of ‘pianbois’ in north Brazil. Trans. R. Soc. Trop. Med. Hyg. 75, 611–612. (doi:10.1016/0035-9203(81)90222-4) [DOI] [PubMed] [Google Scholar]
- 26.Arias JR, Naif RD, Miles MA, de Souza AA. 1981. The opossum, Didelphis marsupialis (Marsupialia: Didelphidae), as a reservoir host of Leishmania braziliensis guyanensis in the Amazon Basin of Brazil. Trans. R. Soc. Trop. Med. Hyg. 75, 537–541. (doi:10.1016/0035-9203(81)90194-2) [DOI] [PubMed] [Google Scholar]
- 27.Dedet JP, Gay F, Chatenay G. 1989. Isolation of Leishmania species from wild mammals in French Guiana. Trans. R. Soc. Trop. Med. Hyg. 83, 613–615. (doi:10.1016/0035-9203(89)90374-X) [DOI] [PubMed] [Google Scholar]
- 28.Quaresma PF, et al. 2011. Wild, synanthropic and domestic hosts of Leishmania in an endemic area of cutaneous leishmaniasis in Minas Gerais State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 105, 579–585. (doi:10.1016/j.trstmh.2011.07.005) [DOI] [PubMed] [Google Scholar]
- 29.Lainson R, Shaw JJ, Ward RD, Ready PD, Naiff RD. 1979. Leishmaniasis in brazil: XIII. Isolation of leishmania from armadillos (Dasypus novemcinctus), and observations on the epidemiology of cutaneous leishmaniasis in north Pará State. Trans. R. Soc. Trop. Med. Hyg. 73, 239–242. (doi:10.1016/0035-9203(79)90225-6) [DOI] [PubMed] [Google Scholar]
- 30.Quinnell RJ, Courtenay O. 2009. Transmission, reservoir hosts and control of zoonotic visceral leishmaniasis. Parasitology 136, 1915–1934. (doi:10.1017/S0031182009991156) [DOI] [PubMed] [Google Scholar]
- 31.Ready PD, Lainson R, Shaw JJ, Ward RD. 1986. The ecology of Lutzomyia umbratilis Ward & Fraiha (Diptera: Psychodidae), the major vector to man of Leishmania braziliensis guyanensis in north-eastern Amazonian Brazil Bull. Entomol. Res. 76, 21–40. (doi:10.1017/S0007485300015248) [Google Scholar]
- 32.Balbino VQ, Marcondes CB, Alexander B, Luna LK, Lucena MM, Mendes AC, Andrade PP. 2001. First report of Lutzomyia (Nyssomyia) umbratilis Ward & Frahia, 1977 outside of Amazonian Region, in Recife, State of Pernambuco, Brazil (Diptera: Psychodidae: Phlebotominae). Mem. Inst. Oswaldo Cruz 96, 315–317. (doi:10.1590/S0074-02762001000300005) [DOI] [PubMed] [Google Scholar]
- 33.Young DG, Duncan MA. 1994. Guide to the identification and geographic distribution of lutzomyia sand flies in Mexico, the West Indies, Central and South America (Diptera: Psychodidae). Gainesville, FL: American Entomological Institute. [Google Scholar]
- 34.Rodríguez-Barraquer I, Góngora R, Prager M, Pacheco R, Montero LM, Navas A, Ferro C, Miranda MC, Saravia NG. 2008. Etiologic agent of an epidemic of cutaneous leishmaniasis in Tolima, Colombia. Am. J. Trop. Med. Hyg. 78, 276–282. [PubMed] [Google Scholar]
- 35.Fouque F, Gaborit P, Issaly J, Carinci R, Gantier JC, Ravel C, Dedet JP. 2007. Phlebotomine sand flies (Diptera: Psychodidae) associated with changing patterns in the transmission of the human cutaneous leishmaniasis in French Guiana. Mem. Inst. Oswaldo Cruz 102, 35–40. (doi:10.1590/S0074-02762007000100005) [DOI] [PubMed] [Google Scholar]
- 36.Lainson R, Shaw JJ, Ready PD, Miles MA, Póvoa M. 1981. Leishmaniasis in Brazil: XVI. Isolation and identification of Leishmania species from sandflies, wild mammals and man in north Para State, with particular reference to L. braziliensis guyanensis causative agent of ‘pian-bois’. Trans. R. Soc. Trop. Med. Hyg. 75, 530–536. (doi:10.1016/0035-9203(81)90192-9) [DOI] [PubMed] [Google Scholar]
- 37.Van der Meide WF, Jensema AJ, Akrum RA, Sabajo LO, Lai A Fat RF, Lambregts L, Schallig HD, van der Paardt M, Faber WR. 2008. Epidemiology of cutaneous leishmaniasis in Suriname: a study performed in 2006. Am. J. Trop. Med. Hyg. 79, 192–197. [PubMed] [Google Scholar]
- 38.Garcia A L, Tellez T, Parrado R, Rojas E, Bermudez H, Dujardin JC. 2007. Epidemiological monitoring of American tegumentary leishmaniasis: molecular characterization of a peridomestic transmission cycle in the Amazonian lowlands of Bolivia. Trans. R. Soc. Trop. Med. Hyg. 101, 1208–1213. (doi:10.1016/j.trstmh.2007.09.002) [DOI] [PubMed] [Google Scholar]
- 39.Rotureau B, Ravel C, Nacher M, Couppié P, Curtet I, Dedet JP, Carme B. 2006. Molecular epidemiology of Leishmania (Viannia) guyanensis in French Guiana. J. Clin. Microbiol. 44, 468–473. (doi:10.1128/JCM.44.2.468-473.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delgado O, Cupolillo E, Bonfante-Garrido R, Silva S, Belfort E, Grimaldi G Jr, Momen H. 1997. Cutaneous leishmaniasis in Venezuela caused by infection with a new hybrid between Leishmania (Viannia) braziliensis and L. (V.) guyanensis. Mem. Inst. Oswaldo Cruz 92, 581–582. (doi:10.1590/S0074-02761997000500002) [DOI] [PubMed] [Google Scholar]
- 41.Bonfante-Garrido R, Meléndez E, Barroeta S, de Alejos MA, Momen H, Cupolillo E, McMahon-Pratt D, Grimaldi G. 1992. Cutaneous leishmaniasis in western Venezuela caused by infection with Leishmania venezuelensis and L. braziliensis variants. Trans. R. Soc. Trop. Med. Hyg. 86, 141–148. (doi:10.1016/0035-9203(92)90544-M) [DOI] [PubMed] [Google Scholar]
- 42.Jennings YL, de Souza AAA, Ishikawa EA, Shaw J, Lainson R, Silveira F. 2014. Phenotypic characterization of Leishmania species causing cutaneous leishmaniasis in the lower Amazon region, western Pará state, Brazil, reveals a putative hybrid parasite, Leishmania (Viannia) guyanensis × Leishmania (Viannia) shawi shawi. Parasite 21, 39 (doi:10.1051/parasite/2014039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.da Silva AC Tojal, Cupolillo E, Volpini AC, Almeida R, Romero GA. 2006. Species diversity causing human cutaneous leishmaniasis in Rio Branco, state of Acre, Brazil. Trop. Med. Int. Health 11, 1388–1398. (doi:10.1111/j.1365-3156.2006.01695.x) [DOI] [PubMed] [Google Scholar]
- 44.Cortes S, Vaz Y, Neves R, Maia C, Cardoso L, Campino L. 2012. Risk factors for canine leishmaniasis in an endemic Mediterranean region. Vet. Parasitol. 189, 189–196. (doi:10.1016/j.vetpar.2012.04.028) [DOI] [PubMed] [Google Scholar]
- 45.Peacock CS, et al. 2007. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 39, 839–847. (doi:10.1038/ng2053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. 2003. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol. Cell 11, 1291–1299. [DOI] [PubMed] [Google Scholar]
- 47.Clayton C, Shapira M. 2007. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 156, 93–101. (doi:10.1016/j.molbiopara.2007.07.007) [DOI] [PubMed] [Google Scholar]
- 48.Rogers MB, et al. 2011. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2, 2129–2142. (doi:10.1101/gr.122945.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wincker P, Ravel C, Blaineau C, Pages M, Jauffret Y, Dedet JP, Bastien P. 1996. The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res. 24, 1688–1694. (doi:10.1093/nar/24.9.1688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Britto C, Ravel C, Bastien P, Blaineau C, Pagès M, Dedet JP, Wincker P. 1998. Conserved linkage groups associated with large-scale chromosomal rearrangements between Old World and New World Leishmania genomes. Gene 222, 107–117. (doi:10.1016/S0378-1119(98)00472-7) [DOI] [PubMed] [Google Scholar]
- 51.Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R. 2015. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 5, 8550 (doi:10.1038/srep08550) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valdivia HO, et al. 2015. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics 16, 715 (doi:10.1186/s12864-015-1928-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harkins KM, Schwartz RS, Cartwright RA, Stone AC. 2016. Phylogenomic reconstruction supports supercontinent origins for Leishmania. Infect. Genet. Evol. 38, 101–109. (doi:10.1016/j.meegid.2015.11.030) [DOI] [PubMed] [Google Scholar]
- 54.Oddone R, et al. 2009. Development of a multilocus microsatellite typing approach for discriminating strains of Leishmania (Viannia) species. J. Clin. Microbiol. 47, 2818–2825. (doi:10.1128/JCM.00645-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lye LF Lye, Owens K, Shi H, Murta SM, Vieira AC, Turco SJ, Tschudi C, Ullu E, Beverley SM. 2010. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 6, e1001161 (doi:10.1371/journal.ppat.1001161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boité MC, Mauricio IL, Miles MA, Cupolillo E. 2012. New insights on taxonomy, phylogeny and population genetics of Leishmania (Viannia) parasites based on multilocus sequence analysis. PLoS Negl. Trop. Dis. 6, e1888 (doi:10.1371/journal.pntd.0001888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. (doi:10.1101/gr.074492.107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. (doi:10.1093/bioinformatics/btq683) [DOI] [PubMed] [Google Scholar]
- 59.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. 2009. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969. (doi:10.1093/bioinformatics/btp347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. 2013. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 14, R47 (doi:10.1186/gb-2013-14-5-r47) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (doi:10.1038/msb.2011.75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. (doi:10.1093/molbev/msj030) [DOI] [PubMed] [Google Scholar]
- 63.Schönian G, Mauricio I, Cupolillo E. 2010. Is it time to revise the nomenclature of Leishmania? Trends Parasitol. 26, 466–469. (doi:10.1016/j.pt.2010.06.013) [DOI] [PubMed] [Google Scholar]
- 64.Kuhls K, et al. 2013. Population structure and evidence for both clonality and recombination among Brazilian strains of the subgenus Leishmania (Viannia). PLoS Negl. Trop. Dis. 7, e2490 (doi:10.1371/journal.pntd.0002490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fraga J, Montalvo AM, De Doncker S, Dujardin JC, Van der Auwera G. 2010. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect. Genet. Evol. 10, 238–245. (doi:10.1016/j.meegid.2009.11.007) [DOI] [PubMed] [Google Scholar]
- 66.Segovia M, Ortiz G. 1997. LDI amplifications in Leishmania. Parasitol. Today 13, 342–348. [DOI] [PubMed] [Google Scholar]
- 67.Fu G, Melville S, Brewster S, Warner J, Barker DC. 1998. Analysis of the genomic organisation of a small chromosome of Leishmania braziliensis M2903 reveals two genes encoding GTP-binding proteins, one of which belongs to a new G-protein family and is an antigen. Gene 210, 325–333. (doi:10.1016/S0378-1119(98)00088-2) [DOI] [PubMed] [Google Scholar]
- 68.Raymond F, et al. 2012. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 40, 1131–1147. (doi:10.1093/nar/gkr834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coughlan S, Mulhair P, Sanders M, Schonian G, Cotton JA, Downing T. 2017. The genome of Leishmania adleri from a mammalian host highlights chromosome fission in Sauroleishmania. Sci. Rep. 7, 43747 (doi:10.1038/srep43747) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hartley MA, Drexler S, Ronet C, Beverley SM, Fasel N. 2014. The immunological, environmental, and phylogenetic perpetrators of metastatic leishmaniasis. Trends Parasitol. 30, 412–422. (doi:10.1016/j.pt.2014.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Acestor N, Masina S, Ives A, Walker J, Saravia NG, Fasel N. 2006. Resistance to oxidative stress is associated with metastasis in mucocutaneous leishmaniasis. J. Infect. Dis. 194, 1160–1167. (doi:10.1086/507646) [DOI] [PubMed] [Google Scholar]
- 72.Eisen JA, et al. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 4, e286 (doi:10.1371/journal.pbio.0040286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steinkraus HB, Greer JM, Stephenson DC, Langer PJ. 1993. Sequence heterogeneity and polymorphic gene arrangements of the Leishmania guyanensis gp63 genes. Mol. Biochem. Parasitol. 62, 173–185. (doi:10.1016/0166-6851(93)90107-9) [DOI] [PubMed] [Google Scholar]
- 74.Joshi PB, Sacks DL, Modi G, McMaster WR. 1998. Targeted gene deletion of Leishmania major genes encoding developmental stage-specific leishmanolysin (GP63). Mol. Microbiol. 27, 519–530. [DOI] [PubMed] [Google Scholar]
- 75.Joshi PB, Kelly BL, Kamhawi S, Sacks DL, McMaster WR. 2002. Targeted gene deletion in Leishmania major identifies leishmanolysin (GP63) as a virulence factor. Mol. Biochem. Parasitol. 120, 33–40. (doi:10.1016/S0166-6851(01)00432-7) [DOI] [PubMed] [Google Scholar]
- 76.Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT. 2012. Leishmania virulence factors: focus on the metalloprotease GP63. Microbes Infect. 14, 1377–1389. (doi:10.1016/j.micinf.2012.05.014) [DOI] [PubMed] [Google Scholar]
- 77.Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. 1995. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J. Immunol. 155, 3102–3111. [PubMed] [Google Scholar]
- 78.Jackson AP. 2010. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol. Biol. Evol. 27, 33–45. (doi:10.1093/molbev/msp214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lakshmi BS, Wang R, Madhubala R. 2014. Leishmania genome analysis and high-throughput immunological screening identifies tuzin as a novel vaccine candidate against visceral leishmaniasis. Vaccine 32, 3816–3822. (doi:10.1016/j.vaccine.2014.04.088) [DOI] [PubMed] [Google Scholar]
- 80.Mannaert A, Downing T, Imamura H, Dujardin JC. 2012. Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania. Trends Parasitol. 28, 370–376. (doi:10.1016/j.pt.2012.06.003) [DOI] [PubMed] [Google Scholar]
- 81.Steinbiss S, Silva-Franco F, Brunk B, Foth B, Hertz-Fowler C, Berriman M, Otto TD. 2016. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 44, W29–W34. (doi:10.1093/nar/gkw292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akhoundi M, et al. 2017. Leishmania infections: molecular targets and diagnosis. Mol. Aspects Med. 57, 1–29. (doi:10.1016/j.mam.2016.11.012) [DOI] [PubMed] [Google Scholar]
- 83.Matta NE, Cysne-Finkelstein L, Machado GM, Da-Cruz AM, Leon L. 2010. Differences in the antigenic profile and infectivity of murine macrophages of Leishmania (Viannia) parasites. J. Parasitol. 96, 509–515. (doi:10.1645/GE-2241.1) [DOI] [PubMed] [Google Scholar]
- 84.Cupolillo E, Grimaldi Júnior G, Momen H, Beverley SM. 1995. Intergenic region typing (IRT): a rapid molecular approach to the characterization and evolution of Leishmania. Mol. Biochem. Parasitol. 73, 145–155. (doi:10.1016/0166-6851(95)00108-D) [DOI] [PubMed] [Google Scholar]
- 85.Berzunza-Cruz M, Cabrera N, Crippa-Rossi M, Sosa Cabrera T, Pérez-Montfort R, Becker I. 2002. Polymorphism analysis of the internal transcribed spacer and small subunit of ribosomal RNA genes of Leishmania mexicana. Parasitol. Res. 88, 918–925. (doi:10.1007/s00436-002-0672-x) [DOI] [PubMed] [Google Scholar]
- 86.Cupolillo E, Grimaldi G, Momen H. 1994. A general classification of new world Leishmania using numerical zymotaxonomy. Am. J. Trop. Med. Hyg. 50, 296–311. (doi:10.4269/ajtmh.1994.50.296) [DOI] [PubMed] [Google Scholar]
- 87.Bañuls A L, Jonquieres R, Guerrini F, Le Pont F, Barrera C, Espinel I, Guderian R, Echeverria R, Tibayrenc M. 1999. Genetic analysis of leishmania parasites in Ecuador: are Leishmania (Viannia) panamensis and Leishmania (V.) Guyanensis distinct taxa? Am. J. Trop. Med. Hyg. 61, 838–845. (doi:10.4269/ajtmh.1999.61.838) [DOI] [PubMed] [Google Scholar]
- 88.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. (doi:10.1093/bioinformatics/btr703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (doi:10.1186/1471-2105-10-421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. (doi:10.1093/bioinformatics/btu170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Otto TD, Sanders M, Berriman M, Newbold C. 2010. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26, 1704–1707. (doi:10.1093/bioinformatics/btq269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. 2005. ACT: the artemis comparison tool. Bioinformatics 21, 3422–3423. (doi:10.1093/bioinformatics/bti553) [DOI] [PubMed] [Google Scholar]
- 93.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. (doi:10.1101/gr.1224503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coughlan S, Taylor A, Feane E, Sanders M, Schonian G, Cotton J, Downing T. 2018. Data from: Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus Dryad Digital Repository. (https://doi.org/10.5061/dryad.4bm23) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Coughlan S, Taylor A, Feane E, Sanders M, Schonian G, Cotton J, Downing T. 2018. Data from: Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus Dryad Digital Repository. (https://doi.org/10.5061/dryad.4bm23) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
The BioProject ID is PRJEB20208 for L. guyanensis LgCL085 and PRJEB20209 for L. naiffi LnCL223. The DNA reads are available at the NCBI Short Read Archive (SRA) and European Nucleotide Archive at ERX180458 for L. guyanensis LgCL085 and ERX180449 for L. naiffi LnCL223 (these are associated with BioProject PRJEB2600). The consensus genome sequence FASTA files are on figshare at https://doi.org/10.6084/m9.figshare.5693290 for L. guyanensis LgCL085 and https://doi.org/10.6084/m9.figshare.5693272 for L. naiffi LnCL223. The chromosome and bin contig annotation EMBL files are at https://doi.org/10.6084/m9.figshare.5693284 for L. guyanensis LgCL085 and https://doi.org/10.6084/m9.figshare.5693278 for L. naiffi LnCL223. The electronic supplementary material tables are on figshare at https://doi.org/10.6084/m9.figshare.5697064. For ease of reader access, the above genome sequence and annotation files, electronic supplementary material, tables and supplementary data are also available on the Dryad Digital Repository at: https://doi.org/10.5061/dryad.4bm23 [94].