

Abstract
Osteosarcomas are the most frequent form of primary bone tumors and mainly affect children, adolescents, and young adults. Despite encouraging progress in therapeutic management, including the advent of multidrug chemotherapy, the survival rates have remained unchanged for more than four decades: 75% at 5 years for localized disease, but two groups of patients are still at high risk: metastatic at diagnosis (overall survival around 40% at 5 years) and/or poor responders to chemotherapy (20% at 5 years). Because these tumors are classified as “complex genomic,” it is extremely difficult to determine the signaling pathways that might be targeted by specific therapies. A hypothesis has thus emerged, stating that the particular microenvironment of these tumors may interfere with the tumor cells that promote chemoresistance and the dissemination of metastases. The stroma is composed of a large number of cell types (immune cells, endothelial cells, mesenchymal stromal cells, etc.) which secrete growth factors, such as transforming growth factor-β (TGF-β), which favors the development of primary tumors and dissemination of metastases by constituting a permissive niche at primary and distant sites. Rather than targeting the tumor cells themselves, which are very heterogeneous in osteosarcoma, the hypothesis is instead to target the key actors secreted in the microenvironment, such as TGF-βs, which play a part in tumor progression. In the last decade, numerous studies have shown that overexpression of TGF-β is a hallmark of many cancers, including primary bone tumors. In this context, TGF-β signaling has emerged as a crucial factor in the cross talk between tumor cells and stroma cells in poor-prognosis cancers. Secretion of TGF-β by tumor cells or stroma cells can effectively act in a paracrine manner to regulate the phenotype and functions of the microenvironment to stimulate protumorigenic microenvironmental changes. TGF-β can thus exert its protumorigenic function in primary bone tumors by promoting angiogenesis, bone remodeling and cell migration, and by inhibiting immunosurveillance. This review focuses on the involvement of TGF-β signaling in primary bone tumor development, and the related therapeutic options that may be possible for these tumors.
Keywords: osteosarcoma, transforming growth factor-β, microenvironment, bone, metastases
Introduction
Osteosarcoma (OS) is the most common malignant primary bone tumor, occurring above all in children, adolescents, or young adults with a median age of onset of 18 years. These tumors occur commonly in the metaphyseal region of the long bones, developing at sites of rapid bone growth (1). The World Health Organization classification of tumors of the soft tissue and bone defines osteosarcoma as a “malignant, bone-forming tumor, divided into several histological subtypes: chondroblastic, fibroblastic, osteoblastic, telangiectasic, or small cells” (2). Some of these histological forms have distinct molecular and biological behaviors. Most osteosarcomas are “conventional OS” (85%), defined as primary intramedullar high-grade malignant tumors in which neoplastic cells produce immature bone or osteoid tissue.
Current treatment associates surgery with combinational chemotherapy which cures at 5 years approximately 70% of patients with localized disease, with response to preoperative chemotherapy as the strongest predictor of overall survival (3). However, survival for patients with metastatic or relapsed disease has remained unchanged over the past 40 years, with an overall survival rate of about 20% at 5 years (4, 5). At the time of diagnosis, 20% of patients present with detectable lung metastases, but it has been estimated that undetectable metastases are present in 80% of cases (6).
New therapeutic options are therefore needed for this type of tumor.
Osteosarcoma Microenvironment: Potential Therapeutic Targets
Conventional high-grade osteosarcomas are generally genomically unstable tumors with complex karyotypes (7). Rarity and genomic complexity, as well as intra-tumoral and intertumoral heterogeneity, have presented challenges for the molecular characterization of osteosarcomas. These tumors are characterized by chromosomal instability, with high levels of somatic structural variations and copy number alterations (8). Somatic mutations in both TP53 and RB1 are the most frequently reported (9, 10). Other mutated genes include RecQ protein-like 4, which encodes a RecQ helicase, and RUNX2. Another contributor to genomic instability is alternative lengthening of telomeres, which prevents telomere shortening and induces senescence (11). To date, the search for common molecular therapeutic targets in osteosarcoma has been disappointing. In this context, rather than targeting tumor cells themselves, the hypothesis is to target the key actors secreted in the microenvironment and which play a part in tumor progression.
Irrespective of their origins, tumors are heterogeneous cellular entities whose progression greatly depends on reciprocal interactions between genetically altered neoplastic cells and their non-neoplastic counterparts present in the microenvironment. Tumor bulk is therefore composed of differentiated tumor cells, and by cancer stem cells that are combined and interact with normal cells. The interplay between them regulates the production and biological activity of many soluble factors and extracellular matrix components that allow the growth and maintenance of solid tumors (12). Therefore, the reactive stroma plays a key role in the development and progression of cancer. Osteosarcoma originates in bone where there is a high concentration of mesenchymal progenitors. Tumor-associated stroma mainly consists of two major categories of component: (i) the extracellular matrix, composed of structural proteins such as collagen and elastin, specialized proteins such as fibronectin, and proteoglycans such as hyaluronan; (ii) cellular elements, composed of cells surrounding the tumor tissue that play a part in the stromal response, i.e., bone cells, vasculature and endothelial cells, pericytes, immune cells such as macrophages [tumor-associated macrophages (TAMs)] and lymphocytes, and mesenchymal stromal cells (MSC). In addition, fibroblasts that differentiate from MSC and usually switch to a tumor-promoting cell phenotype, called the cancer-associated fibroblasts, are also present in the tumor microenvironment (TME) (13).
Mesenchymal stromal cells are involved in osteosarcoma growth and progression, through cross-feeding of the tumor cells via the release of cytokines and soluble growth factors, by helping in migration, proliferation and stemness, membrane cross-talk via microvesicle secretion, metabolic reprogramming of tumor cells, and immune escape. MSC are non-hematopoietic precursors found in the bone marrow. They contribute to the maintenance and regeneration of a variety of tissues of mesodermal lineage, including bone.
One of the main features of osteosarcomas is their influence on bone remodeling as they are characterized by both the formation of osteoid matrix, and by osteolytic lesions. A vicious cyclie between tumor and bone cells occurs during the development of osteosarcoma, promoting tumor growth and metastatic dissemination (Figure 1). In brief, osteosarcoma cells produce soluble osteolytic factors such as interleukin-6 (IL-6), IL-11, tumor necrosis factor-α, or receptor activator of NF-κB ligand (RANKL) that activate osteoclastogenesis, leading to bone degradation. Following this process, growth factors trapped in the bone matrix, such as transforming growth factor-βs (TGF-βs), are released into the bone microenvironment and stimulate tumor growth and metastatic progression (14, 15). The impact of osteoclast activity on osteosarcoma growth and progression has been reported by several studies (16–19). Therefore, therapeutic approaches targeting osteoclasts may be a promising option. Although the use of zoledronate, a strong inhibitor of osteoclast function, in the French randomized OS2006 trial in combination with chemotherapy and surgery, did not show any significant improvement (20), targeting the cytokines released during bone degradation, in particular TGF-β, remains relevant.
Figure 1.

The vicious cycle between tumor and bone cells during osteosarcoma development. Osteosarcoma cells produce soluble osteolytic factors such as receptor activator of nuclear factor kappa-B ligand (RANKL), interleukin-11 (IL-11), IL-6, and tumor necrosis factor-α (TNF-α) that directly activate osteoclastogenesis, leading to bone degradation. Osteosarcoma cells also produce soluble factors, such as bone morphogenetic protein (BMP) or parathyroid hormone-related protein (PTHrP), which stimulate the production of RANKL by osteoblasts and therefore increase osteoclast activity. Osteoblasts are derived from mesenchymal stem cell in response to transcriptional factors such as Runx2 and osterix. Following bone degradation, the growth factors trapped in the bone matrix, such as transforming growth factor-βs (TGF-βs), are released into the bone microenvironment and stimulate both tumor growth and metastatic progression.
TGF-β Signaling Pathways
In humans, the TGF-β family is composed of 33 members, encoded by 33 different genes, including the TGF-βs, activins, nodal, bone morphogenetic proteins, and growth and differentiation factors (21–23). Of these secreted cytokines, three different isoforms of TGF-βs have been identified in mammals: TGF-β1, -β2, and -β3. TGF-β isoforms are secreted as latent precursor molecules, requiring activation into a mature form for receptor binding. Many activators of latent TGF-βs have been described in the last few decades, including integrins, proteases such as MT1-matrix metalloproteinase (MMP) or others MMPs, and physicochemical factors such as detergents, and ionizing and ultraviolet radiation (22, 24–26). Once activated, TGF-β dimers signal from the membrane to the nucleus by binding to two heteromeric cell surface serine/threonine kinase receptors, named type I (TβRI) and type II (TβRII) receptors. Ligand binding induces the assembly of two TβRI and two TβRII receptors into a heterotetrameric complex in which TβRII phosphorylates a specific serine residue of TβRI and in turn activates the serine/theronine kinase of TβRI (27–29).
Transforming growth factor-βs thus activate the Smads cascade (Figure 2), known as the canonical TGF-β signaling pathway. Briefly, receptor-regulated Smads (R-Smads), including Smad2 and Smad3, are phosphorylated and activated by TβRI. Activated R-Smads then dissociate from the Smad anchor for receptor activation protein (30) and recruit the common-mediator Smad (co-Smad), Smad4. This protein complex is translocated into the nucleus to regulate target gene expression. At the regulatory DNA binding sequence of genes, the R-Smad/co-Smad complex activates transcription through physical interaction and functional cooperation of DNA-binding Smads with sequence-specific transcription factors (29). The minimal Smad-binding element contains four base pairs, 5′-AGAC-3′, but binding to other G/C-rich sequences has also been reported (31). Interaction between the R-Smad/co-Smad complex and other transcription factors (either co-activators or co-repressors) generates a high-affinity protein-DNA complex to regulate gene expression. Several inhibitory mechanisms regulate the TGF-β signaling cascade. Of them, Smad7, induced by TGF-β (32), competes with R-Smads for binding to activated TβRI and thus inhibits R-Smad phosphorylation. Smad7 also has the ability to recruit E3-ubiquitin ligases (Smurf1 and Smurf2) to activate TβRI, resulting in receptor degradation (33, 34). Moreover, Smad7 may recruit protein phosphatases to the receptor complex, resulting in its inactivation (35).
Figure 2.
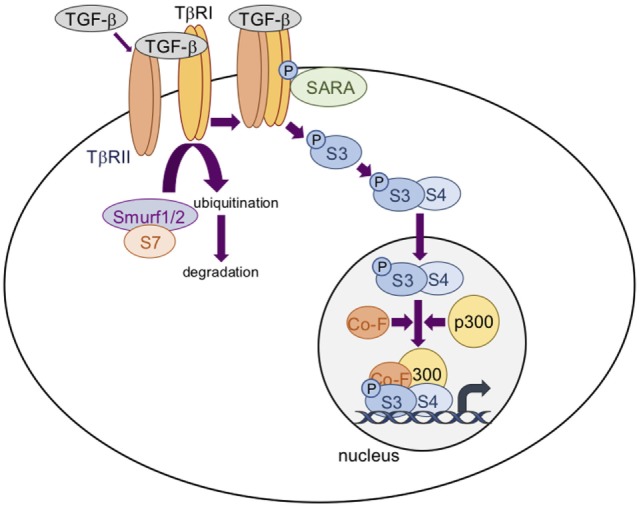
The transforming growth factor-β (TGF-β)/Smad signaling pathway. TGF-β dimers bind to two TβRII receptors that induce the assembly of two TβRI and two TβRII receptors into a heterotetrameric complex in which TβRII phosphorylates and activates TβRI. Smad3 (S3) is then phosphorylated and activated by TβRI. Activated S3 dissociates from the Smad anchor for receptor activation protein (SARA) and recruits Smad4 (S4). This protein complex is translocated into the nucleus to regulate target gene expression in association with cofactors (Co-F) and/or p300. Smad7 (S7) recruits E3-ubiquitin ligases (Smurf1 and Smurf2) to activate TRβI, resulting in receptor degradation.
In addition to this canonical pathway, TGF-βs are also able to activate Smad independent or non-canonical pathways, including mitogen-activated protein kinases and phosphoInositide3-kinase/AKT (PI3K/AKT) signaling pathways (36). In this context, one of the first non-Smad effectors of the TGF-β receptor complex is TRAF6, implicated in the activation of TGF-β-activated kinase 1, capable of activating the SAP/JNK and p38-kinase pathways (37, 38). More recently, it has been shown that TRAF6 favors the formation of a TβRI/p85α complex, leading to activation of the PI3K/AKT cascade (39).
TGF-β and Osteosarcoma
Regarding carcinoma, it is widely accepted that TGF-βs act both as tumor suppressors in premalignant tumors and as tumor promoters in advanced tumors (15, 40–43) (Figure 3).
Figure 3.

The tumor suppressor and protumoral properties of transforming growth factor-β (TGF-β) in carcinoma. Tumor suppressor properties: TGF-βs inhibit cell proliferation largely by inducing the expression of cyclin-dependent kinase (CDK) inhibitors such as p21Cip1 (p21) and p15lnk4b (p15). Protumoral properties: TGF-βs stimulate epithelial–mesenchymal transition (EMT). This process is associated with a loss or downregulation of E-cadherin, claudins, and occludins, and an upregulation of mesenchymal markers such as N-cadherin, fibronectin, and vimentin. These changes in gene expression are regulated by transcription factors such as Snail-1, Snail-2, ZEB-1, and ZEB-2. TGF-βs stimulate angiogenesis in part by stimulating platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) expression. TGF-βs favor cancer cell migration and invasion in part by increasing matrix metalloproteinase-2 (MMP) expression. TGF-βs exert immunosuppressive effects via the modulation of the activity or biology of immune cells such as T-cells, natural killer cells (NK-cells), macrophages, and dendritic cells.
Briefly, TGF-β1 acts as a tumor suppressor mainly through its ability to inhibit cell proliferation both by inducing the expression of cyclin-dependent kinase inhibitors such as p21Cip1 and p15lnk4b, and/or by reducing the expression of proliferative drivers such as c-Myc and cyclin-D (44–46). In this context, alterations or mutations to TGF-β cascade members have been associated with several types of carcinoma (47, 48).
In contrast with carcinoma, it seems that TGF-βs fail to inhibit mesenchymal cell proliferation, particularly in the case of osteosarcoma cells (49, 50) and that TGF-βs exert only protumoral properties in sarcomas through their pro-metastatic effects. In this context, we will focus in this chapter on the pro-metastatic properties of TGF-βs in osteosarcoma. In the last few decades, studies of TGF-βs expression in cancer have correlated TGF-βs levels with the metastatic potential of tumors, suggesting that TGF-βs play a role in tumor progression. In osteosarcoma, TGF-β1 and TGF-β2 expression increase in the sera of patients compared to those of healthy donors (50). This increase in TGF-β production is correlated with high-grade osteosarcoma and associated with the presence of lung metastases (50–52). In addition, our previous results suggest that TGF-β is capable of targeting both tumor cells and their microenvironment. The secretion of TGF-βs by tumor cells or stroma cells can effectively act in an autocrine/paracrine manner to regulate the phenotype and functions of the microenvironment in order to stimulate protumorigenic microenvironmental changes.
TGF-β Exerts Protumorigenic Functions by Targeting Tumor Cells: TGF-β and Epithelial–Mesenchymal Transition (EMT) or “EMT-Like” Phenomena
The switch in TGF-β properties during carcinogenesis has been associated with the ability of TGF-βs to induce the EMT process (53). This multi-step process, characterized by a decrease in epithelial properties and an increase in mesenchymal ones, promotes the invasiveness of cancer cells and contributes to the development of circulating tumor cells (43, 54). This cellular process involves different molecular and cellular modifications, including a loss of cell-to-cell interactions associated with a loss or downregulation of crucial components in the intercellular junction such as E-cadherin, claudins, occludins, and desmosomes. In parallel, an upregulation of mesenchymal marker expression, such as N-cadherin, fibronectin, and vimentin, is observed. These changes in gene expression are regulated by different transcription factors such as Snail-1, Snail-2 (Slug), ZEB-1, and ZEB-2, or Twist (53, 54). Various secreted factors, such as fibroblast growth factors, hepatocyte growth factor, Wnts, Hedgehog proteins, or TGF-βs, induce EMT or are implicated in EMT (55). The regulation of EMT by TGF-βs has been associated with Smad-dependent and Smad-independent signaling pathways (43, 56). In TGF-β-induced EMT, Smad proteins can induce the expression of transcription factors such as Snail, Slug, and Twist involved in the loss of E-cadherin expression, and in turn in the loss of the E-cadherin adhesion complex (55, 57). Interestingly, several other signaling pathways, such as the Wnt, Hippo, and Sonic Hedgehog cascades, cooperate with the Smad cascade to regulate EMT in many cancer cells (55).
Despite the fact that osteosarcoma arises from transformed cells of mesenchymal origin, numerous studies have demonstrated that an overexpression of EMT-transcription factors such as Snails, ZEBs, or Twist is involved in the pathogenesis of osteosarcoma, making possible an “EMT-like” phenomenon (Figure 4) that promotes the invasive properties of osteosarcoma cells and therefore the formation of metastases at distant secondary sites (58). Osteosarcoma tissues thus exhibit elevated Twist expression compared with non-tumorigenic osteochondroma tissue. In addition, metastatic osteosarcoma (grade III) shows an increase in Twist expression compared with non-metastatic osteosarcoma (grade I/II) (59). In this context, in vitro studies have demonstrated that Twist overexpression in SaOS2 osteosarcoma cells is associated with both an increase in cell invasive properties and osteosarcoma cell resistance to cisplatin (60). Similarly, Snail-2 is expressed in the three main histological subtypes of long bone osteosarcoma (osteoblastic, chondroblastic, and fibroblastic), and Snail-2 expression is statistically correlated with tumor grade whatever the osteosarcoma subtype (61). Finally, the transcript and protein levels of ZEB-1 are significantly higher in osteosarcoma tissues when compared with normal bone tissues, ZEB-1 levels being increased in patients with lung metastasis (62).
Figure 4.

The crucial role of transforming growth factor-βs (TGF-βs) in osteosarcoma tumor growth and metastatic dissemination. Following bone degradation, the TGF-βs trapped in the bone matrix are released and promote osteosarcoma growth and metastatic dissemination by targeting both osteosarcoma tumor cells and their microenvironment. TGF-βs target osteosarcoma cells: TGF-βs stimulate “epithelial–mesenchymal transition-like” (EMT), cell migration, and invasion in part by increasing matrix metalloproteinase-2 (MMP-2) expression. TGF-βs target tumor microenvironment: TGF-βs upregulate the expression of osteolytic factors such as receptor activator of nuclear factor kappa-B ligand (RANKL) and interleukin-11 (IL-11) and therefore stimulate bone osteolysis and the secretion of protumoral factors. TGF-βs upregulate platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) expression, and therefore angiogenesis. Finally, TGF-βs exert immunosuppressive effects by regulating immune cell proliferation and activity.
Together, these results demonstrate that an “EMT-like” phenomenon may be associated with the pathogenesis of osteosarcoma. Although the role of TGF-β has not yet been fully defined in this EMT-like process, in vitro experiments have demonstrated that the ability of TGF-βs to promote this EMT-like phenomenon (63) may be associated with the pro-migratory effect of TGF-β1 on several osteosarcoma cell lines (50, 64–66). In vivo experiments, using molecular (overexpression of the Smad inhibitor, Smad7) and pharmacological (SD-208 and/or halofuginone) approaches, have demonstrated that TGF-βs affect the development of lung metastases in osteosarcoma (50, 67). This effect is associated in large part with the ability of Smad7, SD-208, or halofuginone to block the capacity of TGF-β1 to stimulate osteosarcoma migration and invasion (15, 50, 67).
TGF-β Exerts Protumorigenic Functions by Targeting the TME: Angiogenesis, Bone Remodeling, and Immunosurveillance
Tumors are heterogeneous tissues in which tumor cells are surrounded by and interact with a complex TME, composed of both cellular and non-cellular components. This TME plays a critical role in determining the fate of tumor cells during tumorigenesis and metastasis. Aberrant upregulation of TGF-β expression in the TME has thus been implicated in promoting cancer progression and metastasis (68–70). In this context, we will focus the following chapter on the role of TGF-β in tumor angiogenesis, bone remodeling, and modulation of the immune system.
TGF-β and Tumor Angiogenesis
Angiogenesis is a complex biological process that plays a crucial role in sustaining the microenvironment, growth, and metastatic potential of several tumors (71). Schematically, this process favors the formation of blood vessels, increasing the supply of nutriments and providing an entry point for the invasive cells (72). The proliferation, migration, and maturation of endothelial cells are critical steps involved in regulating the angiogenic process. A crucial primary cytokine that drives this process is the secreted vascular endothelial growth factor (VEGF) which stimulates the proliferation and migration of endothelial cells, and the formation of vessels (73–75). Other secreted cytokines, such as platelet-derived growth factor (PDGF), also play a major role in blood vessel formation by inducing vessel maturation mainly by increasing pericyte migration and the induction of pericytes coverage (70, 76). Regarding the role of TGF-β in angiogenesis, TGF-β1 KO mice display a phenotype that is defective in angiogenesis (77, 78). In addition, the loss of endothelial type I or type II TGF-β receptors in mice results in a decrease in vessel formation (79, 80). With regard to tumor angiogenesis, high circulating levels of TGF-β1 are correlated with increased tumor angiogenesis in many forms of cancer (41). Different experimental models for tumor progression, as well as data from human biopsies, have thus demonstrated that a high expression of TGF-β is associated with the expression of angiogenic factors (81) and correlates with the increase in new vessel formation (82, 83). Interestingly, TGF-β1 is able to increase VEGF (84) or PDGF (85) expression in many cancer cells, and therefore to induce tumor angiogenesis (86, 87).
For osteosarcoma pathogenesis (Figure 4), VEGF expression has been associated with microvascular density (88), and patients with high VEGF expression levels exhibit lower disease-free survival (89). In vitro studies revealed that U2OS osteosarcoma cells secrete a PDGF-like growth factor (90), and a malignancy-dependent co-expression of PDGF and PDGF receptors has been observed in the biopsies of osteosarcoma patients (91). Finally, in vivo experiments demonstrated that blocking TGF-β signaling by Smad7 overexpression in osteosarcoma cells or treating mice with the ALK5 inhibitor SD-208, reduces expression of both the endothelial marker CD146 and PDGF (50).
TGF-β and Bone Remodeling
Two cell lineages, the mesenchymal osteoblastic and hematopoietic osteoclastic lineages, are implicated in bone remodeling. Schematically, the osteoblasts and osteoclasts drive bone formation and resorption, respectively. The three mammalian isoforms of TGF-β (TGF-β1, -β2, and -β3) are found in bone (92). The role of these TGF-βs in skeleton development in general, and specifically during bone remodeling, is complex. In vitro, TGF-β1 stimulates the proliferation and migration of mesenchymal stem cells during the early stages of osteoblastogenesis, and inhibits both the differentiation of mesenchymal stem cells into osteoblasts, and the activity of osteoblasts in the late stages of osteoblastogenesis (15, 93–95). For osteoclastogenesis, TGF-β1 affects bone resorption in a dose-dependent manner (15, 92). In vitro, low doses of TGF-β1 stimulate the differentiation of osteoclasts, and high doses of TGF-β1 inhibit the differentiation of osteoclasts through modulation of RANKL and osteoprotegerin expression by osteoblasts (96). As a consequence, in vivo experiments indicate that TGF-βs favor bone resorption and destruction (15).
Interestingly, blocking the TGF-β signaling pathway in osteosarcoma cells reduces the bone osteolysis associated with tumor growth and, in turn, tumor progression. Indeed, in a xenograft murine model of osteosarcoma using human HOS or SaOS2 cells, Smad7 overexpression in tumor cells inhibited the tumor-associated bone destruction by both promoting ectopic bone formation and preventing trabecular bone osteolysis (50). One hypothesis to explain this phenomenon is that blocking the TGF-β cascade in osteosarcoma cells inhibits the expression and secretion of the TGF-β target genes, such as RANKL and IL-11, which stimulate osteoclast activity (50).
One of the hallmarks of the extra cellular matrix in tumor progression is also upregulation of proteolytic enzymes such as MMPs (68, 69, 97). For osteosarcoma, several studies have shown that highly invasive osteosarcomas express higher levels of MMP-2 than weakly invasive tumors, and osteosarcoma cell invasion is associated with MMP-2 expression (98, 99). In addition, MMP-2 expression correlated with prognosis and response to chemotherapy (100, 101). Interestingly, blocking the TGF-β signaling pathway in osteosarcoma cells reduces the formation of lung metastases, mainly by inducing a decrease in MMP-2 expression in tumors. In addition, blocking the TGF-β cascade in tumor cells inhibits the expression and activation of MMP-2 and the ability of TGF-β to stimulate osteosarcoma cell migration and invasion (50) (Figure 4).
TGF-β and the Immune System
Transforming growth factor-βs are secreted cytokines that have multiple immunosuppressive properties (102) via modulation of the activity or biology of several cells in the immune system, such as T-cells, natural killer (NK) cells, macrophages, and dendritic cells (41, 103, 104).
These immunosuppressive abilities of TGF-βs include inhibition of T-cell proliferation, inhibition of T-cell differentiation into cytotoxic T lymphocytes and helper T cells, and inhibition of the T-cell stimulatory functions of antigen-presenting cells (104). For example, in a mouse model expressing a dominant negative form of TβRII restricted to CD4+ and CD8+ T-cells, an antitumor response is observed against melanoma progression (105).
The functional activation of NK cells, which play a crucial role in the antitumor response by recognizing and destroying tumor cells, is inhibited by TGF-β via different mechanisms (105). For example, TGF-βs antagonize the IL-15-induced cell proliferation associated with NK-cell activation and thus block the functional activation of NK-cells (106). The biological functions of the DC cells involved in the activation of the immune response, and therefore in determining the host response to primary tumor cells, are also regulated by TGF-β (104). For example, TGF-β increases the expression of inhibitor of differentiation 1 driving the switch from dendritic cell differentiation to myeloid-derived suppressor cell expansion during tumor progression (107).
Solid tumors are usually invaded by macrophages called TAMs. These TAMs are classically divided into two categories: M1 polarized macrophages, identified as antiumor cells, and M2 polarized macrophages, identified as protumor cells (108, 109). In this context, certain studies have demonstrated the ability of TGF-βs to drive the induction of macrophage polarization from M1 to M2 subtypes (110). All these immunosuppressive properties of TGF-βs induce tumor evasion from immune response (41, 68, 103, 104).
The immune environment of osteosarcoma is mainly composed of myeloid cells (monocytes, macrophages, and dendritic cells) and T-lymphocytes (111). Osteosarcoma cells control the recruitment and differentiation of immune-infiltrating cells, and establish a local immune tolerant microenvironment, allowing the tumor to grow (111).
While initial studies demonstrated that macrophages are associated with reduced metastasis and improved survival in high-grade osteosarcoma (112), recent studies have shown that TAMs are associated with better overall survival (113, 114). T-cells are the other cell population represented in the immune infiltrate in osteosarcoma (111). Recent studies have indicated that the CD8+/FOXP3+-ratio is a strong prognostic factor for osteosarcoma at diagnosis (115).
Conclusion and Clinical Relevance
In carcinoma, TGF-βs exhibit both tumor suppressor and protumoral properties, depending on the stage of the disease. In this context, it seems that the timing of therapies targeting TGF-βs needs to be considered with great precision. For sarcoma, and specifically osteosarcoma, TGF-βs mainly seem to exert protumoral properties by targeting both tumor cells and their microenvironment. It therefore appears that TGF-βs, major drivers for osteosarcoma, could be considered as promising therapeutic targets in this disease. In the last decade, different strategies targeting TGF-βs have been developed (Table 1), including anti-ligand antisense oligonucleotides, which are capable of binding human TGF-β2 mRNA (trabedersen), antibodies that target ligands or receptors, such as fresolimumab, a humanized mAB against TGF-β, and drugs against TGF-β receptor kinases, such as galunisertib (LY2157299, a TβRI inhibitor) [reviewed in Ref. (116–118)].
Table 1.
Transforming growth factor-β (TGF-β) inhibitors in clinical development in cancer (ClinicalTrials.gov).
| Drug | Targets | Trial number | Cancer | Clinical development phase |
|---|---|---|---|---|
| TGF-β ligand inhibitors | ||||
| Fresolimumab (GC-10008) | panTGF-β | NCT00356460 | Advanced Renal Cell Carcinoma or Malignant Melanoma | I |
| NCT00923169 | Advanced Renal Cell Carcinoma or Malignant Melanoma | I | ||
| NCT01472731 | Relapsed Malignant Glioma | II | ||
| NCT01112293 | Relapsed Malignant Pleural Mesothelioma | II | ||
| NCT01401062 | Metastatic Breast Cancer | II | ||
| NCT02581787 | Early Stage Non-small Cell Lung Cancer | I, II | ||
| Trabedersen (AP12009) | TGF-β2 | NCT00844064 | Pancreatic Neoplasms, Melanoma or Colorectal Neoplasms | I |
| NCT00431561 | Glioblastoma or Anaplastic Astrocytoma | II | ||
| NCT00761280 | Anaplastic Astrocytoma or Glioblastoma | III | ||
| Belagenpumatucel-L (Lucanix) | TGF-β2 | NCT00676507 | Advanced Non-small Cell Lung Cancer | III |
| NCT01058785 | Lung Neoplasm, Carcinoma, Bronchogenic | II | ||
| NCT01279798 | Lung Neoplasm or Advanced Carcinoma Non-Small Cell Lung Cancer | III | ||
| Recombinant human granulocyte macrophage-colony stimulating factor (rhGMCSF)/shRNAfurin vaccine | TGF-β1,2 | NCT01309230 | Ovarian Cancer | II |
| NCT01505166 | Colon Cancer | II | ||
| NCT01867086 | Ovarian Cancer | II | ||
| NCT01551745 | Ovarian Cancer | II | ||
| NCT01061840 | Ewings Sarcoma, Non Small Cell Lung Cancer, Liver Cancer | I | ||
| NCT01453361 | Advanced Melanoma | II | ||
| TGF-β receptor inhibitors | ||||
| Galunisertib (LY2157299) | TβRI | NCT02734160 | Metastatic Pancreatic cancer | I |
| NCT02154646 | Pancreatic Neoplasm | I | ||
| NCT01722825 | Advanced or Mestastatic Neoplasm | I | ||
| NCT02452008 | Prostate Cancer | II | ||
| NCT02538471 | Metastaric Breast Cancer | II | ||
| NCT02423343 | Refractory or Recurrent Non-Small Cell Lung Cancer Recurrent, or Hepatocellular Carcinoma | I, II | ||
| NCT02304419 | Neoplasm | I | ||
| NCT01373164 | Pancreatic Cancer | I, II | ||
| NCT02672475 | Triple-Negative Breast Carcinoma | I | ||
| NCT02688712 | Rectal Adenocarcinoma | II | ||
| NCT02240433 | Hepatocellular Carcinoma | I | ||
| NCT03206177 | Carcinosarcoma of the Uterus or Ovary | I | ||
| NCT02906397 | Advanced Hepatocellular Carcinoma | I | ||
| NCT01220271 | Glioma | I, II | ||
| NCT01582269 | Recurrent Glioblastoma | II | ||
| NCT01246986 | Hepatocellular Carcinoma | II | ||
| NCT01682187 | Glioma | I | ||
| NCT02178358 | Hepatocellular Carcinoma | II | ||
| NCT02734160 | Metastatic Pancreatic Cancer | I | ||
| TEW-7197 | TβRI | NCT02160106 | Advanced Stage Solid Tumors | I |
| NCT03143985 | Multiple Myeloma | I | ||
| PF-03446962 | ALK1 | NCT02116894 | Colorectal Cancer | I |
| NCT00557856 | Advanced Solid Tumors | I | ||
| NCT01620970 | Transitional Cell Carcinoma of Bladder | II | ||
| NCT01337050 | Stomach Cancer | I | ||
| NCT01911273 | Advanced or Metastatic Liver Cancer | II | ||
| NCT01486368 | Malignant Pleural Mesothelioma | II | ||
| IMC-TR1 | TβRII | NCT01646203 | Advanced Solid Tumors | I |
Fresolimumab (GC1008) is an antibody capable of neutralizing TGF-β1,2,3. Trabedersen (AP 12009) is an antisense oligodeoxynucleotide specific to human TGF-β2. Belagenpumatucel-L (Lucanix) is a TGF-β2 antisense transgene. rhGMCSF/shRNAfurin vaccine is a dual-modulatory autologous whole cell vaccine (bi-shRNA furin and GMCSF autologous tumor cell vaccine), incorporating the rhGMCSF transgene and the bifunctional shRNA-furin to block pro-protein conversion to active TGF-β1,2. Galunisertib (LY2157299) is a TβRI inhibitor. TEW-7197 is an inhibitor of the protein serine/threonine kinase activity of TβRI. PF-03446962 is a monoclonal antibody against ALK1. IMC-TR1 is a monoclonal antibody against TβRII.
With these strategies, certain trials have shown positive results. For example, trabedersen has been successfully tested in patients with recurrent or refractory high-grade glioma. This randomized, open-label, dose-finding phase IIb study evaluated the efficacy of trabedersen administered intratumorally at doses of 10 or 80 μM compared with standard chemotherapy (temozolomide or procarbazine/lomustine/vincristine) in patients with recurrent/refractory glioblastoma multiform or anaplastic astrocytoma (AA). Analysis of the AA subgroup revealed a significant benefit regarding the 14-month tumor control rate for 10 μM trabedersen vs chemotherapy. In addition, the trend for the 2-year survival rate was for the superiority of 10 μM trabedersen vs chemotherapy (119). Concerning galunisertib, the clinical benefit was observed in 12 of the 56 patients with glioma (21.4%) (120). Most other clinical trials were negative or are still in progress. It should be noted that some drugs that block TGF-β activity have shown cardiotoxicity as side effects.
However, these strategies that target the TGF-β pathway could be considered and proposed in the therapeutic arsenal for osteosarcoma patients.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures were produced with the support of Servier Medical Art (www.servier.com).
Footnotes
Funding. This work was supported by the associations “Etoile de Martin” and “Imagine for Margo,” and by SFCE, the “Societe Française de lutte contre les cancers et leucemies de l’enfant et l’adolescent,” and the Fédération Enfant et Santé.
References
- 1.Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer (2014) 14:722–35. 10.1038/nrc3838 [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg AE. WHO classification of soft tissue and bone, fourth edition: summary and commentary. Curr Opin Oncol (2013) 25:571–3. 10.1097/01.cco.0000432522.16734.2d [DOI] [PubMed] [Google Scholar]
- 3.Collins M, Wilhelm M, Conyers R, Herschtal A, Whelan J, Bielack S, et al. Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J Clin Oncol (2013) 31:2303–12. 10.1200/JCO.2012.43.8598 [DOI] [PubMed] [Google Scholar]
- 4.Link MP, Goorin AM, Miser AW, Green AA, Pratt CB, Belasco JB, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med (1986) 314:1600–6. 10.1056/NEJM198606193142502 [DOI] [PubMed] [Google Scholar]
- 5.Meyers PA, Healey JH, Chou AJ, Wexler LH, Merola PR, Morris CD, et al. Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer (2011) 117:1736–44. 10.1002/cncr.25744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dela Cruz FS. Cancer stem cells in pediatric sarcomas. Front Oncol (2013) 3:168. 10.3389/fonc.2013.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lau CC, Harris CP, Lu XY, Perlaky L, Gogineni S, Chintagumpala M, et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes Cancer (2004) 39:11–21. 10.1002/gcc.10291 [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep (2014) 7:104–12. 10.1016/j.celrep.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toguchida J, Ishizaki K, Sasaki MS, Nakamura Y, Ikenaga M, Kato M, et al. Preferential mutation of paternally derived RB gene as the initial event in sporadic osteosarcoma. Nature (1989) 338:156–8. 10.1038/338156a0 [DOI] [PubMed] [Google Scholar]
- 10.Wunder JS, Gokgoz N, Parkes R, Bull SB, Eskandarian S, Davis AM, et al. TP53 mutations and outcome in osteosarcoma: a prospective, multicenter study. J Clin Oncol (2005) 23:1483–90. 10.1200/JCO.2005.04.074 [DOI] [PubMed] [Google Scholar]
- 11.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet (2010) 11:319–30. 10.1038/nrg2763 [DOI] [PubMed] [Google Scholar]
- 12.Park TS, Donnenberg VS, Donnenberg AD, Zambidis ET, Zimmerlin L. Dynamic interactions between cancer stem cells and their stromal partners. Curr Pathobiol Rep (2014) 2:41–52. 10.1007/s40139-013-0036-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol (2011) 6:209–17. 10.1097/JTO.0b013e3181f8a1bd [DOI] [PubMed] [Google Scholar]
- 14.Wittrant Y, Théoleyre S, Chipoy C, Padrines M, Blanchard F, Heymann D, et al. RANKL/RANK/OPG: new therapeutic targets in bone tumors and associated osteolysis. Biochim Biophys Acta (2004) 1704:49–57. 10.1016/j.bbcan.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 15.Lamora A, Talbot J, Mullard M, Brounais-Le Royer B, Redini F, Verrecchia F. TGF-β signaling in bone remodeling and osteosarcoma progression. J Clin Med (2016) 5(11):E96. 10.3390/jcm5110096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heymann D, Ory B, Blanchard F, Heymann MF, Coipeau P, Charrier C, et al. Enhanced tumor regression and tissue repair when zoledronic acid is combined with ifosfamide in rat osteosarcoma. Bone (2005) 37:74–86. 10.1016/j.bone.2005.02.020 [DOI] [PubMed] [Google Scholar]
- 17.Dass CR, Choong PF. Zoledronic acid inhibits osteosarcoma growth in an orthotopic model. Mol Cancer Ther (2007) 6:3263–70. 10.1158/1535-7163.MCT-07-0546 [DOI] [PubMed] [Google Scholar]
- 18.Costa-Rodrigues J, Fernandes A, Fernandes MH. Reciprocal osteoblastic and osteoclastic modulation in co-cultured MG63 osteosarcoma cells and human osteoclast precursors. J Cell Biochem (2011) 112:3704–13. 10.1002/jcb.23295 [DOI] [PubMed] [Google Scholar]
- 19.Ohba T, Cole HA, Cates JM, Slosky DA, Haro H, Ando T, et al. Bisphosphonates inhibit osteosarcoma-mediated osteolysis via attenuation of tumor expression of MCP-1 and RANKL. J Bone Miner Res (2014) 29:1431–45. 10.1002/jbmr.2182 [DOI] [PubMed] [Google Scholar]
- 20.Piperno-Neumann S, Le Deley MC, Rédini F, Pacquement H, Marec-Bérard P, Petit P, et al. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol (2016) 17:1070–80. 10.1016/S1470-2045(16)30096-1 [DOI] [PubMed] [Google Scholar]
- 21.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol (2005) 21:659–93. 10.1146/annurev.cellbio.21.022404.142018 [DOI] [PubMed] [Google Scholar]
- 22.Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol (2016) 8(5):a021873. 10.1101/cshperspect.a021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang C. Agonists and antagonists of TGF-β family ligands. Cold Spring Harb Perspect Biol (2016) 8(8):a021923. 10.1101/cshperspect.a021923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature (2011) 474:343–9. 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison CA, Al-Musawi SL, Walton KL. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-β superfamily ligands. Growth Factors (2011) 29:174–86. 10.3109/08977194.2011.608666 [DOI] [PubMed] [Google Scholar]
- 26.Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol (2016) 8(6):a021907. 10.1101/cshperspect.a021907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macias MJ, Martin-Malpartida P, Massagué J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem Sci (2015) 40:296–308. 10.1016/j.tibs.2015.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hata A, Chen YG. TGF-β signaling from receptors to Smads. Cold Spring Harb Perspect Biol (2016) 8(9):a022061. 10.1101/cshperspect.a022061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol (2016) 8(8):a022053. 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell (1998) 95:779–91. 10.1016/S0092-8674(00)81701-8 [DOI] [PubMed] [Google Scholar]
- 31.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J (1998) 17:3091–100. 10.1093/emboj/17.11.3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature (1997) 389:631–5. 10.1038/39369 [DOI] [PubMed] [Google Scholar]
- 33.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell (2000) 6:1365–75. 10.1016/S1097-2765(00)00134-9 [DOI] [PubMed] [Google Scholar]
- 34.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem (2001) 276:12477–80. 10.1074/jbc.C100008200 [DOI] [PubMed] [Google Scholar]
- 35.Shi W, Sun C, He B, Xiong W, Shi X, Yao D, et al. GADD34-PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol (2004) 164:291–300. 10.1083/jcb.200307151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang YE. Mechanistic insight into contextual TGF-β signaling. Curr Opin Cell Biol (2017) 51:1–7. 10.1016/j.copbio.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science (1995) 270:2008–11. 10.1126/science.270.5244.2008 [DOI] [PubMed] [Google Scholar]
- 38.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell (2008) 31:918–24. 10.1016/j.molcel.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamidi A, Song J, Thakur N, Itoh S, Marcusson A, Bergh A, et al. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci Signal (2017) 10(486). 10.1126/scisignal.aal4186 [DOI] [PubMed] [Google Scholar]
- 40.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A (2003) 100:8621–3. 10.1073/pnas.1633291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang JJ, Blobe GC. Dichotomous roles of TGF-β in human cancer. Biochem Soc Trans (2016) 44:1441–54. 10.1042/BST20160065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokobori T, Nishiyama M. TGF-β signaling in gastrointestinal cancers: progress in basic and clinical research. J Clin Med (2017) 6(1). 10.3390/jcm6010011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costanza B, Umelo IA, Bellier J, Castronovo V, Turtoi A. Stromal modulators of TGF-β in cancer. J Clin Med (2017) 6(1). 10.3390/jcm6010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pardali K, Kurisaki A, Morén A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J Biol Chem (2000) 275:29244–56. 10.1074/jbc.M909467199 [DOI] [PubMed] [Google Scholar]
- 45.Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol (2001) 3:392–9. 10.1038/35070076 [DOI] [PubMed] [Google Scholar]
- 46.Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol Cancer Res (2010) 8:1375–87. 10.1158/1541-7786.MCR-09-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene (1999) 18:3098–103. 10.1038/sj.onc.1202642 [DOI] [PubMed] [Google Scholar]
- 48.Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res (1999) 59:320–4. [PubMed] [Google Scholar]
- 49.Matsuyama S, Iwadate M, Kondo M, Saitoh M, Hanyu A, Shimizu K, et al. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res (2003) 63:7791–8. [PubMed] [Google Scholar]
- 50.Lamora A, Talbot J, Bougras G, Amiaud J, Leduc M, Chesneau J, et al. Overexpression of smad7 blocks primary tumor growth and lung metastasis development in osteosarcoma. Clin Cancer Res (2014) 20:5097–112. 10.1158/1078-0432.CCR-13-3191 [DOI] [PubMed] [Google Scholar]
- 51.Yang RS, Wu CT, Lin KH, Hong RL, Liu TK, Lin KS. Relation between histological intensity of transforming growth factor-beta isoforms in human osteosarcoma and the rate of lung metastasis. Tohoku J Exp Med (1998) 184:133–42. 10.1620/tjem.184.133 [DOI] [PubMed] [Google Scholar]
- 52.Xu S, Yang S, Sun G, Huang W, Zhang Y. Transforming growth factor-beta polymorphisms and serum level in the development of osteosarcoma. DNA Cell Biol (2014) 33:802–6. 10.1089/dna.2014.2527 [DOI] [PubMed] [Google Scholar]
- 53.Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-β paradox to EMT-MET programs. Cancer Lett (2013) 341:30–40. 10.1016/j.canlet.2013.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jie XX, Zhang XY, Xu CJ. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: mechanisms and clinical applications. Oncotarget (2017) 8:81558–71. 10.18632/oncotarget.18277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr Opin Cell Biol (2014) 31:56–66. 10.1016/j.ceb.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-β: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst (2014) 106(2):djt369. 10.1093/jnci/djt369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang G, Yuan J, Li K. EMT transcription factors: implication in osteosarcoma. Med Oncol (2013) 30(4):697. 10.1007/s12032-013-0697-2 [DOI] [PubMed] [Google Scholar]
- 59.Lei P, Ding D, Xie J, Wang L, Liao Q, Hu Y. Expression profile of Twist, vascular endothelial growth factor and CD34 in patients with different phases of osteosarcoma. Oncol Lett (2015) 10:417–21. 10.3892/ol.2015.3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu J, Liao Q, He H, Zhong D, Yin K. TWIST interacts with β-catenin signaling on osteosarcoma cell survival against cisplatin. Mol Carcinog (2014) 53:440–6. 10.1002/mc.21991 [DOI] [PubMed] [Google Scholar]
- 61.Sharili AS, Allen S, Smith K, Hargreaves J, Price J, McGonnell I. Expression of Snail2 in long bone osteosarcomas correlates with tumor malignancy. Tumour Biol (2011) 32:515–26. 10.1007/s13277-010-0146-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen A, Zhang Y, Yang H, Xu R, Huang G. Overexpression of ZEB1 relates to metastasis and invasion in osteosarcoma. J Surg Oncol (2012) 105:830–4. 10.1002/jso.23012 [DOI] [PubMed] [Google Scholar]
- 63.Sung JY, Park SY, Kim JH, Kang HG, Yoon JH, Na YS, et al. Interferon consensus sequence-binding protein (ICSBP) promotes epithelial-to-mesenchymal transition (EMT)-like phenomena, cell-motility, and invasion via TGF-β signaling in U2OS cells. Cell Death Dis (2014) 5:e1224. 10.1038/cddis.2014.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang Y, Yang Y, Gao R, Yang X, Yan X, Wang C, et al. RLIM interacts with Smurf2 and promotes TGF-β induced U2OS cell migration. Biochem Biophys Res Commun (2011) 414:181–5. 10.1016/j.bbrc.2011.09.053 [DOI] [PubMed] [Google Scholar]
- 65.Kunita A, Kashima TG, Ohazama A, Grigoriadis AE, Fukayama M. Podoplanin is regulated by AP-1 and promotes platelet aggregation and cell migration in osteosarcoma. Am J Pathol (2011) 179:1041–9. 10.1016/j.ajpath.2011.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Song Y, Yang J, Gong L, Zhao P, Zhang Y, et al. The up-regulation of cysteine-rich protein 61 induced by transforming growth factor beta enhances osteosarcoma cell migration. Mol Cell Biochem (2013) 384:269–77. 10.1007/s11010-013-1807-3 [DOI] [PubMed] [Google Scholar]
- 67.Lamora A, Mullard M, Amiaud J, Brion R, Heymann D, Redini F, et al. Anticancer activity of halofuginone in a preclinical model of osteosarcoma: inhibition of tumor growth and lung metastases. Oncotarget (2015) 6:14413–27. 10.18632/oncotarget.3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumor microenvironment. Nat Rev Cancer (2013) 13:788–99. 10.1038/nrc3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caja F, Vannucci L. TGFβ: a player on multiple fronts in the tumor microenvironment. J Immunotoxicol (2015) 12:300–7. 10.3109/1547691X.2014.945667 [DOI] [PubMed] [Google Scholar]
- 70.Pickup MW, Owens P, Moses HL. TGF-β, bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harb Perspect Biol (2017) 9(5). 10.1101/cshperspect.a022285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 72.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov (2011) 10:417–27. 10.1038/nrd3455 [DOI] [PubMed] [Google Scholar]
- 73.Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med (2011) 3:66ra5. 10.1126/scitranslmed.3001542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol (2011) 27:563–84. 10.1146/annurev-cellbio-092910-154002 [DOI] [PubMed] [Google Scholar]
- 75.Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L. Vascular endothelial growth factors and receptors: anti-angiogenic therapy in the treatment of cancer. Mol Aspects Med (2011) 32:88–111. 10.1016/j.mam.2011.04.004 [DOI] [PubMed] [Google Scholar]
- 76.Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal (2013) 11:97. 10.1186/1478-811X-11-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development (1995) 121:1845–54. [DOI] [PubMed] [Google Scholar]
- 78.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest (2005) 128(6 Suppl):585S–90S. 10.1378/chest.128.6_suppl.585S [DOI] [PubMed] [Google Scholar]
- 79.Oshima M, Oshima H, Taketo MM. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol (1996) 179:297–302. 10.1006/dbio.1996.0259 [DOI] [PubMed] [Google Scholar]
- 80.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A (2000) 97:2626–31. 10.1073/pnas.97.6.2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci (Landmark Ed) (2009) 14:4848–61. 10.2741/3573 [DOI] [PubMed] [Google Scholar]
- 82.Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res (2002) 62:6021–5. [PubMed] [Google Scholar]
- 83.Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology (2009) 50:1140–51. 10.1002/hep.23118 [DOI] [PubMed] [Google Scholar]
- 84.Donovan D, Harmey JH, Toomey D, Osborne DH, Redmond HP, Bouchier-Hayes DJ. TGF beta-1 regulation of VEGF production by breast cancer cells. Ann Surg Oncol (1997) 4:621–7. 10.1007/BF02303745 [DOI] [PubMed] [Google Scholar]
- 85.Fischer AN, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene (2007) 26:3395–405. 10.1038/sj.onc.1210121 [DOI] [PubMed] [Google Scholar]
- 86.Xiong B, Gong LL, Zhang F, Hu MB, Yuan HY. TGF beta1 expression and angiogenesis in colorectal cancer tissue. World J Gastroenterol (2002) 8:496–8. 10.3748/wjg.v8.i3.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Petersen M, Pardali E, van der Horst G, Cheung H, van den Hoogen C, van der Pluijm G, et al. Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene (2010) 29:1351–61. 10.1038/onc.2009.426 [DOI] [PubMed] [Google Scholar]
- 88.Yang J, Yang D, Sun Y, Sun B, Wang G, Trent JC, et al. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer (2011) 117:4925–38. 10.1002/cncr.26116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen D, Zhang YJ, Zhu KW, Wang WC. A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumor Biol (2013) 34:1895–9. 10.1007/s13277-013-0733-z [DOI] [PubMed] [Google Scholar]
- 90.Betsholtz C, Westermark B, Ek B, Heldin CH. Coexpression of a PDGF-like growth factor and PDGF receptors in a human osteosarcoma cell line: implications for autocrine receptor activation. Cell (1984) 39(3 Pt 2):447–57. 10.1016/0092-8674(84)90452-5 [DOI] [PubMed] [Google Scholar]
- 91.Kubo T, Piperdi S, Rosenblum J, Antonescu CR, Chen W, Kim HS, et al. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer (2008) 112:2119–29. 10.1002/cncr.23437 [DOI] [PubMed] [Google Scholar]
- 92.Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res (2016) 4:16009. 10.1038/boneres.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev (2005) 26:743–74. 10.1210/er.2004-0001 [DOI] [PubMed] [Google Scholar]
- 94.Alliston T, Choy L, Ducy P, Karsenty G, Derynck R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J (2001) 20:2254–72. 10.1093/emboj/20.9.2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maeda S, Hayashi M, Komiya S, Imamura T, Miyazono K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J (2004) 23:552–63. 10.1038/sj.emboj.7600067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karst M, Gorny G, Galvin RJ, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol (2004) 200:99–106. 10.1002/jcp.20036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B (2015) 5:402–18. 10.1016/j.apsb.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bjørnland K, Flatmark K, Pettersen S, Aaasen AO, Fodstad O, Maelandsmo GM. Matrix metalloproteinases participate in osteosarcoma invasion. J Surg Res (2005) 127:151–6. 10.1016/j.jss.2004.12.016 [DOI] [PubMed] [Google Scholar]
- 99.Fromigué O, Hamidouche Z, Marie PJ. Blockade of the RhoA-JNK-c-Jun-MMP2 cascade by atorvastatin reduces osteosarcoma cell invasion. J Biol Chem (2008) 283:30549–56. 10.1074/jbc.M801436200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang M, Zhang X. Association of MMP-2 expression and prognosis in osteosarcoma patients. Int J Clin Exp Pathol (2015) 8:14965–70. [PMC free article] [PubMed] [Google Scholar]
- 101.Kunz P, Sähr H, Lehner B, Fischer C, Seebach E, Fellenberg J. Elevated ratio of MMP2/MMP9 activity is associated with poor response to chemotherapy in osteosarcoma. BMC Cancer (2016) 16:223. 10.1186/s12885-016-2266-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol (1998) 16:137–61. 10.1146/annurev.immunol.16.1.137 [DOI] [PubMed] [Google Scholar]
- 103.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell (2008) 134:392–404. 10.1016/j.cell.2008.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumor environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. 10.1038/nri2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med (2001) 7:1118–22. 10.1038/nm1001-1118 [DOI] [PubMed] [Google Scholar]
- 106.Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumor immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumor activity. PLoS One (2011) 6(9):e22842. 10.1371/journal.pone.0022842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Papaspyridonos M, Matei I, Huang Y, do Rosario Andre M, Brazier-Mitouart H, Waite JC, et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun (2015) 6:6840. 10.1038/ncomms7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66:605–12. 10.1158/0008-5472.CAN-05-4005 [DOI] [PubMed] [Google Scholar]
- 109.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol (2012) 13:31. 10.1186/1471-2172-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heymann MF, Lézot F, Heymann D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell Immunol (2017). 10.1016/j.cellimm.2017.10.011 [DOI] [PubMed] [Google Scholar]
- 112.Buddingh EP, Kuijjer ML, Duim RA, Bürger H, Agelopoulos K, Myklebost O, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res (2011) 17:2110–9. 10.1158/1078-0432.CCR-10-2047 [DOI] [PubMed] [Google Scholar]
- 113.Dumars C, Ngyuen JM, Gaultier A, Lanel R, Corradini N, Gouin F, et al. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget (2016) 7:78343–54. 10.18632/oncotarget.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gomez-Brouchet A, Illac C, Gilhodes J, Bouvier C, Aubert S, Guinebretiere JM, et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: an immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology (2017) 6:e1331193. 10.1080/2162402X.2017.1331193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fritzsching B, Fellenberg J, Moskovszky L, Sápi Z, Krenacs T, Machado I, et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology (2015) 4(3):e990800. 10.4161/2162402X.2014.990800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Katz LH, Li Y, Chen JS, Muñoz NM, Majumdar A, Chen J, et al. Targeting TGF-β signaling in cancer. Expert Opin Ther Targets (2013) 17:743–60. 10.1517/14728222.2013.782287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther (2015) 147:22–31. 10.1016/j.pharmthera.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 118.Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer (2017) 3:56–71. 10.1016/j.trecan.2016.11.008 [DOI] [PubMed] [Google Scholar]
- 119.Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol (2011) 13:132–42. 10.1093/neuonc/noq142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rodon J, Carducci MA, Sepulveda-Sánchez JM, Azaro A, Calvo E, Seoane J. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res (2015) 21:553–60. 10.1158/1078-0432.CCR-14-1380 [DOI] [PMC free article] [PubMed] [Google Scholar]