

 Steven J. Malcolmson:
Steven J. Malcolmson: Abstract
We report a method for the catalytic, enantioselective intermolecular addition of aliphatic amines to acyclic 1,3-dienes. In most cases, reactions proceed efficiently at or below room temperature in the presence of 5 mol % of a Pd catalyst bearing a PHOX ligand, generating allylic amines in up to 97:3 er. The presence of an electron-deficient phosphine within the ligand not only leads to a more active catalyst but also is critical for achieving high site selectivity in the transformation.
Graphical abstract
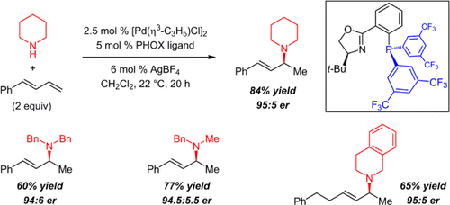
Hydrofunctionalization reactions of unsaturated hydrocarbons are highly atom-economical means of generating complex molecules from simple and readily accessible materials. Strategies to catalyze this broad class of transformations enantioselectively have been greatly sought after. Among this cohort, hydroamination reactions1,2 are prized as an expedient way to generate valuable chiral amines. Despite this, enantioselective intermolecular olefin hydroaminations are rare.
Until recently, successful enantioselective additions of amines have been limited to aryl amines (Scheme 1). In 2001, the Hartwig lab showed that anilines could be added to symmetrical cyclic dienes in the presence of a chiral Pd catalyst prepared from a Trost ligand.3–5 Although a Brønsted acid additive permits a shorter reaction time, it also leads to product racemization. Dong and co-workers demonstrated a Rh-catalyzed 1,2-hydroamination of unsymmetrical acyclic dienes utilizing indoline nucleophiles; in this case, an acid additive positively impacts enantioselectivity.6 Recently, the Hou lab disclosed the first highly enantioselective intermolecular hydroaminations with aliphatic amines, where chiral lanthanide half-sandwich catalysts allow for stereoselective amine additions to cyclopropenes.7–10 In this work, we report the enantioselective 1,2-addition of alkyl amines to unsymmetrical acyclic dienes11–13 catalyzed by a cationic Pd–PHOX complex.14 Transformations afford allylic amines15 in up to 91% yield in 3–24 h without added Brønsted acid. An electron-deficient phosphine enhances both catalyst reactivity and site selectivity for the 1,2-addition product.
Scheme 1.

Catalytic Enantioselective Intermolecular Diene Hydroamination Reactions
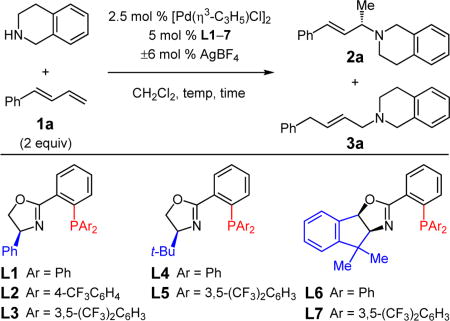
Previous work by the Hartwig group showed that Pd-catalyzed hydroamination of dienes proceeds via Pd–H migratory insertion to generate a Pd–π-allyl complex, followed by outer sphere amine attack.5 The resulting ammonium salt may then oxidatively protonate Pd to regenerate the metal hydride (Scheme 1).16 We therefore began by examining ligands known for promoting enantioselective nucleophilic additions to metal–π-allyl complexes for the hydroamination of 1-phenylbutadiene (1a, Table 1) with tetrahydroisoquinoline (THIQ). Unlike in previously reported aniline additions to cyclohexadiene,3 Trost ligands, in combination with [Pd(η3-C3H5)Cl]2, fail to promote hydroamination of 1a with THIQ. Other classes of bis(phosphine) ligands lead to more reactive catalysts; however, although they provide high selectivity for the desired 1,2-hydroamination product 2a, poor enantioselectivity is observed (<70:30 er).17
Table 1.
Reaction Optimization for Addition of Tetrahydroisoquinoline to 1-Phenylbutadienea
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | ligand | AgBF4 (Y/N) |
temp (°C); time (h) |
2a:3ab | yield (%)c |
erd |
| 1 | L1 | N | 22; 20 | 8.5:1 | 70 | 81:19 |
| 2 | L1 | Y | 22; 20 | 12:1 | 61e | 85:15 |
| 3 | L2 | Y | 22; 20 | 17:1 | 84 | 83:17 |
| 4 | L3 | Y | 22; 20 | >20:1 | 96 | 89:11 |
| 5 | L4 | N | 22; 20 | 4:1 | 63 | 68:32 |
| 6 | L4 | Y | 22; 20 | 2:1 | 76 | 92:8 |
| 7 | L5 | Y | 22; 20 | 19:1 | 89 | 94.5:5.5 |
| 8 | L5 | Y | 22; 0.5 | >20:1 | 79 | 95:5 |
| 9 | L5 | Y | 0; 3 | >20:1 | 89 | 97:3 |
| 10 | L6 | Y | 22; 20 | 6:1 | 87 | 94:6 |
| 11 | L7 | Y | 22; 20 | >20:1 | 64 | 95:5 |
| 12 | L7 | Y | 30; 20 | >20:1 | 79 | 94:6 |
Reactions run with 0.2 mmol tetrahydroisoquinoline (0.8 M). See the Supporting Information for experimental details.
Determined by 400 MHz 1H NMR spectroscopy of the unpurified mixture.
Isolated yield of a 2a/3a mixture unless otherwise noted.
Enantiomeric ratio determined by HPLC analysis of purified products in comparison with authentic racemic standards.
Isolated yield of 2a only.
We then turned our attention to phosphinooxazoline (PHOX) ligands, which have enjoyed a great deal of success in enantioselective allylic substitution,18 but to the best of our knowledge have not been employed in hydrofunctionalization reactions.14 We began by examining the Ph-substituted PHOX ligand L1 (Table 1, entry 1). After 20 h at room temperature with 5 mol % catalyst, a mixture of desired 1,2-hydroamination product 2a and achiral 1,4-hydroamination adduct 3a is generated (70% yield, 8.5:1 2a:3a). The desired allylic amine is formed in 81:19 er. The addition of AgBF4 to the catalyst mixture improves the product ratio and slightly increases the enantioselectivity as well (12:1 2a:3a, 85:15 er, entry 2).19 Switching to a more electron-poor phosphine (L2, entry 3) improves product yield20 and increases the proportion of 2a13l (84% yield, 17:1 site selectivity) without significant change to enantioselectivity. A bis[3,5-(CF3)2]-aryl phosphine (L3, entry 4) further increases yield and site selectivity and also offers enhanced enantioselectivity (89:11 er). Similar trends in site selectivity and enantioselectivity are observed with the t-Bu-substituted oxazolines (L4 and L5, entries 5–7). Once more, the more electron-deficient phosphine increases site selectivity and enantioselectivity (19:1 2a:3a, 94.5:5.5 er, entry 7) relative to the diphenylphosphino ligand (2:1 2a:3a, 92:8 er, entry 6). Ligand L5 provides dramatically increased reactivity as well, at room temperature (entry 8) affording 79% yield of 2a in 0.5 h. Cooling to 0 °C further enhances enantioselectivity: after 3 h, amine 2a is delivered in 89% yield and 97:3 er. Indane-derived PHOX ligands L6 and L7 were also explored (entries 10–12) and lead to similar selectivity trends albeit with overall reduced reactivity. Yield could be improved if the reaction were run at 30 °C without diminishing enantioselectivity (79% yield, 94:6 er, entry 12), but a further increase negatively impacts enantioselectivity.
A number of 1-substituted dienes take part in enantioselective addition of THIQ (Table 2).21 Electronic variation at the arene’s para-position of aryl-substituted butadienes was investigated and proved to have a profound effect on site selectivity (allylic amines 2b–f). More electron-rich substituents lead to high selectivity for 1,2-addition product 2.13l For example, dimethylamino-containing 2b and methoxy-substituted 2c are formed as the exclusive product of the reaction, whereas nitroarene 1f leads to a 3:1 mixture of site isomers. In contrast, enantioselectivity is largely unchanged (91.5:8.5 to 96:4 er). In cases where a mixture is obtained, the two product isomers are separable. Methyl substitution at the meta-position maintains high site selectivity (>20:1) for allylic amine 2g. In comparison, the ortho-tolyl 2h is formed with only 80% selectivity,13l although enantioselectivity is the same in both cases (91:9 er); neither parameter in forming 2h is improved upon cooling to 0 °C. Alkyl-substituted dienes efficiently undergo hydroamination to deliver chiral amines 2i–k as the exclusive product in 89:11–95:5 er. In all cases, olefin migration is not observed.
Table 2.

|
See the Supporting Information for experimental details.
Site-isomer ratio determined by 400 MHz 1H NMR spectroscopy of the unpurified mixture.
Isolated yield of 2 only unless otherwise noted.
Enantiomeric ratio determined by HPLC analysis of purified products in comparison with authentic racemic standards.
Reaction for 9 h.
Reaction at 22 °C.
Isolated yield of a mixture of 2 and 3.
Several aliphatic amines participate in hydroamination of phenylbutadiene 1a with Pd–PHOX catalysts, although the optimal reaction conditions vary considerably depending on the nature of the amine (Table 3). Like THIQ, cyclic amines with inductively electron-withdrawing groups (e.g., morpholine and N-Boc-protected piperazine) are able to furnish allylic amine products at 0 °C with the L5-derived catalyst: 2l and 2m are obtained with ca. 92% site selectivity and 96:4 er. As the cyclic amines become more donating, higher temperature is required to promote reaction, presumably because of increasing competition with the diene for coordination to Pd.1 Piperidine forms allylic amine 2n in 84% yield at room temperature (95:5 er). Pyrrolidine, a challenging amine for many late transition metal catalysts, affords 2o in 91:9 er and 59% yield. Acyclic secondary amines are also competent partners for the diene hydroamination, delivering amines 2p–s as a single site-isomer with good enantioselectivity (88.5:11.5–94.5:5.5 er). Sterically hindered dibenzylamine affords 2q in 94:6 er with higher efficiency with indane-derived ligand L7 at 30 °C.22 Primary alkyl amines undergo a single hydroamination event: secondary amine 2t is generated in 48% yield (94:6 er). Aryl amines are also effective nucleophiles, delivering allylic amines 2u–v in up to 96.5:3.5 er.
Table 3.
Amine Variation in the Hydroamination of 1-Phenylbutadiene.a–d

|
See Table 2.
Isolated as a mixture of 2 and 3; see the Supporting Information.
Ligand L7 used.
Lower yield due to product volatility.
Initial mechanistic studies demonstrate that site selectivity in the reaction is strongly dependent upon diene stereochemistry and isotope effects (Scheme 2). (Z)-Phenylbutadiene (Z)-1a (ca. 88% Z), when combined with morpholine and the Pd–L5 complex under the optimal conditions for (E)-1a (Table 3), yields a dramatically higher proportion of the 1,4-hydroamination product (1:2 2l:3l) compared to reaction of the (E)-isomer, perhaps due to styrenyl olefin insertion now being significantly kinetically preferred. Both hydroamination products are formed exclusively as the (E)-olefin isomer, signifying that olefin isomerization is faster than amine attack in forming 2l. Excess diene 1a is recovered with ca. 82% Z content. Furthermore, 2l is formed as the same enantiomer from (Z)-1a as from the (E)-isomer.
Scheme 2.

Initial Mechanistic Investigations of Pd–PHOX-Catalyzed Diene Hydroaminations
To determine the extent to which the diene/Pd–H insertion might be reversible, reaction of N-deuterated THIQ with diene 1f was examined. At room temperature, the reaction of unlabeled THIQ with 1f generates an 8:1 mixture of 1,2- and 1,4-hydroamination products with L5.17 Unexpectedly, N-deuterated THIQ significantly lowers the site selectivity to 2:1 d-2f:d-3f (Scheme 2). The deuterium distribution in each product, however, demonstrates that the site of migratory insertion largely controls which product forms as indicated by the relatively smaller amount of deuterium incorporation into C1 in d-2f and C4 in d-3f. Whether this analysis extends to unlabeled amines with L5 is ambiguous due to the isotope effect in product distribution. Recovered diene 2f has ca. 12% D-incorporation into the C1 position but <2% at C4.
A Pd complex, bearing an electronically dissymmetric chiral PHOX ligand, is an efficient and highly enantioselective catalyst for the intermolecular hydroamination of dienes with aliphatic amines. Diene and ligand electronics have a dramatic effect on product distribution, and a more electron-deficient phosphine ligand significantly increases catalyst reactivity. Current studies are focused on further elucidating the reaction mechanism and related methods development.
Supplementary Material
Acknowledgments
We are grateful to Duke University for sponsoring this research. N.J.A. was supported by NIGMS (T32GM007105-42). We thank Katherine Wilbur for experimental assistance.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and analytical data for new compounds (PDF)Ligand and product spectral data (PDF)
The authors declare no competing financial interest.
References
- 1.For reviews, see: Reznichenko AL, Nawara-Hultzsch AJ, Hultzsch KC. Top. Curr. Chem. 2013;343:191. doi: 10.1007/128_2013_500.Huang L, Arndt M, Gooßen K, Heydt H, Gooßen LJ. Chem. Rev. 2015;115:2596. doi: 10.1021/cr300389u.Coman SM, Parvulescu VI. Org. Process Res. Dev. 2015;19:1327.
- 2.For reviews on Cu-catalyzed umpolung approaches to hydroamination of unsaturated hydrocarbons, see: Hirano K, Miura M. Pure Appl. Chem. 2014;86:291.Pirnot MT, Wang Y-M, Buchwald SL. Angew. Chem. Int. Ed. 2016;55:48. doi: 10.1002/anie.201507594.
- 3.Löber O, Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 2001;123:4366. doi: 10.1021/ja005881o. [DOI] [PubMed] [Google Scholar]
- 4.For enantioselective Pd-catalyzed aniline additions to styrenes, see: Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 2000;122:9546.Hu A, Ogasawara M, Sakamoto T, Okada A, Nakajima K, Takahashi T, Lin W. Adv. Synth. Catal. 2006;348:2051.
- 5.For mechanistic studies of Pd-catalyzed styrene and diene hydroamination with anilines, see: Nettekoven U, Hartwig JF. J. Am. Chem. Soc. 2002;124:1166. doi: 10.1021/ja017521m.Johns AM, Utsunomiya M, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2006;128:1828. doi: 10.1021/ja056003z. For a study with a Ni catalyst and alkyl amines, see: Pawlas J, Nakao Y, Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 2002;124:3669. doi: 10.1021/ja017575w.
- 6.Yang X-H, Dong VM. J. Am. Chem. Soc. 2017;139:1774. doi: 10.1021/jacs.6b12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teng H-L, Luo Y, Wang B, Zhang L, Nishiura M, Hou Z. Angew. Chem. Int. Ed. 2016;55:15406. doi: 10.1002/anie.201609853. [DOI] [PubMed] [Google Scholar]
- 8.For other intermolecular enantioselective hydroaminations with alkyl amines, see: Utsunomiya M, Hartwig JF. J. Am. Chem. Soc. 2003;125:14286. doi: 10.1021/ja0375535.Reznichenko AL, Nguyen HN, Hultzsch KC. Angew. Chem. Int. Ed. 2010;49:8984. doi: 10.1002/anie.201004570.
- 9.For intermolecular enantioselective hydroaminations with N-nucleophiles other than amines, see: Zhang Z, Lee SD, Widenhoefer RA. J. Am. Chem. Soc. 2009;131:5372. doi: 10.1021/ja9001162.Sevov CS, Zhou JS, Hartwig JF. J. Am. Chem. Soc. 2012;134:11960. doi: 10.1021/ja3052848.
- 10.For other nonenantioselective intermolecular hydroaminations with alkyl amines, see: Ickes AR, Ensign SC, Hull KL, Gupta AK. J. Am. Chem. Soc. 2014;136:11256. doi: 10.1021/ja505794u.Ensign SC, Vanable EP, Kortman GD, Weir LJ, Hull KL. J. Am. Chem. Soc. 2015;137:13748. doi: 10.1021/jacs.5b08500.Musacchio AJ, Lainhart BC, Zhang X, Naguib SG, Sherwood TC, Knowles RR. Science. 2017;355:727. doi: 10.1126/science.aal3010.
- 11.For non-enantioselective intermolecular hydroamination of this class of dienes with anilines and alkyl amines, see: Minami T, Okamoto H, Ikeda S, Tanaka R, Ozawa F, Yoshifuji M. Angew. Chem. Int. Ed. 2001;40:4501. doi: 10.1002/1521-3773(20011203)40:23<4501::aid-anie4501>3.0.co;2-k.Goldfogel MJ, Roberts CC, Meek SJ. J. Am. Chem. Soc. 2014;136:6227. doi: 10.1021/ja502275w.Banerjee D, Junge K, Beller M. Org. Chem. Front. 2014;1:368.
- 12.For intramolecular enantioselective hydroamination of dienes, see: Hong S, Kawaoka AM, Marks TJ. J. Am. Chem. Soc. 2003;125:15878. doi: 10.1021/ja036266y.Deschamp J, Olier C, Schulz E, Guillot R, Hannedouche J, Collin J. Adv. Synth. Catal. 2010;352:2171.Shapiro ND, Rauniyar V, Hamilton GL, Wu J, Toste FD. Nature. 2011;470:245. doi: 10.1038/nature09723.Kanno O, Kuriyama W, Wang ZJ, Toste FD. Angew. Chem. Int. Ed. 2011;50:9919. doi: 10.1002/anie.201104076.
- 13.For select examples of other catalytic enantioselective intermolecular reactions of acyclic 1,3-dienes, see: Shirakura M, Suginome M. Angew. Chem. Int. Ed. 2010;49:3827. doi: 10.1002/anie.201001188.Zbieg JR, Moran J, Krische MJ. J. Am. Chem. Soc. 2011;133:10582. doi: 10.1021/ja2046028.Schuster CH, Li B, Morken JP. Angew. Chem. Int. Ed. 2011;50:7906. doi: 10.1002/anie.201102404.Watkins AL, Landis CR. Org. Lett. 2011;13:164. doi: 10.1021/ol102797t.Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274.Kliman LT, Mlynarski SN, Ferris GE, Morken JP. Angew. Chem. Int. Ed. 2012;51:521. doi: 10.1002/anie.201105716.McInturff EL, Yamaguchi E, Krische MJ. J. Am. Chem. Soc. 2012;134:20628. doi: 10.1021/ja311208a.Ebe Y, Nishimura T. J. Am. Chem. Soc. 2014;136:9284. doi: 10.1021/ja504990a.Stokes BJ, Liao L, de Andrade AM, Wang Q, Sigman MS. Org. Lett. 2014;16:4666. doi: 10.1021/ol502279u.Timsina YN, Sharma RK, RajanBabu TV. Chem. Sci. 2015;6:3994. doi: 10.1039/c5sc00929d.Wu X, Lin H-C, Li M-L, Li L-L, Han Z-Y, Gong L-Z. J. Am. Chem. Soc. 2015;137:13476. doi: 10.1021/jacs.5b08734.Liu Y, Xie Y, Wang H, Huang H. J. Am. Chem. Soc. 2016;138:4314. doi: 10.1021/jacs.6b00976.
- 14.For Pd-catalyzed hydroamination with achiral P,N-ligands, see: Shaffer AR, Schmidt JAR. Organometallics. 2008;27:1259.Kuchenbeiser G, Shaffer AR, Zingales NC, Beck JF, Schmidt JAR. J. Organomet. Chem. 2011;696:179.Beck JF, Samblanet DC, Schmidt JAR. RSC Adv. 2013;3:20708.
- 15.For intermolecular enantioselective hydroamination of alkynes to generate allylic amines, see: Chen Q-A, Chen Z, Dong VM. J. Am. Chem. Soc. 2015;137:8392. doi: 10.1021/jacs.5b05200. For intermolecular enantioselective hydroamination of allenes to generate allylic amines, see: Butler KL, Tragni M, Widenhoefer RA. Angew. Chem. Int. Ed. 2012;51:5175. doi: 10.1002/anie.201201584.Kim H, Lim W, Im D, Kim D-g, Rhee YH. Angew. Chem. Int. Ed. 2012;51:12055. doi: 10.1002/anie.201206967. For a review concerning other examples, see: Koschker P, Breit B. Acc. Chem. Res. 2016;49:1524. doi: 10.1021/acs.accounts.6b00252.
- 16.Amine attack upon the original η3-allyl Pd salt generates an ammonium salt that oxidatively protonates Pd, initiating catalysis.
- 17.See the Supporting Information for details.
- 18.For reviews, see: Helmchen G, Pfaltz A. Acc. Chem. Res. 2000;33:336. doi: 10.1021/ar9900865.Bausch CC, Pfaltz A. PHOX Ligands. In: Zhou Q-L, editor. Privileged Chiral Ligands and Catalysts. Wiley-VCH; Weinheim, Germany: 2011. Chapter 6.Liu Y, Han S-J, Liu W-B, Stoltz BM. Acc. Chem. Res. 2015;48:740. doi: 10.1021/ar5004658.
- 19.Other Ag salts afford greater quantities of achiral 3a, although the er of 2a is largely invariant. Additionally, polar solvents lead to greater product yields, with CH2Cl2 proving optimal.
- 20.Tani K, Behenna DC, McFadden RM, Stoltz BM. Org. Lett. 2007;9:2529. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- 21.1,4-Disubstituted dienes fail to generate hydroamination product. Other substitution patterns deliver 1,4-hydroamination or a complex mixture. See the Supporting Information.
- 22.Product 2q is obtained in 53% yield, 12:1 2q:3q, and 93:7 er after 17 h at 0 °C with L5.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.