


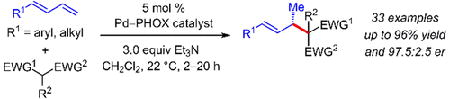
Abstract
We report a highly enantioselective Pd−PHOX-catalyzed intermolecular hydroalkylation of acyclic 1,3-dienes. Meldrum’s acid derivatives and other activated C-pronucleophiles, such as β-diketones and malononitriles, react with a variety of aryl- and alkyl-substituted dienes in ≤20 h at room temperature. The coupled products, obtained in up to 96% yield and 97.5:2.5 er, are easily transformed into useful chemical building blocks for downstream synthesis.
Graphical abstract
The discovery of new methods for the enantioselective construction of C−C bonds is a critical objective in chemical synthesis, especially if novel transformations additionally offer access to new or expanded chemical space. Catalytic protocols are particularly desirable. A steadfast approach in C−C bond formation involves the enantioselective addition of polarized electrophiles to preformed enolates or their analogs. Alternatively, the direct addition of enolate precursors (pronucleophiles) to polarized reaction partners have also been developed. Aldol and Mannich reactions,1 conjugate additions,1a,b,e,2 allylic substitutions,3 and alkylation with alkyl halides4 comprise several examples.
Far less common are catalytic enantioselective additions of enols/enolates to simple unsaturated hydrocarbons. The Trost5 and Luo6 laboratories have reported Pd-catalyzed transformations involving terminal allenes. The Breit group has illustrated Rh-catalyzed reactions with 1,1-disubstituted allenes7,8 and Dong and co-workers have described Rh-catalyzed reactions of methyl-substituted alkynes (Scheme 1).9,10 These transformations involve the intermediacy of a terminal metal−π-allyl complex. The resulting products contain a terminal olefin with an allylic stereogenic center or internal olefins with a homoallylic stereogenic center. Additionally, the Hartwig group has demonstrated Pd-catalyzed additions of two β-diketones to cyclohexadiene and 2,3-dimethylbutadiene.11–14
Scheme 1.

Previous and Present Work in Intermolecular Enantioselective Hydroalkylation
Our lab has recently disclosed highly enantioselective and efficient intermolecular additions of aliphatic amines to 1,3- dienes.15 The reactions, promoted by an electron deficient Pd−PHOX catalyst, proceed via a 1,3-disubstituted Pd−π-allyl intermediate to generate myriad allylic amines. Herein, we demonstrate that Pd−PHOX catalysts permit the addition of a variety of activated C-pronucleophiles to several aryl- or alkyl-substituted acyclic dienes (Scheme 1). Reactions take place within 20 h at room temperature to generate products bearing internal olefins with allylic stereogenic centers by the formal addition of an enol across the diene’s terminal olefin. Several transformations of the carbonyls and/or the olefins within the coupled products highlight the synthetic utility of the process.
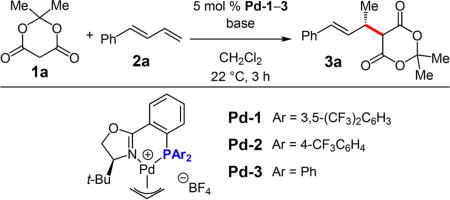
We began by examining the coupling of Meldrum’s acid 1a and phenylbutadiene 2a under previously established conditions for hydroamination (Table 1); however, none of the desired addition product 3a was observed with Pd-1 (entry 1). Reasoning that an ammonium salt might be needed as the acid source for Pd−H formation within the catalytic cycle, we explored the addition of amine base additives, which upon deprotonating 1a would generate the corresponding ammonium enolate. Pleasingly, with Et3N (2.0 equiv), 3a is formed as the sole site isomer in 72% yield and 96.5:3.5 er (entry 2). Hünig’s base (entry 3) offers identical enantioselectivity but higher yield of 3a than Et3N. DABCO also shows improved product yield and similar selectivity (entry 4). However, both bases consistently lead to lower product yields with other nucleophile classes.16 DBU is ineffective (entry 5). We therefore chose to pursue Et3N as the base of choice due to its generality. As little as 5 mol % Et3N generates 3a, but increasing the quantity of the base raises the reaction yield and enantioselectivity (entries 6−8). By introducing 1.5 equiv 1a with 3.0 equiv Et3N and increasing the reaction time to 15 h, 3a was isolated in 81% yield and 97.5:2.5 er (entry 9). Neither lower temperature nor electronically modified phosphines (Pd-2–3) were able to improve upon this result (entries 10–13).17
Table 1.
Reaction Optimization for Meldrum’s Acid Addition to Phenylbutadienea
| |||||
|---|---|---|---|---|---|
| entry | catalyst | 1a (equiv) | base (equiv) | yield (%)b | erc |
| 1 | Pd-1 | 1.1 | none | <2 | – |
| 2 | Pd-1 | 1.1 | Et3N (2.0) | 72 | 96.5:3.5 |
| 3 | Pd-1 | 1.1 | i-Pr2NEt (2.0) | 82 | 96.5:3.5 |
| 4 | Pd-1 | 1.1 | DABCO (2.0) | 88 | 95:5 |
| 5 | Pd-1 | 1.1 | DBU (2.0) | <2 | – |
| 6 | Pd-1 | 1.1 | Et3N (0.05) | 46 | 94:6 |
| 7 | Pd-1 | 1.1 | Et3N (0.50) | 51 | 95:5 |
| 8 | Pd-1 | 1.1 | Et3N (3.0) | 72 | 98:2 |
| 9d | Pd-1 | 1.5 | Et3N (3.0) | 81 | 97.5:2.5 |
| 10d,e | Pd-1 | 1.5 | Et3N (3.0) | 84 | 97.5:2.5 |
| 11 | Pd-2 | 1.5 | Et3N (3.0) | 95 | 93:7 |
| 12e,f | Pd-2 | 1.5 | Et3N (3.0) | 89 | 96:4 |
| 13d | Pd-3 | 1.5 | Et3N (3.0) | 80 | 94:6 |
Reactions run with 0.2 mmol 2a in 0.25 mL CH2Cl2.
Isolated yield of 3a.
Enantiomeric ratio determined by HPLC analysis of purified products.
15 h reaction.
0 °C reaction.
16 h reaction.
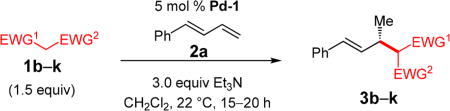
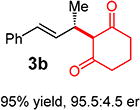
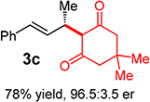
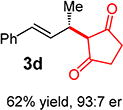
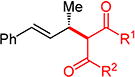
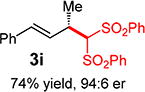
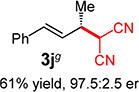
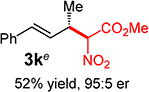
With conditions determined for enantioselective hydroalkylation of 2a, we next sought to discover the range of pronucleophiles that were amenable to the coupling (Table 2). Cyclic β-diketones5b efficiently undergo addition to phenylbutadiene to afford adducts 3b–d in up to 95% yield and 96.5:3.5 er. Acyclic diketones5b,7 also participate, delivering diones 3e–h in 66–96% yield and 90.5:9.5 to 92:8 er and illustrating that both alkyl and aryl ketones are competent partners. Bis(sulfones) (3i), malononitrile (3j), and α-nitroesters (3k) all take part in diene hydroalkylation reactions. Dimethylmalonate (pKa in DMSO = 15.9),18a however, fails to add to phenylbutadiene with the present catalytic system, suggesting an upper limit in pronucleophile acidity to between 14.2 (benzoylacetone 2h)18b and 15.9. It should also be noted that products 3h and 3k, formed by the addition of prochiral nucleophiles to 2a, are generated as a 1:1 mixture of diastereomers at the nucleophilic carbon; however, both stereoisomers are obtained with identical enantioselectivity in each case. In all cases, addition occurs across the diene’s terminal olefin to form the illustrated product exclusively; the site selectivity is likely at least somewhat attributable to the PHOX ligand.12b
Table 2.
Addition of Unsubstituted Pronucleophiles to Phenylbutadienea
| |||
|---|---|---|---|
|
|
|
|
|
| |||
|
3ed | R1, R2 = Me: | 90% yield, 92:8 er |
| 3f | R1, R2 = Ph: | 96% yield, 91.5:8.5 er | |
| 3g | R1, R2 = 4-MeOC6H4: | 70% yield, 90.5:9.5 er | |
| 3he,f | R1 = Me, R2 = Ph: | 66% yield, 90.5:9.5 er | |
|
| |||
|
|
|
|
See the Supporting Information for experimental details.
Isolated yield of 3.
Enantiomeric ratio determined by HPLC analysis of purified products.
BArF4 counterion used in place of BF4 for Pd-1.
1:1 dr at carbonyls’ α-position; both diastereomers have the same er.
2.0 mmol scale reaction.
2 h reaction; ca. 20% double alkylation product.
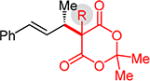
Substituted Meldrum’s acid derivatives5a,b and malononitriles also participate in coupling with phenylbutadiene to deliver products that contain quaternary centers adjacent to the allylic stereogenic center (Table 3). The former’s products 3l–o are generated with modest efficiency but good enantioselectivity. These sterically congested pronucleophiles afford ca. 5% 1,4-addition product16 with C–C bond formation occurring at the terminus of the diene. The enantioselectivity dependence on Et3N equivalents is magnified in several cases with the Meldrum’s acids 1l–o compared to unsubstituted 1a: with fewer equivalents of Et3N, lower enantioselectivity is obtained, becoming even lower with longer reaction times.16 In contrast, enantioselectivity is largely constant over the course of the reaction with 3.0 equiv Et3N, suggesting less reaction reversibility under the optimized conditions.
Table 3.
Quaternary Center Formation by Addition of Substituted Pronucleophiles to Phenylbutadienea–c
| |||
|---|---|---|---|
|
3ld,e | R = Me: | 48% yield, 94:6 er |
| 3md,e | R = Et: | 37% yield, 91:9 er | |
| 3nd,e | R = n-Bu: | 33% yield, 91:9 er | |
| 3od,e | R = i-Pr: | 29% yield, 88:12 er | |
|
| |||
|
3pf | R= Me: | 90% yield, 95:5 er |
| 3qf | R = n-Bu: | 79% yield, 96:4 er | |
| 3rf | R = Bn: | >98% yield, 94.5:5.5 er | |
|
|
81% yield, 97.5:2.5 er | ||
|
| |||
|
3tf | 80% yield, 1.5:1 dr, 10% 1,4-addition | |
| major: 94.5:5.5 er | |||
| minor: 86:14 er | |||
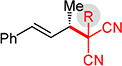
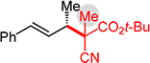
Conversely, the less hindered substituted malononitriles couple with 2a to deliver products 3p–s in 77 to >98% yield within 2 h. In general, enantioselectivity is enhanced and the reactions show perfect site selectivity. Notably, unlike with Pd–bis(phosphine) catalysts, deallylation of malononitrile 1s, which ultimately yields a statistical distribution of allylation products, does not occur with Pd-1. Prochiral pronucleophiles, such as tert-butyl cyanopropionate 1t, also effectively add to diene 1a but with little stereocontrol at the nucleophile’s carbon (1.5:1 dr in forming 3t); enantioselectivity of each diastereomer is substantial but unequal. Approximately 10% 1,4-addition accompanies the major product.
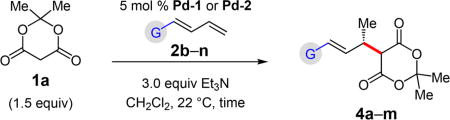
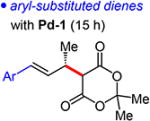
Several dienes have been examined for additions of Meldrum’s acid (Table 4). Aryl-substituted dienes lead to styrenyl products 4a–d in good yields (66–78%) within 15 h with Pd-1 (96:4–97:3 er); however, an o-methyl group significantly slows the reaction (28% yield of 4e; 97:3 er).19 A furyl-substituted diene affords 4f in 54% yield and 93:7 er. Just as in Pd–PHOX-catalyzed hydroaminations of these dienes,15 the reaction is fastest with electron rich substrates, but unlike in amine–diene couplings, electron deficient or sterically hindered aromatic rings do not lead to 1,4-addition.
Table 4.
Meldrum’s Acid Addition to Various Aryl- and Alkyl-Substituted Dienesa–c
| |||
|---|---|---|---|
|
4a | Ar = 4-MeOC6H4 | 78% yield, 97:3 er |
| 4b | Ar = 4-CIC6H4 | 70% yield, 96.5:3.5 er | |
| 4c | Ar = 4-F3CC6H4: | 66% yield, 96:4 er | |
| 4d | Ar = 3-MeC6H4: | 75% yield, 96.5:3.5 er | |
| 4e | Ar = 2-MeC6H4 | 28% yield, 97:3 er | |
| 4f | Ar = 2-furyl: | 54% yield, 93:7 er | |
|
| |||
|
4g R = n-Bu: | 68% yield, 95:5 er | |
| 4h R = Ph: | 89% yield, 93.5:6.5 er | ||
| 4i R = OTBS: | 76% yield, 95:5 er | ||
| 4j R = OH: | 70% yield, 95.5:4.5 er | ||
| 4kd R = NPhth: | 85% yield, 95:5 er | ||
|
| |||
See Table 2.
1.0 equiv Meldrum’s acid and 1.1 equiv diene 2l.
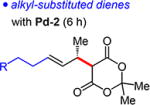
Alkyl-substituted dienes react sluggishly with Meldrum’s acid when Pd-1 is employed (ca. 50% yield in 15 h). Contrastingly, with the sterically less hindered Pd-2, reactions are complete within 6 h, affording unsaturated Meldrum’s acids 4g–m in 68–89% yield (Table 4). Unlike in reactions of aryl-substituted dienes, Pd-2 is equally as enantioselective as Pd-1 (93:7 to 95:5 er). Several functional groups are tolerated, including ethers (4i), imides (4k), and even free alcohols (4j,l).
The Meldrum’s acid addition products provide a useful platform for accessing a number of β-methyl-γ,δ-unsaturated carbonyls. Ethanol addition to 3a, prepared on 4.0 mmol scale, generates carboxylic ester 5a in 96% yield (Scheme 2A).20 Similarly, N-hydroxyphthalimide ester21 5b (91% yield), carboxylic acid 5c (79% yield), and Weinreb amide 5d (74% yield) may be obtained. Products resulting from the addition of the unsubstituted malononitrile (e.g., 3j, Scheme 2B) may undergo oxidation with MMPP and conversion to the methyl ester to furnish 6 in 63% yield,22 which now bears a stereogenic center at the carbonyl’s α-position. The transformation takes place with minimal erosion of enantiopurity. Additionally, the β-diketone scaffold may be utilized to generate heterocycles with α-stereogenic centers.7,10c For example, hydroxylamine condensation with diketone 3h affords isoxazole 7 in 84% yield (Scheme 2C). The transformation significantly favors initial amine attack upon the alkyl ketone (13:1 regioselectivity)16 and ameliorates the lack of stereochemical control at the carbonyl’s α-position in the hydroalkylation reaction by erasing the stereochemistry at the offending center.
Scheme 2.

Carbonyl Functionalization within Coupled Products
aReaction at 80 °C.
The presence of both carbonyl and olefin functionality within the products may be leveraged to build molecular complexity quickly. The allylic hydroxyl group of 4l may be selectively acylated and subjected to Pd-catalyzed allylic substitution in the presence of ethanol,23 yielding γ-lactone 8 in 66% yield as a 7:1:1 mixture of diastereomers with the major isomer shown (Scheme 3A).16 Additionally, Sharpless dihydroxylation of unsaturated ester 5a with AD-mix β leads to spontaneous lactonization to afford γ-lactone 9 as the sole product of the reaction (70% yield, 19:1 dr, Scheme 3B).16
Scheme 3.

Simultaneous Carbonyl and Olefin Derivatization within Hydroalkylation Products
Highly efficient and enantioselective intermolecular addition of activated C-pronucleophiles to acyclic dienes is enabled by Pd catalysts bearing electron deficient phosphines within a PHOX ligand scaffold. A range of aryl- and alkyl-substituted dienes may be coupled with a number of β-dicarbonyl-like nucleophiles to generate allylic stereogenic centers at the carbonyl’s β-position. The olefin and carbonyl functional groups provide handles for subsequent complexity-building product elaboration and access to useful synthetic motifs. Application of Pd–PHOX catalysts to other enantioselective hydrofunctionalizations is underway.
Supplementary Material
Acknowledgments
We are grateful to Duke University for sponsoring this research. N.J.A. was supported by NIGMS (T32GM007105-42). K.C.E.W. thanks the Duke Chemistry Department for a summer research fellowship. NMR spectroscopic assistance was provided by Dr. Benjamin Bobay and the Duke NMR center. We thank Prof. Alex Grenning (University of Florida) for suggesting the malononitrile oxidation to methyl ester 6.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b13300.
Experimental procedures and analytical data for new compounds (PDF)
NMR spectra (PDF)
The authors declare no competing financial interest.
References
- 1.For reviews, see: Johnson JS, Evans Da. Acc. Chem. Res. 2000;33:325. doi: 10.1021/ar960062n.Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471. doi: 10.1021/cr0684016.Kobayashi S, Mori Y, Fossey JS, Salter MM. Chem. Rev. 2011;111:2626. doi: 10.1021/cr100204f.Beutner GL, Denmark SE. Angew. Chem., Int. Ed. 2013;52:9086. doi: 10.1002/anie.201302084.Pellissier H. Chem. Rev. 2016;116:14868. doi: 10.1021/acs.chemrev.6b00639.
- 2.For reviews, see: Cohen DT, Scheidt KA. Chem. Sci. 2012;3:53. doi: 10.1039/C1SC00621E.Hui C, Pu F, Xu J. Chem. - Eur. J. 2017;23:4023. doi: 10.1002/chem.201604110.
- 3.For reviews, see: Helmchen G, Pfaltz A. Acc. Chem. Res. 2000;33:336. doi: 10.1021/ar9900865.Trost BM, Machacek MR, Aponick A. Acc. Chem. Res. 2006;39:747. doi: 10.1021/ar040063c.
- 4.For select examples, see: Ooi T, Miki T, Taniguchi M, Shiraishi M, Takeuchi M, Maruoka K. Angew. Chem., Int. Ed. 2003;42:3796. doi: 10.1002/anie.200351469.Hong S, Lee J, Kim M, Park Y, Park C, Kim M-h, Jew S-s, Park H-g. J. Am. Chem. Soc. 2011;133:4924. doi: 10.1021/ja110349a.Kanemitsu T, Koga S, Nagano D, Miyazaki M, Nagata K, Itoh T. ACS Catal. 2011;1:1331.Kano T, Hayashi Y, Maruoka K. J. Am. Chem. Soc. 2013;135:7134. doi: 10.1021/ja403340r.Teng B, Chen W, Dong S, Kee CW, Gandamana DA, Zong L, Tan C-H. J. Am. Chem. Soc. 2016;138:9935. doi: 10.1021/jacs.6b05053.
- 5.(a) Trost BM, Jäkel C, Plietker B. J. Am. Chem. Soc. 2003;125:4438. doi: 10.1021/ja029190z. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Simas ABC, Plietker B, Jäkel C, Xie J. Chem. - Eur. J. 2005;11:7075. doi: 10.1002/chem.200500826. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Xie J, Sieber JD. J. Am. Chem. Soc. 2011;133:20611. doi: 10.1021/ja209244m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou H, Wang Y, Zhang L, Cai M, Luo S. J. Am. Chem. Soc. 2017;139:3631. doi: 10.1021/jacs.7b00437. [DOI] [PubMed] [Google Scholar]
- 7.Beck TM, Breit B. Angew. Chem., Int. Ed. 2017;56:1903. doi: 10.1002/anie.201610577. [DOI] [PubMed] [Google Scholar]
- 8.For nonenantioselective intermolecular hydroalkylation of allenes, see: Yamamoto Y, Al-Masum M, Asao N. J. Am. Chem. Soc. 1994;116:6019.Trost BM, Gerusz VJ. J. Am. Chem. Soc. 1995;117:5156.Li C, Breit B. J. Am. Chem. Soc. 2014;136:862. doi: 10.1021/ja411397g.
- 9.Cruz FA, Dong VM. J. Am. Chem. Soc. 2017;139:1029. doi: 10.1021/jacs.6b10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For nonenantioselective intermolecular hydroalkylation of alkynes, see: Cruz FA, Chen Z, Kurtoic SI, Dong VM. Chem. Commun. 2016;52:5836. doi: 10.1039/c6cc02522f.Li C, Grugel C, Breit B. Chem. Commun. 2016;52:5840. doi: 10.1039/c6cc02272c.Beck T, Breit B. Org. Lett. 2016;18:124. doi: 10.1021/acs.orglett.5b03391.
- 11.Leitner A, Larsen J, Steffens C, Hartwig JF. J. Org. Chem. 2004;69:7552. doi: 10.1021/jo0490999. [DOI] [PubMed] [Google Scholar]
- 12.For nonenantioselective intermolecular hydroalkylation of dienes, see: Takahashi K, Miyake A, Hata G. Bull. Chem. Soc. Jpn. 1972;45:1183.Trost BM, Zhi L. Tetrahedron Lett. 1992;33:1831.
- 13.For enantioselective intermolecular hydroarylation reactions, see: Bexrud J, Lautens M. Org. Lett. 2010;12:3160. doi: 10.1021/ol101067d.Pattison G, Piraux G, Lam HW. J. Am. Chem. Soc. 2010;132:14373. doi: 10.1021/ja106809p.Podhajsky SM, Iwai Y, Cook-Sneathen A, Sigman MS. Tetrahedron. 2011;67:4435. doi: 10.1016/j.tet.2011.02.027.Saxena A, Lam HW. Chem. Sci. 2011;2:2326.So CM, Kume S, Hayashi T. J. Am. Chem. Soc. 2013;135:10990. doi: 10.1021/ja406169s.Friis SD, Pirnot MT, Buchwald SL. J. Am. Chem. Soc. 2016;138:8372. doi: 10.1021/jacs.6b04566.Cruz FA, Zhu Y, Tercenio QD, Shen Z, Dong VM. J. Am. Chem. Soc. 2017;139:10641. doi: 10.1021/jacs.7b05893.Marcum JS, Roberts CC, Manan RS, Cervarich TN, Meek SJ. J. Am. Chem. Soc. 2017;139:15580. doi: 10.1021/jacs.7b08575.
- 14.For a related transformation, see: Wu X, Lin H-C, Li M-L, Li L-L, Han Z-Y, Gong L-Z. J. Am. Chem. Soc. 2015;137:13476. doi: 10.1021/jacs.5b08734.
- 15.Adamson NJ, Hull E, Malcolmson SJ. J. Am. Chem. Soc. 2017;139:7180. doi: 10.1021/jacs.7b03480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For details, see the Supporting Information.
- 17.Dienes with other substitution patterns, such as isoprene and 2,3-dimethylbutadiene, fail to undergo reaction with Meldrum’s acid.
- 18.(a) Arnett EM, Maroldo SG, Schilling SL, Harrelson JA. J. Am. Chem. Soc. 1984;106:6759. [Google Scholar]; (b) Bordwell FG, Harrelson JA., Jr Can. J. Chem. 1990;68:1714. [Google Scholar]
- 19.Product 4e is generated in 82% yield and 82.5:17.5 er with Pd-2.
- 20.Adapted from: Sato M, Ban H, Kaneko C. Tetrahedron Lett. 1997;38:6689.
- 21.For potential applications, see: Toriyama F, Cornella J, Wimmer L, Chen T-G, Dixon DD, Creech G, Baran PS. J. Am Chem. Soc. 2016;138:11132. doi: 10.1021/jacs.6b07172.Li C, Wang J, Barton LM, Yu S, Tian M, Peters DS, Kumar M, Yu AW, Johnson KA, Chatterjee AK, Yan M, Baran PS. Science. 2017;356 doi: 10.1126/science.aam7355. No. eaam7355.
- 22.Adapted from: Förster S, Tverskoy O, Helmchen G. Synlett. 2008:2803.
- 23.Adapted from: Fillion E, Carret S, Mercier LG, Trépanier VÉ. Org. Lett. 2008;10:437. doi: 10.1021/ol702613f.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.