

Abstract
A genetic etiology for autism spectrum disorders (ASDs) was first suggested from twin studies reported in the 1970s. The identification of gene mutations in syndromic ASDs provided evidence to support a genetic cause of ASDs. More recently, genome-wide copy number variant and sequence analyses have uncovered a list of rare and highly penetrant copy number variants (CNVs) or single nucleotide variants (SNVs) associated with ASDs, which has strengthened the claim of a genetic etiology for ASDs. Findings from research studies in the genetics of ASD now support an important role for molecular diagnostics in the clinical genetics evaluation of ASDs. Various molecular diagnostic assays including single gene tests, targeted multiple gene panels and copy number analysis should all be considered in the clinical genetics evaluation of ASDs. Whole exome sequencing could also be considered in selected clinical cases. However, the challenge that remains is to determine the causal role of genetic variants identified through molecular testing. Variable expressivity, pleiotropic effects and incomplete penetrance associated with CNVs and SNVs also present significant challenges for genetic counseling and prenatal diagnosis.
Keywords: Autism spectrum disorders, array comparative genomic hybridization, copy number variation, single nucleotide variant, whole exome sequencing, whole genome sequencing, prenatal diagnosis
Introduction
The suggestion of a biological mechanism in autism spectrum disorders (ASDs) was first raised by Leo Kanner who along with Hans Asperger coined the term “autism”1. The landmark paper by Kanner in 1943 described a group of 11 children with “autism” and concluded with an insightful statement: “We must, then, assume that these children have come into the world with innate inability to form the usually biologically provided affective contact with people, just as other children come into the world with innate physical or intellectual handicaps… For here, we seem to have pure-culture examples of inborn autistic disturbances of affective contact”. The use of “inborn” to describe the possible cause of autism is explicit to the suggestion of a biological mechanism. It should be noted that the term “inborn error” was coined by the British physician, Archibald Garrod, during the same time period to describe biochemical defects in metabolism. Kanner also drew a comparison between autism and physical and intellectual disability. For the latter, the etiological role of biological mechanisms was well recognized. However, evidence supporting a biological mechanism in “autism” was not reported until the late 1970s when the first twin study was published by Folstein and Rutter2. Despite the small number of cases in this first twin study, the finding was seminal because for the first time a genetic factor was implicated in the etiology of autism. Subsequently, this finding has been replicated in numerous studies that have validated the initial observation3-6. The identification of gene mutations in several syndromic ASDs such as fragile X and Rett syndrome in the 1990s rendered the first direct evidence for a genetic etiology or single gene defect in the clinical presentation of autistic disorders7,8. With the rapid development of new genome technologies, the past decade has seen tremendous advances in the understanding of the genetics of ASDs9-11. Findings from these more recent genetic and genomic studies provide good evidence to not only support the role of a genetic etiology in ASDs but also underscore the challenge in understanding the molecular basis underlying ASDs. In fact, the cause in the majority of ASD cases (~70%) remains unknown12.
The diagnostic criteria of ASDs have evolved and are the subject of frequent debate in both professional and public forums. In the Diagnostic and Statistical Manual of Mental Disorders IV (DSM-IV-TR)13, published in 2000, a diagnosis of autism was made based on recognizing impairments in three core domains in social interaction and communication as well as restricted, repetitive and stereotyped patterns of behavior, interests and activities. Furthermore, patients with autism could be diagnosed with four separate disorders: autistic disorder, Asperger’s syndrome, pervasive developmental disorder – not otherwise specified (PDD-NOS), and childhood disintegrative disorder. In DSM-5, published in 2013, an umbrella term of autism spectrum disorders is used to cover all the separate diagnoses defined in DSM-IV14; thus an individual diagnosis such as Asperger’s syndrome is no longer used. The diagnostic criteria of ASDs are based on impairment in two domains: (1) persistent deficits in social communication and social interaction across multiple contexts and (2) restricted, repetitive patterns of behavior, interests or activities. The severity is based on the level of support that is required to meet the needs of social communication impairment and restricted and repetitive patterns of behavior. In current clinical practice, the diagnosis of an ASD is primarily a behavioral diagnosis. The comorbidity with other neurological presentations such as intellectual disability and seizures is not considered in the diagnostic criteria of ASDs, nor are non-behavioral features such as dysmorphic features and congenital anomalies taken into account. However, in the clinical evaluation of ASDs15, the probability of uncovering a genetic defect is significantly higher in ASD cases with dysmorphic features, congenital anomalies, seizures and significant intellectual disability16,17. ASDs are referred to as syndromic if the autistic diagnosis is part of the clinical presentation of a known genetic syndrome, or idiopathic if the cause is unknown.
The diagnosis of ASDs has been increasing steadily over the last decade. The reported prevalence in children was 1 in 88 in the US in 201218 compared to 1 in 110 as reported in 200919; the male to female ratio is 4 to 1. The underlying reasons for the increase in prevalence are not known and are also the subject of debate. The gender difference is also not well understood. There is no clear evidence that supports mutations in a major gene(s) in the sex chromosomes as significant contributors to the cause of idiopathic ASDs. The alarming increase in the number of new ASD cases has intensified the debate about the role of non-genetic factors or the interaction between genes and the environment in the etiology of ASDs4,20.
In this review, we will summarize key findings from recent ASD genetic and genomic studies using genome-wide copy number variant (CNV) analysis and next generation sequencing (NGS) techniques, describe the application of these techniques in the clinical genetics evaluation of ASDs, discuss challenges in determining a causal role of copy number and sequence variants, and outline genetic counseling dilemmas associated with ASDs.
Genetic basis of ASDs
Despite high heritability, the genetic inheritance implicated in the majority of cases of ASD remains elusive. The knowledge gained from syndromic ASDs indicates that autistic disorders can be caused by a defect in a single gene. The inheritance of syndromic ASDs becomes clear once the gene implicated in the genetic syndrome is identified. Classic examples in this category are fragile X syndrome and Rett syndrome7,8. With increasing numbers of reports of autism as a clinical feature of different genetic syndromes, the list of syndromic ASDs is growing (Table 1). However, because systematic natural history studies have not been conducted for most of these genetic syndromes, these claims may be overstated in some cases or underappreciated in others. Regardless, knowledge of syndromic ASDs provides a framework for differential diagnosis in the clinical evaluation of ASDs.
Table 1.
Syndromic ASD genes.
| Chromosomes | Genes | Syndromes |
|---|---|---|
| 2 | MBD5 | 2q31 microdeletion syndrome |
| 2 | SOS1 | Noonan syndrome |
| 5 | CDKL5 | Rett-like syndrome |
| 5 | NSD1 | Sotos syndrome |
| 7 | CHD7 | CHARGE |
| 7 | CNTNAP2 | Cortical dysplasia-focal epilepsy syndrome |
| 7 | RAF1 | Noonan syndrome |
| 7 | BRAF | Noonan syndrome |
| 9 | TSC1 | Tuberous sclerosis complex |
| 12 | CACNA1C | Timothy syndrome |
| 12 | PTPN11 | PTEN associated disorder |
| 12 | KRAS | Noonan syndrome |
| 14 | PTEN | PTEN associated disorders |
| 14 | FOXG1 | Angelman-like syndrome |
| 15 | UBE3A | Angelman syndrome |
| 15 | MAP2K1 | Noonan syndrome |
| 16 | TSC2 | Tuberous sclerosis complex |
| 17 | RA1 | Smith-Magenis syndrome |
| 18 | TCF4 | Pitt-Hopkins syndrome |
| 22 | SHANK3 | Related to Phelan-McDermid syndrome |
| X | MECP2 | Rett syndrome |
| X | FMR1 | Fragile X syndrome |
| X | SLC6A8 | Creatine transporter |
| X | SLC9A6 | X-linked Angelman-like syndrome |
| X | HPRT1 | Lesch-Nyhan syndrome |
| X | ARX | ARX related disorders |
| X | MED12 | Lujan-Fryns syndrome |
Discussion of the genetic basis in idiopathic ASD cases is more complicated because the cause for most cases has not been determined and a single gene cause accounts for less than 5% of cases in all the studies in the literature9,10,12,21. Two generation pedigrees with typical ASD have rarely been reported in the literature. The reduced reproduction fitness in individuals with ASD is often cited as the reason. Although families with more than one affected child, i.e. multiplex families, are frequently encountered, the clinical presentations among affected siblings usually have more differences than similarities, which poses the intriguing question of whether recessive inheritance is a good fit in general.
De novo genetic defects including CNVs and single nucleotide variants (SNV) have been the main focus for autism genetic studies conducted over the last decade. As described below, the results from these studies strongly support an important role of de novo and rare genetic mutations in the etiology of ASDs9,22.
Copy number variants
Although the term copy number variant (CNV) is relatively new, the impact of chromosome dosage, i.e. chromosomal deletion and duplication, in ASDs and other neurodevelopmental disorders has long been recognized23. Autistic behaviors have been described in several well-characterized microdeletion and duplication syndromes including Angelman and Prader–Willi, Smith–Magenis, Potoki–Lupski, velocadiofacial syndrome (VCFS) and Phelan–McDermid syndromes (PMS)24-27. In the case of idiopathic ASD, one of the first and still the best example of an associated chromosomal rearrangement is the maternally-inherited duplication of the chromosomal 15q11–q13 Angelman and Prader–Willi syndrome region observed using traditional chromosome analysis28. Although a variable phenotype has been reported with this particular duplication, autism, developmental delay, seizures and hypotonia are common features29-31. This rearrangement has been validated in numerous subsequent case reports and large scale genome-wide CNV studies using chromosome microarray analysis (CMA)32-36.
In an extensive review of 33 studies that included 22 698 patients, the International Standard Cytogenomic Array Consortium found that CMA offered a diagnostic yield of 15–20% as compared to 3% for G-banded chromosome analysis37 in patients with idiopathic ASDs or intellectual disability37. A set of CNVs highly penetrant for ASDs that were identified through research studies using genome-wide CNV analyses in a large set of ASD cases is listed in Table 233,38-40. These studies clearly suggest an important role of CNVs in the genetic etiology of ASDs.
Table 2.
Clinically relevant ASD-associated CNVs.
| Locus | Candidate Gene | Other associated disorders |
|---|---|---|
| 1q21.1 | ? | ID/SCZ/BD |
| 2p16.3 | NRXN1 | ID/SCZ |
| 2q23.1 | MBD5 | ID/SE |
| 3q29 | DLG1/PAK2? | ID/SCZ |
| 7q11.23 | LIMK | ID |
| 11q13.3 | SHNAK2 | ID/SE |
| 15q11.2 | UBE3A | ID/SE |
| 15q13.3 | CHRNA7 | ID/SCZ |
| 16p11.2 | KCD13? | ID/BD/SCZ |
| 22q11.2 | ? | ID/SCZ |
| 22q13.3 | SHANK3 | ID/SE/BD |
| Xp22.1 | PTCHD1 | ID/SE |
| Xp22.3 | NLGN3 | ID |
| Xq13.1 | NLGN4X | ID |
ID: intellectual disability; SCZ: Schizophrenia; BD: bipolar disorder; SE: seizure disorder.
It has been suggested that rare CNVs containing genes involved in neural plasticity or synapse formation and maintenance are associated with ASDs and intellectual disability and that disruption of these shared biological pathways can result in neurodevelopmental disorders21,41. In a study comparing 996 ASD individuals of European ancestry to 1287 matched controls, ASD cases were found to carry a higher global burden of rare CNVs, especially for loci previously implicated in either ASD and/or intellectual disability38. Among the CNVs identified, there were numerous de novo and inherited events, sometimes in combination in a given family, that implicated many novel ASD genes.
Recently, an exon-targeted oligo array was used to detect intragenic CNVs in a cohort of 10 362 patients including those with a clinical indication of ASDs42. The more commonly observed CNVs detected in this cohort include those affecting known and candidate genes for ASDs such as NRXN1, CNTNAP2, NLGN4X, A2BP1(RBOX1), CNTN4, CDH18 and TMEM195. A more detailed clinical and molecular characterization of 24 patients who have intragenic deletions of NRXN1 revealed that the 17 patients with deletions involving exons manifested developmental delay/intellectual disability (93%), infantile hypotonia (59%) and ASDs (56%)43. These results indicate that a small intragenic deletion is significant enough to contribute to neurodevelop-mental disorders, including ASDs, but the detection of these defects could be easily missed by array designs that lack exonic coverage.
Detection of recurring intragenic CNVs in this patient population will support reclassifying candidate genes implicated in neurodevelopmental disorders as disease-causing genes. For example, two patients with developmental delay and ASDs were identified with small duplication CNVs involving single exons that disrupt AUTS244. This finding was further verified by another study comparing 17 well-characterized individuals with microdeletions that affected at least one exon of AUTS2; this enabled the identification of a variable syndromic phenotype that included intellectual disability, autism, short stature, microcephaly, cerebral palsy and facial dysmorphisms45. These results illustrate the different sensitivities for detecting genetic defects based on the resolution of different arrays. However, different array designs to detect disease-causing defects in a clinical setting have not been fully evaluated.
The results from research studies and clinical tests not only have confirmed the previous observations from individual case reports but also have consolidated several major findings related to the role of CNVs in ASDs: (1) a number of new CNVs are strongly implicated in ASDs but also show both variable expressivity and pleiotropic effects; (2) between 5 and 10% of previous idiopathic ASD cases will carry rare CNVs; (3) both de novo and inherited CNVs confer a risk of ASD; and (4) there is a 3-fold increase in large and rare de novo CNVs in probands compared to their siblings10,22. These findings support the clinical application of copy number analysis in ASDs as the recommended first tier molecular test in the clinical genetics evaluation of ASDs15,37. However, the translation of these findings into clinical practice remains a challenge because the causal role of these CNVs cannot always be established (see below).
Single nucleotide variants
The identification of genes implicated in syndromic ASDs indicates that a sequence change of a single nucleotide in a single gene can confer a significant genetic risk for ASDs. This principle has been validated in idiopathic ASDs using a candidate gene sequencing approach. One of the best examples is the identification of mutations in the SHANK3 gene46. SHANK3 was mapped to the critical region of chromosome 22q13.3 in Phelan–McDermid syndrome in which autism is a prominent feature46-48. Subsequently, pathogenic point mutations in SHANK3 have been identified in idiopathic ASD in many independent reports49,50. Similar scenarios have also contributed to the discovery of other genes such as MBD5, CNTNAP2, neuroligins and neurexins, all of which are implicated in ASDs51-54. The low frequency of these sequence variants posed a challenge in experimental design for discovering new candidate genes until the recent emergence of the NGS technique. Whole exome sequencing (WES) and whole genome sequencing (WGS) by NGS have provided an unprecedented opportunity to discover rare variants throughout the genome in ASDs. To date, there are multiple large-scale ASD WES and several small scale WGS studies that have been carried out in trios, namely the proband plus the unaffected biological parents55-63. The majority of these studies were focused on the analysis of de novo mutations. Several studies analyzed both rare inherited and de novo mutations55,64,65. Among more than 1000 families assessed by these studies, the presence of de novo loss-of-function (LOF) mutations was consistently significantly higher in probands compared to controls. These studies have led to the discovery of more than a dozen genes with a high confidence of a causal role in ASDs (Table 3). A large number of de novo missense variants (10-fold higher than LOF alleles) have also been identified in ASDs from WES studies. Some of these missense changes are certain to contribute to the risk of ASD susceptibility. However, only a 5–10% excess of such mutations was found in ASD cohorts compared to controls, which did not reach statistical significance collectively. It is not possible to assign risk confidently for these missense variants without functional studies. Only three ASD WGS studies have been reported so far59,60,66. In contrast to WES, the sample sizes of those evaluated by WGS are relatively small. In one study, WGS of 32 trios detected clinical relevant variants in 16 probands60. This study supports the feasibility of using WGS as a clinical test and validates the improved sensitivity of WGS compared to WES in detecting pathogenic sequence variants.
Table 3.
ASD candidate genes from WES/WGS studies.
| Chromosome | Gene symbol | Description | Mutations |
|---|---|---|---|
| 1 | POMGNT1 | Protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase | p.R367H (64) |
| 1 | NTNG1 | Netrin G1 | p.T135I (58); p.Y23C (58) |
| 1 | POGZ | Pogo transposable element with ZNF domain | Frame shift (57); Frame shift (56) |
| 1 | USH2A | Usher syndrome 2A (autosomal recessive, mild) | p.W2075X, p.Y4238X, compound heterozygous (65) |
| 2 | DNMT3A | DNA (cytosine-5-)-methyltransferase 3 alpha | p.R635W (55) |
| 2 | ARID5A | AT rich interactive domain 5A (MRF1-like) | p.G120V (55) |
| 2 | IFIH1 | Interferon induced with helicase C domain 1 | Splicing site (65) |
| 2 | SCN2A | Sodium channel, voltage-gated, type II, alpha subunit | p.G1013X (61); p.C959X (61); deletion (60) |
| 2 | SCN1A | Sodium channel, voltage-gated, type I, alpha subunit | p.P1894L (1 mis-sense in additional cases) (58) |
| 3 | AMT | Aminomethyltransferase | p.I308F (64); p.D198G (64) |
| 5 | GPR98 | G protein-coupled receptor 98 | p.D6252N (3 point mutations in additional cases) (59) |
| 6 | PEX7 | Peroxisomal biogenesis factor 7 | p.W75C (64) |
| 6 | SYNE1 | Spectrin repeat containing, nuclear envelope 1 | p.L3206M (64) |
| 6 | VIP | Vasoactive intestinal peptide | p.Y73X (60) |
| 8 | VPS13B | Vacuolar protein sorting 13 homolog B (yeast) | Frame shift (64); p.S824A (64) |
| 8 | PKHD1L1 | Polycystic kidney and hepatic disease 1 (autosomal recessive)-like 1 | Splicing site (65) |
| 9 | LAMC3 | Laminin, gamma 3 | p.D339G (1 missense in additional cases) (58) |
| 10 | ANK3 | Ankyrin 3, node of Ranvier (ankyrin G) | p.G3690R (63) |
| 10 | USP54 | Ubiquitin specific peptidase 54 | Frame shift (60) |
| 11 | MICALCL | MICAL C-terminal like | Frame shift (60) |
| 11 | CAPRIN1 | Cell cycle associated protein 1 | p.Q399X (60) |
| 11 | KIRREL3 | Kin of IRRE like 3 (Drosophila) | Downstream (3 point mutations in additional cases) (59) |
| 12 | GRIN2B | Glutamate receptor, ionotropic, N-methyl D-aspar-tate 2B | Splicing site (1 nonsense and 1 frame shift in additional cases) (58) |
| 12 | NCKAP5L | NCK-associated protein 5-like | p.G11D (55) |
| 12 | PAH | Phenylalanine hydroxylase | Deletion (64); p.Q235X (64) |
| 12 | UBE3B | Ubiquitin protein ligase E3B | p.R40C (55) |
| 14 | CHD8 | Chromodomain helicase DNA binding protein 8 | 3 LOF mutations in additional cases (57); p.Q959X (58); Frame shift (58) |
| 14 | KIAA0284 | Centrosomal protein 170B | p.R1122H (55) |
| 16 | ABCC12 | ATP-binding cassette, sub-family C (CFTR/MRP), member 12 | p.W1024X (65) |
| 17 | ZNF18 | Zinc finger protein 18 | p.H377N (55) |
| 18 | KATNAL2 | Katanin p60 subunit A-like 2 | Splicing site (61); 3 LOF mutations in additional cases (57) |
| 20 | KCNQ2 | Potassium voltage-gated channel, KQT-like sub-family, member 2 | Frame shift (60) |
| 21 | DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A | Splicing site (58); Frame shift (56) |
| 22 | CLTCL1 | Clathrin, heavy chain-like 1 | p.R125C (55) |
| X | NLGN4X | Neuroligin 4, X-linked | p.Q329X (64) |
| X | KAL1 | Kallmann syndrome 1 sequence | p.R423X (60) |
| X | WWC3 | WWC family member 3] | p.R940Q (55) |
| X | ZC3H12B | Zinc finger CCCH-type containing 12B | p.R318Q (55) |
| X | DGAT2L6 | Diacylglycerol O-acyltransferase 2-like 6 | p.R151X (65); p.Q214X (65) |
| X | KIAA2022 | KIAA2022 | p.Q1471X (65) |
| X | PCDH11X | Protocadherin 11 X-linked | Splicing site (65) |
| X | SRPX2 | Sushi-repeat containing protein, X-linked 2 | Splicing site (65) |
| X | LUZP4 | Leucine zipper protein 4 | p.R161X (65); p.R163X (65) |
| X | BCORL1 | BCL6 corepressor-like 1 | p.R1090P (55) |
| X | AFF2 | AF4/FMR2 family, member 2 | p.A283X (65) |
| X | MECP2 | Methyl CpG binding protein 2 | p.E495X (65); p.E483X (64) |
| X | TMLHE | Trimethyllysine hydroxylase, epsilon | Splicing site (65) |
Shading represents recurrent in multiple studies.
In contrast to the convincing evidence supporting the pathogenicity of rare CNVs and SNVs in ASDs, the implication of common variants remains tenuous22,67. Several large and independent genome-wide association studies have been conducted to identify common variants that exert significant risk for ASDs67-70. Two earlier large studies (>3000 subjects) reported a significant association of two different loci, at 5p14.1 and 5p15.2, respectively68,71. However, these loci have not been replicated in other studies that included WES analysis. Although further investigation may be necessary to clarify the reasons underlying the differences among the studies, it is probably fair to conclude that common variants may not have a significant impact in ASDs66,72.
Biochemical defects
Numerous metabolic abnormalities have been reported in the context of an ASD phenotype but these are usually not specific enough to determine a diagnosis15,73. Metabolic disorders associated with an ASD phenotype are relatively rare (Table 4). Most metabolic disorders in a classical form, including mitochondrial diseases, are associated with other clinical symptomatology such as seizures and extrapyramidal signs. However, it is not known whether atypical or mild presentations of metabolic disorders may present more commonly as an ASD phenotype and whether they are under-detected.
Table 4.
Metabolic diseases with ASD clinical presentation.
| (1) Untreated phenylketonuriac |
| (2) MTHFR deficiency (methylenetetrahydrofolate reductase) |
| (3) 3β-hydroxycholesterol-7-reductase deficiency |
| (4) 6-N-trimethyllysine dioxygenase deficiency |
| (5) Adenylosuccinate lyase deficiency |
| (6) Cerebral folate deficiency |
| (7) Disorders of creatine transportor |
| (8) Mitochondrial diseases? |
Diagnostic techniques
Currently in the US, a clinical genetics evaluation is recommended for all children with a confirmed diagnosis of ASD15,74,75. It is estimated that a specific genetic etiology can be determined in about 15–20% of individuals with an ASD15. However, high quality or unbiased clinical data to validate the real yield of a clinical genetics evaluation in ASD is lacking76,77. The variable practice models among clinics and different diagnostic platforms used in molecular diagnostic laboratories complicate the assessment of validity. A flowchart for the proposed clinical genetics evaluation of ASDs is presented in Figure 1, which highlights the incorporation of new testing methodologies for determining a molecular defect.
Figure 1.

The flowchart for clinical genetics evaluation of ASDs. *Proceeding with WES should be dictated by clinical judgment at this stage.
Copy number variant analysis
Detection of CNVs is usually achieved through array technology, which is now the recommended first tier genetic test for the evaluation of ASDs15,37. Initially, CMA via array comparative genomic hybridization (CGH) was used for CNV analysis. Single nucleotide polymorphism (SNP) arrays were then introduced and enabled detection of copy number neutral regions of absence of heterozygosity (AOH) in addition to copy number analysis. The level of genomic resolution achieved by array analysis depends on the number, size and distance between the interrogating probes. Array CGH utilizes oligonucleotides (60–85 mers) while SNPs (25–50 mers) are the probes for SNP arrays; millions of both probe types can be synthesized on one glass slide78. There are also customized arrays that can detect single exon CNVs involving only a few hundred base pairs42. However, because SNPs predominate in non-coding regions, it is possible that certain regions of the genome, such as single exons, may not be represented on SNP arrays. In addition, SNP arrays demonstrate a lower signal-to-noise ratio per probe than CMA, which can result in less sensitivity for detecting CNVs. Therefore, to maximum clinical sensitivity, platforms that combine array CGH and SNP genotyping are becoming the predominant type of array used clinically79-82.
An advantage of SNP arrays is that they can reveal regions within the genome that lack heterozygosity, i.e. AOH. Identification of these regions is useful clinically in detecting polyploidy, uniparental disomy, consanguinity and recessive diseases, although these are thought to be infrequent causes of ASDs83. However, a recent study reported that ASD individuals with intellectual disability have more regions of AOH compared to their unaffected siblings84. Furthermore, analysis of specific ASD AOH regions has aided in the discovery of autism candidate genes by identifying single genes within an AOH interval that either are recurrent in ASD or harbor a homozygous, rare deleterious variant upon analysis of exome-sequencing data84.
The value of CGH/SNP combination arrays is to detect both CNV and copy-neutral AOH in a single assay. As shown in Figure 2, an intragenic deletion of 8q involving exons in the VPS13B gene for Cohen syndrome was detected by CNV analysis (Figure 2A), and the SNP data plot revealed an AOH block corresponding to the region on 8q that includes the VPS13B gene (Figure 2B). Together, the results indicate a homozygous intragenic deletion in VPS13B.
Figure 2.
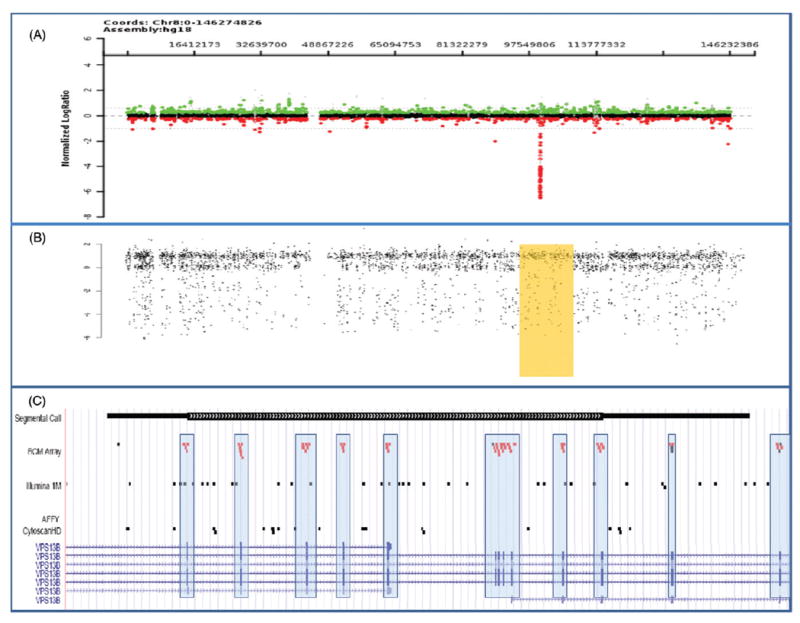
Comparison of different array CGH platforms. (A) Array results for chromosome 8 shows an intragenic deletion of exons 4–14 in the VPS13B gene which is associated with Cohen syndrome. The log2 ratio suggests the deletion is homozygous. (B) The SNP data plot reveals an AOH block (high-lighted rectangle) corresponding to the region on 8q that includes the VPS13B gene. Together, these results confirm a homozygous intragenic deletion. (C) Comparison of the probe distribution within the VPS13B gene between the exon-targeted BCM array and two commercial SNP arrays (Illumina 1M and Affy CytoscanHD). Note that the oligonucleotide probes (red dots [gray dots in print version]) on the BCM array are strategically selected for their location within the exons (hatch marks at bottom of figure and included in the boxes) whereas the SNP probes (black dots) predominate outside the exons and thus are unable to detect single exonic CNVs.
While Table 2 lists large-scale, recurrent CNVs that were detected from the first generation of clinical arrays, the increased resolution of arrays currently being used has promise for discovering more ASD-associated CNVs or individual candidate genes. For example, the combined analyses of CNVs in exonic regions and sequencing of coding exons has led to a diagnosis of an autosomal recessive disorder by unmasking a recessive allele in a patient with autism. In this case, a deletion of exons 22–25 in the VPS13B gene, which is associated with Cohen syndrome, was first detected by CNV analysis using an exon-targeted array (BCM V8); subsequent sequence analysis of the VPS13B gene identified a missense mutation in the other allele79,85.
These examples highlight that, to discover the precise molecular etiology, it may be necessary to combine different technologies within one test (array CGH + SNP) or use a combination of molecular diagnostic tests (CNV analysis plus medical re-sequencing of candidate genes or WES).
Single nucleotide variant analysis
Single gene test for syndromic ASDs
If the patient’s clinical presentation strongly suggests a syndromic ASD, the most effective approach is to request the specific gene test for the suspected syndrome. A list of single genes associated with syndromic ASDs is found in Table 1. There are numerous molecular diagnostic tests utilized for single gene testing, depending on the type of causative mutation usually observed for that specific gene. Testing methodologies include sequencing analysis (point mutations), FISH (microdeletions/duplications), Southern blotting (large repeat expansions) and multiplex ligation-dependent probe amplification (MLPA) (small deletions/duplications). According to the practice guidelines of the American College of Medical Genetics (ACMG) and American Academy of Pediatrics, DNA testing for fragile X is recommended for all children suspected of an ASD15,86. With the application of CMA, FISH analysis has been phased out in clinical diagnostic practice. However, for a defined microdeletion syndrome with characteristic clinical features, FISH still has value because of its relative low cost compared to CMA.
Targeted gene panels
The development of targeted gene panels (TGP) using NGS technology has been a popular approach in molecular diagnosis practice87. TGP allows a greater depth of coverage compared to a single gene test and better analytical sensitivity and specificity compared to WES (described below). The basic design is to group genes that are implicated in a disorder with significant genetic heterogeneity or are relevant to a particular clinical phenotype that shares the same molecular pathophysiology. TGP is particularly attractive for a disorder with extensive locus heterogeneity that is difficult to differentiate based on clinical presentation. Representative examples include, but are not limited to, retinitis pigmentosa, Bardet–Bidel syndrome, hearing loss and Noonan syndrome.
In the case of ASDs, extensive genetic heterogeneity is well recognized despite the fact that the molecular basis for the majority of cases is still not known. Therefore, TGP is an attractive diagnostic strategy. Several ASD-related gene panels are offered by clinical molecular diagnostic laboratories and include genes implicated in syndromic ASDs or candidate genes from recent medical re-sequencing studies. The results of WES and WGS have not yet been incorporated into these diagnostic panels. While the clinical validity of these panels has not been fully evaluated, it is expected that they will become a useful tool in the clinical setting.
The development of an updated ASD TGP that incorporates ASD candidate genes from recent WES and WGS studies is expected to be the next logical step. Although many candidate genes suggested from WES or WGS research studies remain to be validated, it is probably reasonable to include them in the updated ASD TGP. This design is a good transitional strategy until the use of WES or WGS techniques (described below) are fully validated for ASDs. The findings from the use of clinical TGP may speed the validation of research findings if clinical laboratories share results and clinical phenotypes.
Whole exome sequencing
The application of whole exome sequencing (WES) in clinical genetics practice has recently gained momentum88,89. A proof of principle study using WES in a clinical application was first published in 200919,90; a disease-causing mutation in a patient with a known genetic syndrome was successfully identified by WES in a research protocol. Subsequently, the success of using WES to discover new disease-causing genes was reported in 201019,89,91,92. In 2011, clinical molecular diagnostic laboratories started to offer WES as a clinical test using different array capture platforms and analytic paradigms (Figure 3). Currently, indications for using WES as a clinical test are quite variable among clinical geneticists or physicians in other specialties. Patients with significant intellectual disability, seizure disorders, multiple congenital anomalies and other unusual clinical presentations are typically good candidates for WES. Because a significant sub-set of individuals with ASDs, particular severe ASDs, usually present with comorbidity of severe intellectual disability, seizure disorders and other minor congenital anomalies, it is reasonable to argue that clinical WES is indicated in these cases if first tier testing is negative (Figure 1). The exact sensitivity of WES in clinical applications cannot be easily obtained. However, accumulated evidence in one clinical laboratory in the US reported a success rate of approximately 25% in determining the cause for ~250 cases with variable clinical presentations referred for WES93. Another group in the Middle East reported finding 37 potential variants in 100 probands for known Mendelian diseases (37%)94. In these cases, it is more difficult to predict the detection rate for causal variants. The results from recent ASD WES/WGS research studies could not be used to estimate the positive predictive value for WES in a clinical setting as the study subjects were not carefully selected and the focus was to identify de novo mutations.
Figure 3.
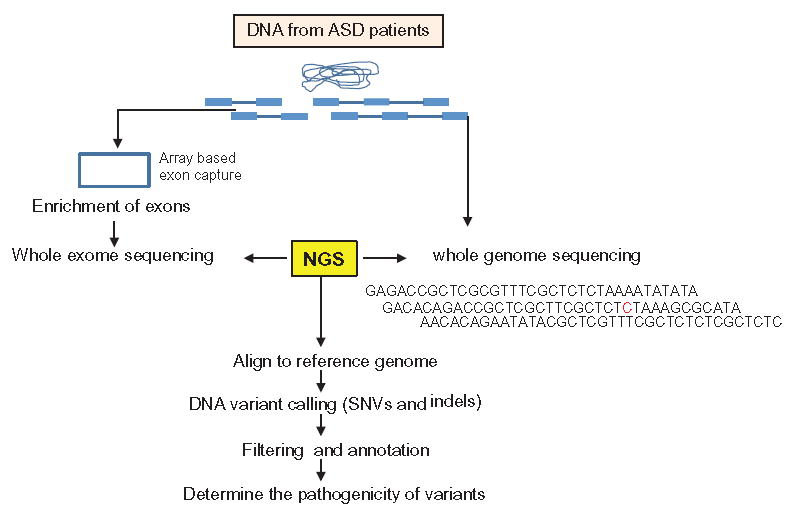
The workflow of WES and WGS in clinical diagnostic applications.
The ACMG recently published a laboratory standard on the use of WES in clinical testing87. Several issues regarding experimental design and the clinical application of WES in ASDs are worth discussing. The sequencing of trios, i.e. proband and both parents, has been the common experimental design in most WES research studies in ASDs. However, clinical laboratories vary regarding sequencing of the parents; some laboratories sequence the trio while others sequence only the proband and follow this with validation in the parents if necessary. The advantage of the proband only model is lower cost. The difference between these two designs is apparent because the sensitivity to detect de novo sequence variants is predicted to be higher in the trio design. In addition, the algorithm of the variant calling and filtering process varies among clinical laboratories. All laboratories have an equal opportunity to access public reference genomes such as the NHLBI exome server, 1000 genomes and dbSNP database. However, individual laboratories may have accumulated their own internal references genomes. In addition, different array platforms may be used for the exon enrichment or capture step. Technically, not all coding exons can be captured for sequencing because some fraction of the genome is difficult to capture by design. Therefore, there are inherent differences in each laboratory that may contribute to differences in sensitivity or success rate. However, it is certain that technique and sequence analytic ability will improve over time and the diagnostic success rate for WES will steadily increase. One development that is urgently needed in the genetics community is a curated database to share clinical phenotype and exome data world-wide.
Whole genome sequencing
Compared to WES, WGS offers many distinct advantages except for cost and the complexity of data analysis. DNA sample preparation is straightforward because no DNA enrichment step is needed. WGS has better genome coverage for the coding sequences based on the design and also has been validated60. In addition, non-coding regions that are not covered by WES are included in WGS. WGS also allows the detection of CNVs in high resolution that current array CGH platforms are not able to detect. For these reasons, it is reasonable to predict that WGS will eventually replace WES and CMA for routine testing as long as costs are reduced and data management and analysis are streamlined.
Biochemical testing
There have been no systematic studies examining the diagnostic yield of metabolic testing in an unselected cohort of patients with ASDs. The general opinion expressed about metabolic disorders in ASDs is that they are “low incidence, yet high impact” because some are treatable. Although no consensus has been reached on the level of testing that should be recommended, a baseline biochemical screen (Table 5) as a first tier evaluation is probably indicated (Figure 1), particularly in countries or regions where newborn screening for inborn errors of metabolism is not mandatory in medical practice. For clinical settings with good clinical expertise in metabolic disorders, a targeted biochemical test after clinical evaluation is probably more cost effective.
Table 5.
Biochemical screening for ASD clinical evaluation.
| (1) Plasma amino acid profile |
| (2) Plasma homocystine |
| (3) Urine organic acid |
| (4) Acylcarnitine profile |
| (5) Urine carnitine biosynthesis panel |
| (6) Plasma carnitine biosynthesis panel |
| (7) Urine creatine/guanidinoacetate analysis |
| (8) Plasma acylcarnitine profile |
| (9) Urine purine analysis |
| (10) Urine pyrimidine analysis |
| (11) Lactate |
Clinical interpretation
Copy number variants
A major challenge of using a whole genome analysis approach for clinical application is to determine the causal role of the genetic variants identified from these tests. ACMG standards and guidelines for interpretation and reporting postnatal constitutional CNVs have recently been updated95. In general, careful consideration is given to the size of the CNV, genomic position, gene(s) involved and patient’s reported phenotype. If the implicated region contains gene(s) with functions compatible with the abnormal clinical findings or previously described patients with a similar imbalance and phenotype, the CNV should be regarded as likely pathogenic. If the clinical significance is still unclear, investigation of the parents and additional family members by FISH analysis or CMA (depending on the size of the CNV) may be necessary to interpret and clarify the results. Review of the family history can often provide clues to the interpretation of these variants. Although the presence of the CNV in healthy family members suggests that it is benign, low penetrance and variable expressivity of the phenotype can complicate the interpretation. A larger, rare CNV that is determined to be de novo in origin is more likely to be pathogenic. The de novo occurrence of a CNV is, however, not absolute evidence of its pathogenicity, and caution must be exercised for possible non-paternity.
Interpretation of the clinical significance of CNVs remains challenging and requires time-consuming extensive searches of databases as well as the literature, and collaboration between the laboratory and the referring clinician96. A guide for utilizing public databases can be found in the Diagnostic Interpretation of Array Data Using Public Databases and Internet Sources. Examples of such databases are the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), UCSC Genome Browser (http://genome.ucsc.edu), Simons Foundation Autism Research Initiative (https://sfari.org) and Autism Chromosome Rearrangement Database (http://projects.tcag.ca/autism). As the number of samples screened by copy number analysis has grown significantly over the past several years, increased experience has revealed many subtleties and complexities of CNV interpretation that has resulted in a better understanding of the contribution of CNVs to pathogenicity97.
Even in cases where the role of the CNV is known to confer a significant risk for an ASD, it is still difficult to pinpoint the exact causative gene or genes because there are usually more than 10 genes embedded in a single CNV. In some cases, genes adjacent to or outside the deleted or duplicated intervals may also be affected due to position effect or disruption of a regulatory sequence element. The difficulty is to determine which gene or genes within the CNV are responsible for key features observed in these cases. In some cases, the clue may come from the function of a known gene(s) in the interval that has been elucidated from in vitro study or other model organisms. For example, mapping SHANK3, a gene known to encode a synaptic scaffolding protein at postsynaptic density98 to the 22q13.3 interval and which is deleted in Phelan–McDermid syndrome led to the hypothesis that SHANK3 is the important gene for ASD in this critical region48. This conclusion was supported by the subsequent discovery of point mutations in SHANK3 in idiopathic ASD patients46,99. However, in most cases, genes within the CNV are new and their function is unknown. For example, there are more than 25 genes within the 16p11.2 ASD-associated CNV100 and the functions of these genes are not known. Using a zebra fish model, Golzio et al. conducted a functional screen for the individual genes mapped within the interval and presented evidence suggesting that KCTD13 is a major driver of mirrored neuroanatomical phenotypes associated with copy number gain or loss in the 16p11.2 region in humans. Increase in the dosage of KCTD13 resulted in a small brain and loss of the same gene resulted in a large brain in fish101. However, translation of this finding from zebra fish to humans is not straightforward because neither microcephaly nor macrocephaly are features consistently reported in human ASD patients with CNVs of 16p11.2. Many other phenotypes associated with a gain or loss of 16p11.2 cannot be assessed reliably in a fish model. Despite this caveat, this finding represents a first and important step in elucidating the contribution of individual genes to the etiology of human ASD.
Single nucleotide variants
Interpretation challenges are similar for SNVs identified from targeted gene panels, WES or WGS. For the protein disrupting mutations in homozygotes or compound heterozygotes, the causal role for these mutations can be made with reasonable confidence, although the rarity of these sequence variants is still a cause for concern. For missense variants, it is not possible to determine causality based on reading the sequence information alone. Evidence from functional studies in cellular and in vivo models is frequently cited in the research literature to support the causal role of these variants.
Another common complication for interpretation of SNVs identified from TGP, WES or WGS is that multiple variants of unknown significance (VUS) may be identified in one individual. In most cases, it is difficult to determine a causal role based on reading sequencing information. The newly developed genome editing system of CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat) has showed great promise in rapidly manipulating the human genome by introducing SNVs or small inDels102,103. The application of CRISPR/Cas9 may also facilitate the dissection of the role of individual genes within a CNV rapidly by inactivating the individual genes within the interval. The much needed development of databases to catalog variant annotation for standard mutation nomenclature and a comprehensive reference database of medically important variants that is easily cross referenced to exome and genome sequence data is underway.
A growing body of evidence suggests that multiple genetic “hits” ultimately lead to ASDs12,104. These results support the suggestion that ASD is a complex genetic disorder resulting from simultaneous genetic variation in a few, several or even multiple genes105. Overall, the term “clan genomics” is introduced to remind us not to focus disproportionately on specific variants but rather to integrate across all classes of risk-associated variants. In some individuals, risk may be caused by an unusual combination of common variants whereas in others it may be due to a smaller number of large effect rare variants106. Furthermore, as observed in several studies, epigenetic dysregulation of synaptic genes at the transcriptional level may also contribute to ASD susceptibility107-109.
Incidental findings
Many unintended consequences occur when a WGS approach is used in clinical practice. One of the current, contentious debates is how to handle “incidental findings” from WES/WGS sequencing. The term “incidental findings” in this review refers to results of a deliberate search for pathogenic or likely pathogenic alterations in genes that are not apparently relevant to the diagnostic indication for which the sequencing test was ordered. The incidental finding may apply to the probands or to the parents. The ACMG has recently published a guideline for the reporting of incidental findings in clinical exome and genome sequencing that recommends that laboratories performing clinical sequencing seek and report mutations of the specified classes or types in the genes listed in the article110. This is certainly the subject of ongoing debate regarding many different aspects of genetic testing. Practically, concern over incidental findings may indeed increase rather than decrease anxiety.
Genetic counseling
The observed variable expressivity, pleiotropic effect and incomplete penetrance associated with CNVs and SNVs pose challenges for genetic counseling and determining recurrence risks. For example, the copy number gain and loss of chromosome 16p11.2 was first identified from studies of several large cohorts of ASD patients99,100,111. Subsequent studies have implicated 16q11.2 in both a wide spectrum of neuropsychiatric phenotypes and metabolic conditions such as childhood obesity, and it is also present in asymptomatic or unaffected parents or siblings112-119. The presence of the same CNV in unaffected family members clearly poses a dilemma in counseling families who are interested in using this information for prenatal diagnosis. The extremely variable expressivity and penetrance is not entirely unexpected. However, in many other microdeletion syndromes, such as Angelman and Prader–Willi, William, 1p36, 22q11.2 and Phelan–McDermid syndromes, these causative CNVs have never been reported in healthy individuals. Even in the case of 22q11.2 deletion syndrome in which the pleiotropic effect has been well documented and 10% are inherited, the penetrance is complete. The interesting question is whether ASD-associated CNVs operate under molecular mechanisms that are more amenable to genetic modifiers or environmental factors during evolution. In clinical practice, the identification of novel, de novo or inherited CNVs and SNVs has been encountered frequently. They are usually unique and not present in reference databases and have not been reported by research studies. Therefore, the pathogenicity of these variants with respect to ASD cannot be easily determined.
Given the current complications with interpreting results and providing counseling, genome-wide testing should occur in conjunction with a comprehensive medical genetics evaluation that includes a detailed family history and dysmorphology exam of the patient and relevant family members. In addition, with the quickly changing genomic landscape, it is important for families to return for reevaluation at recommended time intervals, both to review the current interpretation of previously identified variants of unclear clinical significance and to determine if any new molecular diagnostic studies are appropriate.
Prenatal diagnosis
Requests for prenatal diagnosis for ASDs have been uncommon because only rarely has the genetic etiology for the proband been known. With more widespread use of genetic testing for ASDs and the discovery of new genetic etiologies, it is expected that requests for prenatal diagnosis will become more common. In addition, technologies such as CMA are being used increasingly in prenatal specimens120 and have the potential to uncover ASD-related variants in fetuses with no prior risk. However, there have been only a few studies in pregnant women that document the prenatal identification of 16p13.11 and 15q26 deletions, which are predicted to be associated with an ASD phenotype121,122. Without long term follow-up, there is insufficient data to use in counseling when such findings are unexpectedly identified prenatally.
Prenatal testing for ASD is technically feasible for at-risk pregnancies if there has been prior identification of the causative alteration in the proband. However, it is not always possible to reliably predict the phenotype even if the alteration is present in the fetus. Furthermore, prenatal ultrasonography is of limited use to determine if the fetus is affected because there are no pathognomonic signs for diagnosis of an ASD. For example, the 16p11.2 microdeletion is often de novo. Based on current literature reports117, while ASD is not diagnosed in most individuals with a 16p11.2 microdeletion, it still occurs much more commonly among individuals with this microdeletion than in the general population. First, an important issue that causes difficulties for prenatal diagnosis of ASD is that (i) most pathogenic variants are not found exclusively in individuals with ASD but also occur in unaffected controls, although with a much lower frequency or (ii) they are inherited from a parent with or without a diagnosis of ASD. For example, Sanders et al.39 reported de novo CNVs in 6% of simplex ASD cases that were also present in 2% of unaffected siblings. Second, a current theory regarding the genotype–phenotype correlation in ASDs is determined by the degree of mutational events/burden (CNVs or SNPs) that a given individual carries, which may not be able to be determined in a prenatal setting123.
In summary, in many cases, the appropriateness of prenatal diagnosis for ASDs is uncertain because of the intrinsic difficulty in accurately detecting and predicting the ASD phenotype associated with a given genotype. Incomplete penetrance and variable expressivity represent the biggest challenges in prenatal testing for ASDs.
Conclusion and future directions
A decade of genetic studies of ASDs has produced evidence to support a major conclusion that rare genetic variants including CNVs and SNVs are strongly implicated in the etiology of ASDs. The development of new molecular diagnostic technologies such as array CGH/CMA and NGS has provided an unprecedented opportunity to uncover rare genetic variants in ASDs. Although these studies have not yet led to a major breakthrough in the understanding of the molecular basis in the majority of ASD cases, these findings do provide an opportunity for clinical applications.
Many challenges remain in the development of a robust ASD genetic testing paradigm in clinical practice and for counseling the families about genetic findings. Several important steps need to be taken in this direction. There is an urgent need for the development of a reliable functional assay to assist with the interpretation of the clinical relevance of genetic variants identified from clinical tests. To facilitate this process, it would be extremely valuable for different laboratories to share the clinical WES sequence database and clinical phenotype data. To establish evidence-based practice, it is essential to know the validity or yield of each test modality. This may be obtained from a systematically designed study or accumulated clinical experience. In addition, a cost and effect analysis should also be conducted to firmly establish a standard of care for the clinical genetics evaluation of ASDs. Despite these many challenges, there are good reasons to believe that the clinical genetics evaluation of ASDs is an important part of clinical care for ASD children and their families.
Acknowledgments
We would like to thank Ping Wang for the preparation of the Tables.
SWC and ANP are faculty members of the Department of Molecular and Human Genetics at Baylor College of Medicine, which derives revenue from the CMA offered in the Medical Genetics Laboratory. YHJ is supported by the National Institute of Health (Grant MH-1089441), Autism Speaks, and the Ruth K. Broad Foundation and Prader-Willi Syndrome Research Foundations. YW is supported by the Nature Science Foundation of China (NSFC 81071116), the Shanghai Science and Technology Commission (12XD1401100), and the China Ministry of Health Special Scientific Research Funds (201302002).
Abbreviations
- AOH
absence of heterozygosity
- ACMG
American College of Medical Genetics
- ASD
autism spectrum disorder
- CGH
comparative genomic hybridization
- CMA
chromosomal microarray analysis
- CNV
copy number variant
- CRISPR
clustered regularly interspaced short palindromic repeats
- DSM
Diagnostic and Statistical Manual of Mental Disorders
- FISH
fluorescence in situ hybridization
- LOF
loss of function
- MLPA
multiplex ligation-dependent probe amplification
- NGS
next generation sequencing, high-throughput sequencing
- PMS
Phelan–McDermid syndrome
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
- TGP
targeted gene panel
- VCFS
velocardiofacial syndrome
- VUS
variant of unknown significance
- WES
whole exome sequencing
- WGS
whole genome sequencing
Footnotes
Declaration of interest
The other authors have no conflicts to declare.
Referees: Dr. Judith Miles, Thompson Center for Autism and Neurodevelopmental Disorders and Department of Child Health, University of Missouri Hospitals and Clinics, Columbia, MO, USA. Dr. Marwan Shinawi, Department of Pediatrics, Division of Genetics and Genomic Medicine, Washington University School of Medicine, St. Louis, MO, USA.
References
- 1.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–53. [PubMed] [Google Scholar]
- 2.Folstein S, R M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;197:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 3.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 4.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallmayer J, Glasson EJ, Bower C, et al. On the twin risk in autism. Am J Hum Genet. 2002;71:941–6. doi: 10.1086/342990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steffenburg S, Gillberg C, Hellgren L, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–16. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 7.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 8.Pieretti M, Zhang FP, Fu YH, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–22. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 9.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14:1499–506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Daly MJ, Devlin B, et al. The Autism Sequencing Consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron. 2012;76:1052–6. doi: 10.1016/j.neuron.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–5. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaaf CP, Goin-Kochel RP, Nowell KP, et al. Expanding the clinical spectrum of the 16p11.2 chromosomal rearrangements: three patients with syringomyelia. Eur J Hum Genet. 2011;19:152–6. doi: 10.1038/ejhg.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. Washington, DC: American Psychiatric Association; 2000. pp. 70–5. [Google Scholar]
- 14.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013. pp. 50–9. [Google Scholar]
- 15.Schaefer GB, Mendelsohn NJ, Professional P, et al. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 2013;15:399–407. doi: 10.1038/gim.2013.32. [DOI] [PubMed] [Google Scholar]
- 16.Jacquemont ML, Sanlaville D, Redon R, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43:843–9. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaudet AL. Autism: highly heritable but not inherited. Nat Med. 2007;13:534–6. doi: 10.1038/nm0507-534. [DOI] [PubMed] [Google Scholar]
- 18.Prevalence of autism spectrum disorders – Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19. [PubMed] [Google Scholar]
- 19.Prevalence of autism spectrum disorders – Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ. 2009;58:1–20. [PubMed] [Google Scholar]
- 20.Gregory SG, Anthopolos R, Osgood CE, et al. Association of autism with induced or augmented childbirth in North Carolina Birth Record (1990-1998) and Education Research (1997-2007) databases. JAMA Pediatr. 2013;167:959–66. doi: 10.1001/jamapediatrics.2013.2904. [DOI] [PubMed] [Google Scholar]
- 21.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Ann Rev Genom Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 22.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22:229–37. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Stankiewicz P, Inoue K, Bi W, et al. Genomic disorders: genome architecture results in susceptibility to DNA rearrangements causing common human traits. Cold Spring Harb Symp Quant Biol. 2003;68:445–54. doi: 10.1101/sqb.2003.68.445. [DOI] [PubMed] [Google Scholar]
- 24.Williams CA, Lossie A, Driscoll D. Angelman syndrome: mimicking conditions and phenotypes. Am J Med Genet. 2001;101:59–64. doi: 10.1002/ajmg.1316. [DOI] [PubMed] [Google Scholar]
- 25.Phelan MC. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008;3:14–20. doi: 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laje G, Morse R, Richter W, et al. Autism spectrum features in Smith-Magenis syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:456–62. doi: 10.1002/ajmg.c.30275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potocki L, Bi W, Treadwell-Deering D, et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet. 2007;80:633–49. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook EH, Jr, Lindgren V, Leventhal BL, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–34. [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts SE, Dennis NR, Browne CE, et al. Characterisation of interstitial duplications and triplications of chromosome 15q11-q13. Hum Genet. 2002;110:227–34. doi: 10.1007/s00439-002-0678-6. [DOI] [PubMed] [Google Scholar]
- 30.Ingason A, Kirov G, Giegling I, et al. Maternally derived microduplications at 15q11-q13: implication of imprinted genes in psychotic illness. Am J Psychiatry. 2011;168:408–17. doi: 10.1176/appi.ajp.2010.09111660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas NS, Durkie M, Potts G, et al. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11.23, 15q11-q13 and 22q11. Eur J Hum Genet. 2006;14:831–7. doi: 10.1038/sj.ejhg.5201617. [DOI] [PubMed] [Google Scholar]
- 32.Boyar FZ, Whitney MM, Lossie AC, et al. A family with a grand-maternally derived interstitial duplication of proximal 15q. Clin Genet. 2001;60:421–30. doi: 10.1034/j.1399-0004.2001.600604.x. [DOI] [PubMed] [Google Scholar]
- 33.Bucan M, Abrahams BS, Wang K, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang NJ, Liu D, Parokonny AS, et al. High-resolution molecular characterization of 15q11-q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage. Am J Hum Genet. 2004;75:267–81. doi: 10.1086/422854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurrieri F, Battaglia A, Torrisi L, et al. Pervasive developmental disorder and epilepsy due to maternally derived duplication of 15q11-q13. Neurology. 1999;52:1694–7. doi: 10.1212/wnl.52.8.1694. [DOI] [PubMed] [Google Scholar]
- 36.Depienne C, Moreno-De-Luca D, Heron D, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66:349–59. doi: 10.1016/j.biopsych.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinto D, Pagnamenta AT, Klei L, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanders SJ, Ercan-Sencicek AG, Hus V, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Celestino-Soper PB, Shaw CA, Sanders SJ, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011;20:4360–70. doi: 10.1093/hmg/ddr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toro R, Konyukh M, Delorme R, et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–72. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Boone PM, Bacino CA, Shaw CA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–42. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaaf CP, Boone PM, Sampath S, et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet. 2012;20:1240–7. doi: 10.1038/ejhg.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagamani SC, Erez A, Ben-Zeev B, et al. Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur J Hum Genet. 2013;21:343–6. doi: 10.1038/ejhg.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beunders G, Voorhoeve E, Golzio C, et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet. 2013;92:210–20. doi: 10.1016/j.ajhg.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–7. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonaglia MC, Giorda R, Mani E, et al. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet. 2006;43:822–8. doi: 10.1136/jmg.2005.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson HL, Wong AC, Shaw SR, et al. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–84. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boccuto L, Lauri M, Sarasua SM, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet. 2013;21:310–16. doi: 10.1038/ejhg.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moessner R, Marshall CR, Sutcliffe JS, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–97. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Talkowski ME, Mullegama SV, Rosenfeld JA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–63. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zweier C, de Jong EK, Zweier M, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85:655–66. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laumonnier F, Bonnet-Brilhault F, Gomot M, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–7. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arking DE, Cutler DJ, Brune CW, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–4. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chahrour MH, Yu TW, Lim ET, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michaelson JJ, Shi Y, Gujral M, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–42. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang YH, Yuen RK, Jin X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–63. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bi C, Wu J, Jiang T, et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat. 2012;33:1635–8. doi: 10.1002/humu.22174. [DOI] [PubMed] [Google Scholar]
- 63.Shi L, Zhang X, Golhar R, et al. Whole-genome sequencing in an autism multiplex family. Mol Autism. 2013;4:8. doi: 10.1186/2040-2392-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu TW, Chahrour MH, Coulter ME, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–73. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim ET, Raychaudhuri S, Sanders SJ, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–42. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szatmari P, Paterson AD, Zwaigenbaum L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–28. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–33. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weiss LA. Autism genetics: emerging data from genome-wide copy-number and single nucleotide polymorphism scans. Expert Rev Mol Diagn. 2009;9:795–803. doi: 10.1586/erm.09.59. [DOI] [PubMed] [Google Scholar]
- 70.Anney R, Klei L, Pinto D, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–82. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weiss LA, Arking DE, Daly MJ, et al. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–8. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anney R, Klei L, Pinto D, et al. Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Hum Mol Genet. 2012;21:4781–92. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zecavati N, Spence SJ. Neurometabolic disorders and dysfunction in autism spectrum disorders. Curr Neurol Neurosci Rep. 2009;9:129–36. doi: 10.1007/s11910-009-0021-x. [DOI] [PubMed] [Google Scholar]
- 74.Johnson CP, Myers SM American Academy of Pediatrics Council on Children with Disabilities. Identification and evaluation of children with autism spectrum disorders. Pediatrics. 2007;120:1183–215. doi: 10.1542/peds.2007-2361. [DOI] [PubMed] [Google Scholar]
- 75.Zwaigenbaum L, Bryson S, Lord C, et al. Clinical assessment and management of toddlers with suspected autism spectrum disorder: insights from studies of high-risk infants. Pediatrics. 2009;123:1383–91. doi: 10.1542/peds.2008-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen Y, Dies KA, Holm IA, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–35. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herman GE, Henninger N, Ratliff-Schaub K, et al. Genetic testing in autism: how much is enough? Genet Med. 2007;9:268–74. doi: 10.1097/gim.0b013e31804d683b. [DOI] [PubMed] [Google Scholar]
- 78.Ou Z, Kang SH, Shaw CA, et al. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med. 2008;10:278–89. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiszniewska J, Bi W, Shaw C, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet. 2014;22:79–87. doi: 10.1038/ejhg.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peiffer DA, Le JM, Steemers FJ, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–48. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwartz S. Clinical utility of single nucleotide polymorphism arrays. Clin Lab Med. 2011;31:581–94. doi: 10.1016/j.cll.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A. 2011;155A:757–68. doi: 10.1002/ajmg.a.33939. [DOI] [PubMed] [Google Scholar]
- 83.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–76. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gamsiz ED, Viscidi EW, Frederick AM, et al. Intellectual disability is associated with increased runs of homozygosity in simplex autism. Am J Hum Genet. 2013;93:103–9. doi: 10.1016/j.ajhg.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen D, Pichard N, Tordjman S, et al. Specific genetic disorders and autism: clinical contribution towards their identification. J Autism Dev Disord. 2005;35:103–16. doi: 10.1007/s10803-004-1038-2. [DOI] [PubMed] [Google Scholar]
- 86.Myers SM, Johnson CP American Academy of Pediatrics Council on Children with Disabilities. Management of children with autism spectrum disorders. Pediatrics. 2007;120:1162–82. doi: 10.1542/peds.2007-2362. [DOI] [PubMed] [Google Scholar]
- 87.Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–47. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jacob HJ. Next-generation sequencing for clinical diagnostics. N Engl J Med. 2013;369:1557–8. doi: 10.1056/NEJMe1310846. [DOI] [PubMed] [Google Scholar]
- 89.Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–9. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoischen A, van Bon BW, Rodriguez-Santiago B, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43:729–31. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- 92.Gilissen C, Hoischen A, Brunner HG, et al. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228–239. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodriguez-Flores JL, Fakhro K, Hackett NR, et al. Exome sequencing identifies potential risk variants for Mendelian disorders at high prevalence in Qatar. Hum Mutat. 2014;35:105–16. doi: 10.1002/humu.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel AB, Hays SA, Bureau I, et al. A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J Neurosci. 2013;33:2593–604. doi: 10.1523/JNEUROSCI.2447-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stankiewicz P, Pursley AN, Cheung SW. Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am J Med Genet A. 2010;152A:1089–100. doi: 10.1002/ajmg.a.33216. [DOI] [PubMed] [Google Scholar]
- 97.Hehir-Kwa JY, Pfundt R, Veltman JA, et al. Pathogenic or not?. Assessing the clinical relevance of copy number variants. Clin Genet. 2013;84:415–21. doi: 10.1111/cge.12242. [DOI] [PubMed] [Google Scholar]
- 98.Grabrucker AM, Knight MJ, Proepper C, et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011;30:569–81. doi: 10.1038/emboj.2010.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 101.Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–7. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leblond CS, Heinrich J, Delorme R, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.El-Fishawy P, State MW. The genetics of autism: key issues, recent findings, and clinical implications. Psychiatr Clin North Am. 2010;33:83–105. doi: 10.1016/j.psc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lupski JR, Belmont JW, Boerwinkle E, et al. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhu L, Wang X, Li XL, et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum Mol Genet. 2014;23:1563–78. doi: 10.1093/hmg/ddt547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jiang YH, Sahoo T, Michaelis RC, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet A. 2004;131:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- 109.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–92. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Green RC, MD, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–38. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 112.Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–5. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bochukova EG, Huang N, Keogh J, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–70. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shen Y, Chen X, Wang L, et al. Intra-family phenotypic heterogeneity of 16p11.2 deletion carriers in a three-generation Chinese family. Am J Med Genet B Neuropsychiatr Genet. 2011;156:225–32. doi: 10.1002/ajmg.b.31147. [DOI] [PubMed] [Google Scholar]
- 115.Shinawi M, Liu P, Kang SH, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–41. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Puvabanditsin S, Nagar MS, Joshi M, et al. Microdeletion of 16p11.2 associated with endocardial fibroelastosis. Am J Med Genet A. 2010;152A:2383–6. doi: 10.1002/ajmg.a.33562. [DOI] [PubMed] [Google Scholar]
- 117.Miller DTNR, Sobeih MM, Shen Y, Wu BL, Hanson E. 16p11.2 microdeletion. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews [Internet] Seattle (WA): University of Washington; 1993–2014; 2009. Sep 22, updated 2011 Oct 27. [Google Scholar]
- 118.McCarthy SE, Makarov V, Kirov G, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–7. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Simons Vip Consortium. Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron. 2012;73:1063–7. doi: 10.1016/j.neuron.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 120.Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–84. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Law LW, Lau TK, Fung TY, et al. De novo 16p13.11 microdeletion identified by high-resolution array CGH in a fetus with increased nuchal translucency. BJOG. 2009;116:339–43. doi: 10.1111/j.1471-0528.2008.01948.x. [DOI] [PubMed] [Google Scholar]
- 122.Poot M, Verrijn Stuart AA, van Daalen E, et al. Variable behavioural phenotypes of patients with monosomies of 15q26 and a review of 16 cases. Eur J Med Genet. 2013;56:346–50. doi: 10.1016/j.ejmg.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 123.Choy KW, Setlur SR, Lee C, et al. The impact of human copy number variation on a new era of genetic testing. BJOG. 2010;117:391–8. doi: 10.1111/j.1471-0528.2009.02470.x. [DOI] [PubMed] [Google Scholar]