

Abstract
Peptides synthesized in the likeness of their native interaction domain(s) are natural choices to target protein–protein interactions (PPIs) due to their fidelity of orthostatic contact points between binding partners. Despite therapeutic promise, intracellular delivery of biofunctional peptides at concentrations necessary for efficacy remains a formidable challenge. Peptide amphiphiles (PAs) provide a facile method of intracellular delivery and stabilization of bioactive peptides. PAs consisting of biofunctional peptide headgroups linked to hydrophobic alkyl lipid-like tails prevent peptide hydrolysis and proteolysis in circulation, and PA monomers are internalized via endocytosis. However, endocytotic sequestration and steric hindrance from the lipid tail are two major mechanisms that limit PA efficacy to target intracellular PPIs. To address these problems, we have constructed a PA platform consisting of cathepsin-B cleavable PAs in which a selective p53-based inhibitory peptide is cleaved from its lipid tail within endosomes, allowing for intracellular peptide accumulation and extracellular recycling of the lipid moiety. We monitor for cleavage and follow individual PA components in real time using a Förster resonance energy transfer (FRET)-based tracking system. Using this platform, we provide a better understanding and quantification of cellular internalization, trafficking, and endosomal cleavage of PAs and of the ultimate fates of each component.
Graphical abstract
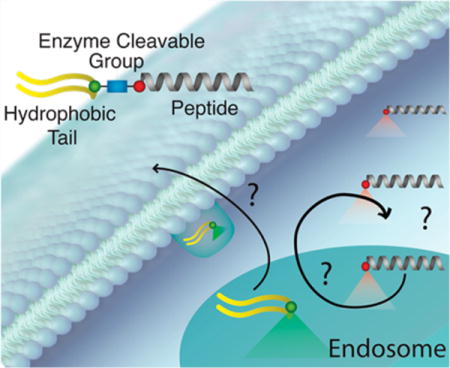
INTRODUCTION
Recent advances in genomics and computational methods have identified ~650,000 essential intracellular protein–protein interactions (PPIs) within the human interactome responsible for normal cellular homeostasis, and of these, many also perpetuate malignant transformation.1,2 One example of such a protein is p53, a tumor suppressor essential for regulating cell stress response through the induction of cell cycle arrest and apoptosis at the level of the mitochondrion.3 Defects in the p53 pathway occur in ~22 million cancer patients, with approximately 50% of these defects being due to the inactivation of p53 itself and the remaining defects due to aberrancies in other p53 signaling or effector proteins.4–6 A pair of these proteins are MDM2 and MDM4, both of which nonredundantly target p53 for degradation.7,8 MDM2 is an E3 ubiquitin ligase that targets p53 for ubiquitin-dependent degradation, while MDM4 inhibits p53 through PPI-mediated sequestration.9–11 In many cancers, the signaling pathway of wild-type TP53 is corrupted by the over-expression of these two proteins.5,12 As a result, there is an urgent need to reactivate p53, particularly in those patients with “complex” p53-pathway copy-number alterations who have significantly shorter overall survival when treated with conventional chemotherapies.5,12,13
Small-molecule and peptide-based PPI inhibition of p53 binding to MDM2/4 has been shown to reactivate cell death in vitro and in preclinical animal models of chemoresistant cancers.14–18 Leading the way are compounds that, through protein binding mimicry, displace p53 from MDM2, allowing free p53 to reactivate apoptosis. Both hydrocarbon-stapled α-helical p53(14–29) peptides and p53(14–29) peptide amphiphiles (PAs) are examples of peptide-based therapeutics that inhibit p53–MDM2/4 interactions and have shown preclinical promise.15,19,20 A hydrocarbon-stapled peptide MDM2/4 inhibitor is currently in phase I and II clinical trials for advanced solid tumors.6,21
Although focus has classically centered on using small molecules for inhibition of PPIs, small molecules are best at targeting PPIs with defined “hot spot” binding residues or concentrated binding foci and often fail to target PPIs with large, diffuse interfaces (>800 Å2) where binding is the summation of geographically distinct low-affinity interactions.1,22 Therefore, peptides are highly desirable choices to target PPIs due to their fidelity of orthostatic contact points between binding partners.23,24 Despite increasing interest, obstacles remain with peptide-based compared to small-molecule therapeutics, namely lower metabolic stability, endosomal entrapment, and cell-membrane impermeability.14 PAs represent one strategy with which to increase the cellular impermeability and serum stability of biofunctional peptides. PAs consist of a peptide headgroup linked to a hydrophobic alkyl lipid-like tail that self-assembles into molecules with distinct hydrophobic and hydrophilic ends, akin to natural lipids within the cellular membrane.25,26 PAs self-assemble into micellar structures in aqueous medium in which the hydrophobic tails are buried within the core, while the peptide head-groups remain on the periphery.26,27 PAs also stabilize peptide secondary structure, protect peptides from proteolytic degradation, and delay plasma clearance because of their nanoscale size and shape while simultaneously enhancing intracellular internalization.20,28,29 Examples of preclinical PAs can be found in diverse areas, including tissue targeting, diagnostic imaging, and cancer therapy.30
Despite the advantages of micellar PA-based systems, one major barrier to using PAs for intracellular PPI disruption is endosomal sequestration.29,31 Most endosomal vesicles recycle back to the cell surface quickly in the early state. Some, however, become long-lived perinuclear late endo- and lysosomal compartments within 30–60 min following internalization, during which peptides have been shown to survive for up to 24 h.20,30,32 As a result, the bulky hydrophobic tail, which is advantageous outside of the cell and during cellular internalization, becomes a membrane “anchor” within the endosome. Thus, enhancing endosomal escape is critical for meaningful clinical transition of PA-based intracellular peptide-based PPI targeting agents.
Endo- and lysosomes degrade their contents using amino-acid sequence-specific proteases, such as cathepsins, that are activated in low pH. Cathepsin cleavage sequences have been extensively studied as linkers for antibody–drug conjugates, with cathepsin-B (catB)-specific sequences being the most commonly used.33 CatB is rarely found in the extracellular matrix, and therefore, conjugates produced with catB cleavable linkers remain remarkably stable in circulation.34 Valine–citrulline–PABC (para-amino benzyl carbamate) has been used as an eflective endosomally responsive cleavable sequence (and spacer) for anticancer prodrugs and antibody-based drug conjugates.35–37 CatB cleavage occurs C-terminally to the valine–citrulline dipeptide linker, while the PABC spacer allows for improved enzyme binding and kinetics and, due to its strong aromatic ring 1,6-elimination, ultimately self-immolates following cleavage.38,39 Given its excellent stability in human plasma, robust cleavage after endocytosis, and potent antigen-specific cytotoxicity, we aimed to use a similar strategy with PA-based therapeutics.
The present study used p53-based therapeutic peptide (p53(14–29)) PAs prepared with a double palmitic acid (diC16) hydrophobic tail and valine–citrulline–PABA (para-amino benzoic acid) (VC–PABA) synthesized between the peptide and the hydrophobic tail to allow for intracellular transport and peptide accumulation. Because the ultimate fate of PA components following endocytosis are poorly understood, we coupled p53(14–29) and diC16 with Forster resonance energy transfer (FRET)-compatible chromophores to monitor intracellular PA cleavage in real time. We were also able to individually track diC16 tails and p53(14–29) peptides using these fluorophores to gain a better understanding of PA cellular internalization, peptide accumulation, lipid tail-mediated membrane sequestration, and intact PA intracellular and extracellular movements that can be extended to other PA-based systems (Figure 1).
Figure 1.
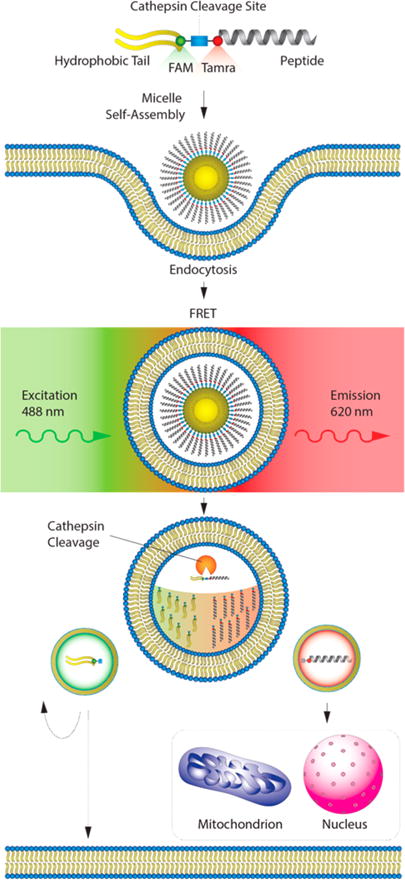
Evaluation and trafficking of enzyme-cleavable peptide amphiphiles with FRET-compatible fluorophores. The ultimate goal of this study is to facilitate ceullar penetration and cleavage of peptides from their lipid carriers in an affort to amplify the intracellular availability of therapeutic peptides. To evalulate this in real time, we used confocal imaging and intracellular FRET analysis with one dye located on the biofunctional peptide and the other on the hydrophobic tail. To begin the process, PAs self-assemble into micelles that are incorporated into the cell through endocytosis. Following endocytosis, cathepsin-B (catB) cleaves the PA through a specific amino-acid linker group between the peptide and the hydrophobic tail (blue square). Following cleavage, the peptide and tail are trafficked throughout the cell, with the ultimate goal of targeting diseased protein–protein interactions (PPIs) in specific intracellular compartments and organelles, depending on the specificity of the peptide being used.
RESULTS AND DISCUSSION
CatB-Cleavable Linker Evaluation
A variety of enzyme-cleavable peptide sequences used in antibody–drug and peptide–drug conjugates were initially tested for efficacy in our system.35,40–43 We ultimately chose the enzyme-cleavable peptide sequence, valine–citrulline (VC), as it gave us the fastest and most complete cleavage (Figure S1; data not shown). We substituted PABC for PABA because PABA has equivalent functional cleavage characteristics in preclinical testing and contains a moderately electron withdrawing carboxylic acid group, making it more stable during solid-phase peptide synthesis.44
To determine if our cathepsin cleavage and intracellular mapping strategy would allow for the complete dissociation of p53(14–29) from diC16, we measured catB-specific cleavage kinetics in situ using recombinant human catB. To do this, we conjugated the experimentally cleavable VC–PABA sequence and a control, noncleavable triple glycine (GGG) linker to a 7-amino-4-methylcoumarin (AMC) dye, useful in studying protease activity and specificity (VC–PABA–AMC and GGG–AMC) (Figures S1A and B).45 The electron group of the AMC fluorophore is localized, and thus remains quenched, when linked to the VC–PABA or GGG peptide substrate. When the covalent bond between the peptide and AMC is cleaved, this group delocalizes, resulting in fluorescence detected at 440 nm (excitation: 348 nm), allowing for the real-time measurement of enzyme and substrate kinetics. The addition of PABA improved the catB-mediated cleavage of VC from AMC, likely through its well-established spacer eflect, allowing catB ample access to the peptide substrate (Figure S1C).46 The fluorescence intensity of VC–PABA–AMC was rapidly increased compared to VC alone, supporting previous findings (Figure S1C).34,36 Neither GGG–AMC nor GG–PABA–AMC showed any change in fluorophore intensity, indicating that PABA alone does not facilitate catB cleavage. We chose to use GGG as a control sequence in following studies due to its similar peptide length with VC–PABA and its nonreactive side groups.
CatB-Cleavable PAs with FRET Chromophores
We next sought to determine if catB cleavage fidelity and kinetics would transfer to intact PA monomers. To determine the transit time and location of individual PA components, FAM (fluorescein) and Tamra (rhodamine) were placed on either side of the VC–PABA or GGG spacers. The fluorophores were located approximately 35.5 and 35.1 Å apart, respectively, with Tamra labeling the N-terminus of p53(14–29) and FAM labeling the N-terminus of either valine or glycine (Figures 2A, B and S2). FAM (donor) excitation at 488 nm causes emission at 520 nm that in turn excites Tamra (acceptor) that emits a FRET wavelength of 620 nm. The efficiency of this energy transfer (FRET efficiency) is extremely sensitive to the small changes in distance within 10 nm of one another.47 A change in intensity of the emitted light at 620 nm after excitation at 488 nm would thus reflect dissociation of p53(14–29) from the PA hydrophobic tail (Figure 1).
Figure 2.
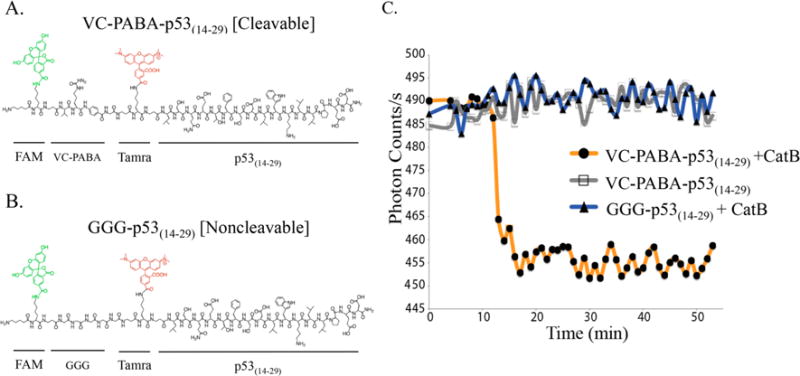
Chemical structures of (A) cleavable and (B) noncleavable p53(14–29) peptides on resin with FAM and Tamra fluorophores used for FRET analysis. (C) Recombinant catB was added the side of the chamber, and FRET signal is rapidly lost as the enzyme reaches VC-PABA targets (orange) within the focused viewing area of the microscope. No loss of FRET signal was measured against GGG-p53(14–29) targets (blue) or without the addition of catB (gray).
To ultimately monitor the enzymatic cleavage of intact PA monomers, we first measured FRET efficiency using wide-field microscopy on p53(14–29) peptides with N-terminally located catB sequences with corresponding fluorochromes. To ensure that the p53(14–29) peptide did not induce apoptosis in later cellular trafficking studies, we chose to use the native conformer of p53(14–29). Native p53(14–29) cannot enter cells and binds MDM2/4 less avidly than α-helical-reinforced peptides, thus ensuring that the driving force for intracellular PA translocation is diC16 and that native p53 is not activated, thereby allowing treated cells to live long enough for trafficking analysis (Figure 2A,B).15,19,33 Peptides on resin were incubated on a platform with incoming light fixed at a diameter of 200 nm, allowing the measurement of a localized bead area using 100× magnification (Figure S3). Using this method, the acceptor intensity (Tamra) diminished significantly following catB addition in the VC–PABA–p53(14–29), while there was no difference in FRET signal when catB was not added or when added to the noncleavable control peptide (Figure 2C). To ensure that catB did not significantly affect p53(14–29), media samples were collected at 3 and 24 h following the addition of catB and individual FAM and Tamra fluorescence measured. Increased FAM emission (N-terminal to the cleavage site) would indicate successful FAM dissociation, while Tamra emission would indicate enzymolysis of internal p53(14–29) amino acids. FAM intensity in the media following catB addition to VC–PABA–p53(14–29) was significantly higher than that measured in the supernatant from GGG–p53(14–29), indicating efficient catB-directed cleavage from PA monomers (Figure S4). Importantly, Tamra fluorescence in the media was minimal for both compounds, indicating relative in vitro stability of the p53(14–29) peptide (Figure S4). We next tested if this p53(14–29) catB resistance would enable the measurement of peptide accumulation inside cells.
Intracellular Accumulation of PA Components
Building from our in vitro testing, we synthesized p53(14–29) cleavable (diC16–VC–PABA–p53(14–29)) and noncleavable (diC16–GGG–p53(14–29)) PAs using the diC16 hydrophobic tail (Figures 3, S5, and S6). Both PA micelles were of similar size and critical micellar concentrations (CMC), allowing for valid comparisons of treatment doses (Figure S7). Unlike previously reported diC16–p53(14–29) PA, which formed rod-like, elongated micelles, these PAs formed spherical micelles between 20 and 40 nm (with occasional larger aggregates), as measured by transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Figures 3 and S7).29 Although the DLS size distribution suggests that the PAs could exist in either micelle or rod-like transition, only round micelles were seen in the TEM images. This difference in structure likely resulted from the additional amino acids and fluorochromes between diC16 and p53(14–29) elongating the polar PA headgroup and driving round micelle formation through electrostatic repulson.26,48–50 We repeated our catB cleavage analysis of these PA monomers and confirmed separation of diC16 from p53(14–29) only after the addition of recombinant catB using HPLC (Figure S8).
Figure 3.
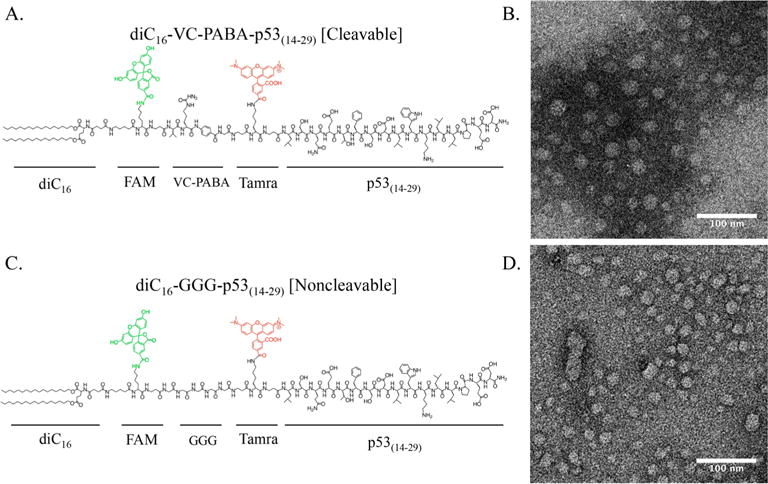
Chemical structures of (A) cleavable and (C) noncleavable PAs. Transmission electron microscopy images of each PA reveal rounded micelles of similar shape and size are shown in panels B and D.
To determine long-term diC16–VC–PABA–p53(14–29) and diC16–GGG–p53(14–29) intracellular accumulation under continuous PA incubation, we strategically moved FAM and Tamra out of FRET overlap range (Figure 4). By moving the fluorochromes away from one another, we were able to determine individual component accumulation without FRET interference. DLS found these PAs similar to the those detailed in Figures 3 and S7 (although slightly larger) between 50 and 100 nm, and with CMCs of 4.7 and 5.8 μM, respectively (data not shown). HeLa cells were incubated with 10 μM PA, such that p53(14–29) was C-terminally labeled with FAM and diC16 C-terminally labeled with Tamra. Intracellular accumulation of diC16–VC–PABA-p53(14–29) PA at 16 and 24 h was far greater than noncleavable control PAs (Figure 4). Cells incubated with cleavable PAs accumulated diC16 diffusely throughout the cells, while the discrete punctae of p53(14–29) overlapped considerably with diC16 in cells incubated with diC16–GGG–p53(14–29) (Figure 4). However, due to the intense accumulation over time, it was impossible to determine if p53(14–29) had been cleaved from diC16 in cells incubated with diC16–VC–PABA–p53(14–29). Although initially internalized, noncleavable PAs did not intracellularly accumulate over time (data not shown; Figure 4). This may have been because of recycling out of the cell, as has been previously demonstrated, or sequestration by fetal bovine serum (FBS) in the culture serum.29,51 Because therapeutic peptide accumulation within target cells is necessary to obtain effective clinical responses, we next sought to determine if the p53(14–29) peptide was cleaved from PA monomers and at what time this had occurred following internalization.
Figure 4.
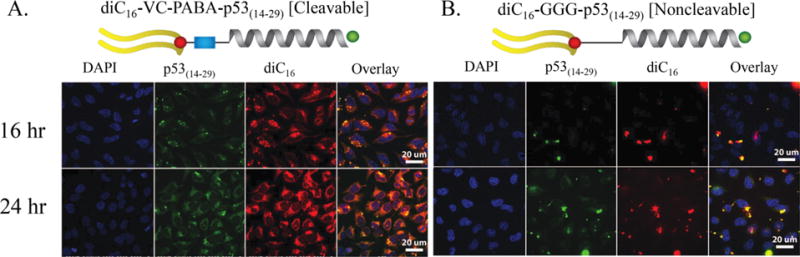
Intracellular accumulation of PAs in HeLa cells. HeLa cells under constant incubation with 10 μM (A) cleavable or (B) noncleavable PAs. FRET incompatible spacing of FAM (labeling p53(14–29)) and Tamra (labeling diC16) allowed for evaluation of PA sequestration over time. Cells incubated with diC16–VC–PABA–p53(14–29) showed PA accumulation, while cells incubated with diC16–GGG–p53(14–29) did not. (The nucleus of each cell was marked with DAPI; p53(14–29), green; diC16, red). Scale bars: 20 μm.
PA Component Intracellular Cleavage and Trafficking
To better understand trafficking of p53(14–29) inside cells, we returned to our FRET capable PA constructs (Figure 3). HeLa cells were pulsed with 2.5 μM PA for 1 h and washed rather than allow for continuous PA exposure that would complicate our visualization of catB-mediated cleavage and intracellular trafficking. Both PAs were equivalently internalized within 1 h of incubation (Figure 5). Each was taken in through endocytosis with substantial compartmental colocalization with transferrin-positive intracellular vesicles, reflective of early and late endosomal trafficking (Figure 6). Despite most intact PAs being associated with transferrin-positive early endosomes, there was also evidence of dissociation and movement of diC16 out of these endosomes and into other areas of the cell as early as 1 h following treatment with diC16–VC–PABA–p53(14–29) (Figure 6). Transferrin labels early endosomes that ultimately transition to sorting endosomes or endocytic recycling compartments where transferrin is released from the transferrin receptor at low pH.52–54 Because this process can take as little as 10 min, it is unclear if these diC16 tail fragments were located within late endosomes or were being recycled back to the cell surface.54 Regardless, unlike diC16–VC–PABA–p53(14–29), control diC16–GGG–p53(14–29) PAs were universally found in vesicles as one unit (Figures 5 and 6). Cleavage of p53(14–29) from diC16 and loss of FRET signal in cells treated diC16–VC–PABA–p53(14–29) occurred almost completely by 3 h following incubation, whereas the FRET signal was retained in cells treated with diC16–GGG–p53(14–29) PAs (Figure 5). p53(14–29) peptide appeared to accumulate in discrete locations within the cell by 6 h, in contrast to control PAs. Additionally, diC16–GGG–p53(14–29) PA-treated cells lost overall intensity over time, as was observed previously (Figures 4). The diffuse spreading of diC16 throughout the cell after treatment with diC16–VC–PABA–p53(14–29) suggests that these compartments are destined for exocytosis and membrane recycling (Figure 5A).20,29,51,54 Supporting this hypthesis is the apparent decrease in tail and peptide signal intensity in cells treated with diC16–GGG–p53(14–29) PAs, suggesting that intact PA monomers are ejected from the cell over time (Figure 5B).
Figure 5.
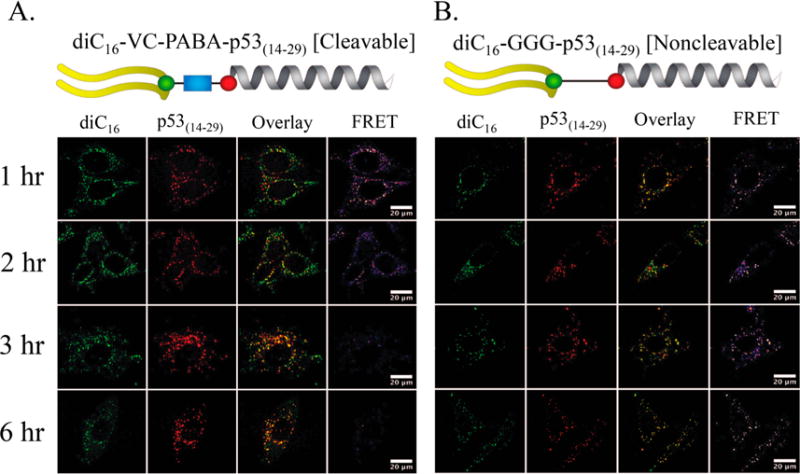
Real-time measurement of catB-mediated cleavage of (A) diC16–VC–PABA–p53(14–29) and (B) diC16–GGG–p53(14–29). HeLa cells were incubated for 1 h with respective PAs and then washed. The location of diC16 (green) and p53(14–29) (red) were followed for 6 h. FRET signaling was lower within 2 h in cells treated with the cleavable diC16–VC–PABA–p53(14–29) PA and almost undetectable by 6 h after incubation. Conversely, FRET signaling decreased but still remained in cells treated with noncleavable diC16–GGG–p53(14–29) PAs. Interestingly, the amount of diC16 and p53(14–29) in cells treated with diC16–GGG–p53(14–29) decreased at the same rate as FRET signaling in these cells suggesting the trafficking of diC16–GGG–p53(14–29) out of the cells. Scale bars: 20 μm.
Figure 6.
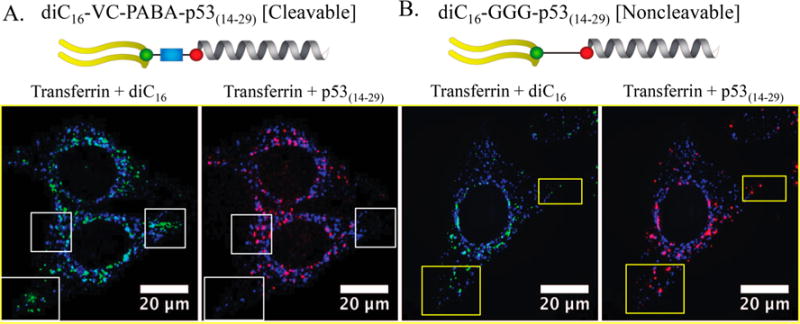
(A, B) Both diC16–VC–PABA–p53(14–29) and diC16–GGG–p53(14–29), localized in HeLa cells to transferrin-positive early endosomes after 1 h following incubation. However, diC16 and p53(14–29) from diC16–VC–PABA–p53(14–29) quickly dissociate and can be found in separate locations within the cells (white boxes) with diC16 in transferrin-negative compartments. Components from diC16–GGG–p53(14–29) PAs are located in identical locations (yellow boxes). Transferrin, blue; diC16, green; and p53(14–29), red. Scale bars: 20 μm.
To confirm that loss of FRET signal was due to cleavage of p53(14–29) from diC16 and not loss of FRET efficiency (due to loss of fluorescent intensity, photobleaching, etc.), HeLa cells were treated as above but with an increased PA concentration of 10 μM. Extracellular PA was washed away after 1 h, and cells were allowed to incubate for 6 and 24 h, followed by super-resolution laser scanning confocal microscopy. Raw images were analyzed for FRET signaling at each time point, comparing diC16–VC–PABA–p53(14–29) to diC16–GGG–p53(14–29) and nontreated cells (Figure 7). While FRET signaling decreased from 6 to 24 h in cells treated with diC16–VC–PABA–p53(14–29) (Figure 5), there was no change in FRET efficiency of diC16–GGG–p53(14–29)-treated cells. Therefore, cleavage of p53(14–29) from diC16 and movement of p53(14–29) to spatially distinct areas of the cell occurred only in relation to diC16–VC–PABA–p53(14–29) and was not due to loss of the ability of intact PAs to provide a quantifiable FRET signal over time (Figure 7A).
Figure 7.
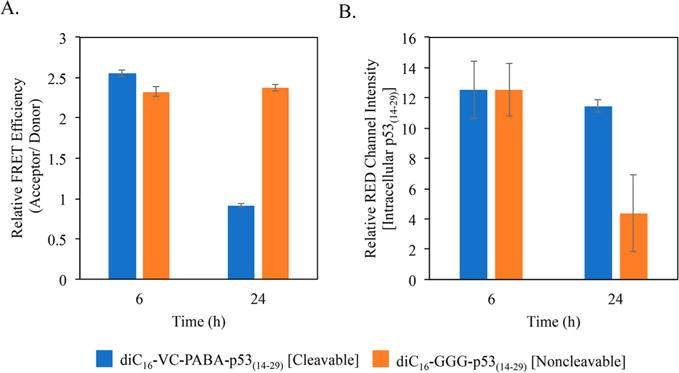
Superresolution laser-scanning confocal microscopy measurements of the ability of diC16–VC–PABA–p53(14–29) and diC16–GGG–p53(14–29) to provide a FRET signal (FRET Efficiency) and to quantify the amount of intracellular p53(14–29) peptide. (A) Although the FRET signal decreased in cells incubated with both PAs over time (Figure 5), the remaining diC16–GGG–p53(14–29) within HeLa cells retained its ability to provide a FRET signal up to 24 h, indicating that the loss of FRET signal in cells treated with diC16–VC–PABA–p53(14–29) was not due to photobleaching during analysis. (B) Superresolution confocal microscopy measurements of the loss of p53(14–29) peptide in cells incubated with diC16–GGG–p53(14–29) compared with those incubated with diC16–VC–PABA–p53(14–29) by 24 h, indicating extracellular loss.
While FRET intensity changes indicate catB-mediated cleavage, it lacks quantifiable information regarding the amount of either PA component in individual cells. To quantify the amount of p53(14–29) peptide in individual cells following incubation for 24 h, we measured Tamra intensity alone at 520 nm excitation and fluorescence at 580–660 nm. Using this method, the relative amount of intracellular p53(14–29) peptide was similar between diC16–VC–PABA–p53(14–29) and diC16–GGG–p53(14–29)-treated cells at 6 h (Figure 7B). However, by 24 h, p53(14–29) peptide levels dropped significantly in cells treated with diC16–GGG–p53(14–29), confirming our previous results under continuous treatment conditions (Figure 4). One explanation for this decrease over time is that diC16 leads to endosomal membrane tethering and facilitates recycling of the intact diC16–GGG–p53(14–29) PA monomers out of the cells.
Extracellular Trafficking of Intact PA Monomers and Individual PA Components
We next sought to determine if the overall loss of diC16–GGG–p53(14–29) monomers or diC16 from diC16–VC–PABA–p53(14–29)-treated cells was due to membrane recycling and extrusion via extracellular vesicles. The hydrophobic tails of PAs are thought to promote lipid membrane anchoring of PA monomers and subsequent membrane tethering.20,27,51,55 Membrane invaginations during endocytosis therefore contain these and other extracellular lipids that are transported through the endolysomal pathway and either metabolized through autophagocytosis or refluxed out of the cell within extracellular vesicles.56,57 Given the rapid intracellular trafficking of PAs, we wondered if the loss of diC16–GGG–p53(14–29) signal over time was due to the active exocytosis of noncleaved PA monomers.
To measure extracellular vesicles, HeLa cells were incubated for 1 h followed by washing and replacement with PA-free media. The cells were then allowed to incubate, and media samples were collected at 6 h following incubation. Vesicles within the media were analyzed using a Nanosight N300 with fluorescence filters, and nanoparticle tracking analysis (NTA) software was used to determine the number of total and red particles per frame over a threshold of a constant intensity (Figure 8). The number of red extracellular particles were lower in media from cells incubated with diC16–VC–PABA–p53(14–29) compared with cells treated with diC16–GGG–p53(14–29) PAs supporting efflux of intact diC16–GGG–p53(14–29) and either intracellular accumulation (as seen in Figures 4–6) or the metabolism of p53(14–29) peptides. The number of peptides within each vesicle could not be determined using this technique. Coincident measurement of diC16-laded vesicles could not be performed accurately due to limitations in the Nanosight laser and detector thresholds. Despite these limitations, these results support our earlier findings and indicate that the hydrophobic diC16 tails drive the excretion and recycling of intact PA monomers in our system.
Figure 8.
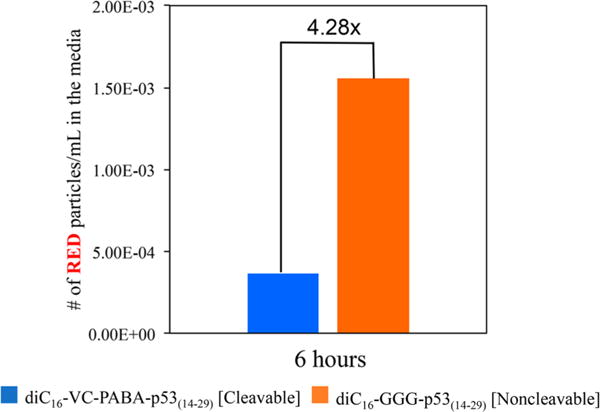
Extracellular vesicle examination of media from HeLa cells incubated with diC16–VC–PABA-p53(14–29) or diC16–GGG–p53(14–29). The number of extracellar vesicles that are filtered by the red channel (p53(14–29)-containing fraction), as measured by red fluorescence, is 4.28× greater from cells incubated with diC16–GGG–p53(14–29) compared with those incubated with diC16–VC–PABA–p53(14–29). This supports that more p53(14–29) remained intracellular when treated with diC16–VC–PABA–p53(14–29) and that intact diC16–GGG–p53(14–29) PAs were shuttled in an extracellular manner. (A representative analysis of three independent experiments is shown.)
CONCLUSIONS
The ultimate goal of this study was to allow for controlled release of biologic peptides following PA cellular penetration and prevent rapid extracellular excretion. This is the first time, to our knowledge, that a PA has been designed with two dyes to individually follow long-term intracellular and extracellular trafficking of the functional peptide group and the hydrophobic tail. The FRET effect of the two dyes allows the visualization of effective early cleavage soon following treatment. Our results indicate that catB-mediated cleavage between p53(14–29) and diC16 is feasible and that therapeutic peptides are able to accumulate inside target cells, greatly improving their chance at biologic efficacy. It remains unclear if the p53(14–29) peptide that accumulates within the cell remains entrapped within endosomal vesicles or is able to pass through the endosome and localize to other organelles (Figure 1).
Peptide accumulation within target cells remains a formidable obstacle in using macromolecules, peptides, and nucleotide-based nanoparticle therapeutics, and endosomal escape remains the largest rate-limiting step in delivering therapeutic peptides to target cellular compartments.28,58–60 Reinforcement of the peptide secondary structure or addition of cationic membrane-penetrating amino acids could theoretically facilitate cytoplasmic trafficking.15,59–61 Future studies aim to measure p53(14–29) movement within treated cells and enhance endosomal escape and organelle trafficking.
Removal of the hydrophobic lipid tail may also be ultimately necessary for efficient target PPI engagement by way of decreasing the expected steric obstruction at the binding interface (manuscript in preparation).62–64 Through relieving hindrance, cathepsin-cleavable PA designs can be applied to peptides targeting any number of intracellular or extracellular PPIs and thus can be adapted for use as therapeutics, diagnostics, or molecular tools to elucidate PPI mechanisms of disease.
EXPERIMENTAL PROCEDURES
Peptide Amphiphile Synthesis
All amino acids were purchased from Protein Technologies Inc. Peptide p53(14–29) (LSQETFSDLWKLLPEN) was synthesized on Rink amide resin (Novabiochem) using a standard Fmoc solid-phase peptide synthesis strategy on an automated peptide synthesizer (Protein Technologies Inc.), as previously described.51 The coupling of 5-carboxyfluorescein (FAM) and 5(6)-carboxyte-tramethylrhodamine (Tamra; Novabiochem) were performed through the orthogonal side chain protections of Fmoc–Lys(Mtt)–OH and Fmoc–Lys(Dde)–OH (Novabiochem), respectively. A total of 2 equiv (with respect to resin substitution) of each dye dissolved in dimethylformamide (DMF) with 4× N,N-diisopropylethylamine (DIPEA) and 1.95× 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) were used for coupling to the ε-amine of lysine for 24 h at room temperature. The dialkyl lipid acid 4-(1,5-bis(hexadecyloxy)-1,5-dioxopentan-2-ylamino)-4-oxobutanoic (diC16COOH) was synthesized as described previously.65 The Fmoc group of the N-terminal lysine was cleaved with 20% piperidine in DMF, and the free α-amine group of the lysine-containing peptides were conjugated with 2× diC16COOH hydrophobic tail in DMF with 4× DIPEA and 1.95× HATU, as previously reported.66 The coupling reaction shook for 24 h at room temperature. Complete cleavage from the resin was achieved using a trifluoroacetic acid (TFA)/triisopropylsilane/water (98:1:1) solution. The resulting product was precipitated in cold diethyl ether prior to purification.
Modified peptides were purified by reverse-phase preparative high-performance liquid chromatography (RP-HPLC; Prominence, Shimadzu Corporation, Kyoto, Japan) with an XBridge Prep C8 OBD column (Waters Corporation, Milford, MA) at 50 °C (flow rate: 10 mL/min from 10% to 100% within 55 min). Product identity was confirmed using matrix-assisted laser desorption–ionization (MALDI) mass spectrometry (Bruker Ultraflextreme MALDI-TOF), as previously described.29
Micelle Formation
PAs were dissolved in chloroform and the solvent evaporated under N2 gas to form a layer on the wall of the eppendorf tube. Milli-Q water (or PBS for cell culture experiments; pH 7.4) was added to the PAs, sonicated for 1 h at 60 °C, and then incubated in a hot bath without sonication for 1 h for 60 °C. After cooling to room temperature, the micelle solutions were filtered through a 0.45 μm polycarbonate syringe filter (Millipore).
Critical Micelle Concentration
CMC was performed as previously described.67 A range of PA concentrations (from 512 to 0.01 μM in half-increments) were prepared in a 1 μM DPH aqueous solution and equilibrated for 1 h at room temperature. Solutions were plated in triplicates in a black 96-well plate, and their fluorescent intensity was measured using a Tecan Infinite M200 PRO plate reader (Mannedorf, Switzerland). Data were fit with two trend lines for low- and high-intensity measurements, and CMC was calculated as the inflection point where the two trend lines meet.68
Dynamic Light Scattering
Micelle size was assessed using dynamic light scattering (Brookhaven Instruments, Holtzville, NY) as previously described. Stock solutions of 0.5 mM micelles were prepared in water as described above, and DLS measurements were performed at a 90° angle and in a 637 nm system consisting of a BI-200SM goniometer and a BI-9000AT autocorrelator. Hydrodynamic radii were determined via the Stokes–Einstein equation using the diffusion coefficient determined from the auto correlation function.
Transmission Electron Microscopy
Ultrathin carbon type-A 400 mesh copper grids (Ted Pella, Redding, CA) were loaded with 5 μL of 0.5 mM PA micelles and allowed to dry. Grids were washed with several drops of water and then negatively stained with 1% aqueous phosphotungstic acid for 3 min. The excess solution was then removed, and grids were left to dry. Grids were imaged on a FEI Tecnai 12 TEM using an accelerating voltage of 120 kV.
Synthesis of Peptide–AMC
Fmoc–Lys(carbamate Wang resin)–AMC (Novabiochem) was used to synthesize the cleavable groups on the resin individually. Basic solid-phase peptide synthesis, as detailed above, was used to synthesize VC–PABA–AMC and GGG–AMC peptides. TFA cleavage of 98% was used to cleave the peptide–AMC from the resin.
Cathepsin-B Cleavage Testing
Peptide–AMC
Recombinant human liver cathepsin-B (Sigma-Aldrich) was used for the in vitro experiments. DTT (0.25 μM) was used in 0.25 μM HEPES in PBS (pH 5) as activation buffer. Peptides were dissolved in activation buffer to a final concentration as 1 mM. A total of 0.5 μL of catB enzyme or vehicle control was added into the peptide solution. Plate-reader analysis with Tecan Infinite M200 PRO plate reader (Mannedorf, Switzerland) with triplicates of each sample were performed in 96-well plates. The intensity of the excited dye at 388 nm was measured at 440 nm.
Dual-Dye-Labeled Peptide on Resin
CatB and activation buffer were prepared as described above. Dye-labeled peptides were again left on the resin. Resin was washed with methanol, dried in a vacuum overnight, and then immersed in PBS (pH 7.4) for 1 h at 37 °C. PBS was then drained followed by addition of 1 mL of activation buffer and 5 μL of catB for 100 mg of resin. Control testing was performed in the same solution without the addition of catB. Supernatant was collected after 3 and 24 h. The florescence intensity of triplicates of each sample was measured with plate reader FAM (λexc = 485 nm and λem = 535 nm) and Tamra (λexc = 520 nm and λem = 620 nm).
FRET Measurements with Wide-Field Microscopy
Peptide-laden resins were washed with methanol, vacuum-dried overnight, and then immersed in PBS (pH 7.4) for 1 h at 37 °C. After the PBS was drained, 100 μL of activation buffer and 5 μL of enzyme was added to the edge of the well containing peptide -resin at 37 °C. FRET change based on enzyme cleavage was measured by a home-built two-channel FRET imaging system. Figure S3 depicts the experimental setup. The system is based on an inverted microscope (Nikon Ti) with differential interference contrast (DIC) imaging components. The excitation light from a CW-laser source (λex = 488 nm; Cobalt) is combined with a fiber optics and sent to the total internal reflection fluorescence (TIRF) illumination combiner attached to the back port of the microscope. Light was reflected by a dichroic beam splitter (quadband) and focused onto the resin beads attached to the two dye-labeled peptides by a high numerical-aperture oil-immersion objective (1.4 NA, 100×). The fluorescence signal emitted from the FRET donor (FAM) and acceptor (Tamra) was nonpolarized and relayed to the camera with combination two 200 mm achormatic doublet lenses applying the 4f relay system methods. The emission signals were passed through a 500 nm long-pass filter to obtain the fluorescence images and intensity trajectories. A dichroic beam-splitter (555 nm long pass) at an orientation of a 45° angle on the direction of the signal separates out the beam, depending on the color of light. A dichroic beam splitter transmits acceptor signal and reflect the donor signal. Donor (FAM) and acceptor (Tamra) signals were passed through band-pass filters at 525/50 and 605/50 nm, respectively. The donor and acceptor channels were then reflected by two mirrors and focused to a 1024 × 1024 pixel electron-multiplying charge-coupled device (EMCCD) camera (Andore iXon 888) through a 2 in. achromatic doublet lens. The fluorescence signals were recorded using a time lapse-video with acquisition times of 10 ms and interval times of 30 or 60 s.
Cell Culture
HeLa cells were maintained in Dulbeccos modified Eagle medium (DMEM; Invitrogen) supplemented with 10% FBS, 100 U mL−1 penicillin–streptomycin, 2 mM L-glutamine, and 0.1 mM MEM nonessential amino acids. Cells were grown at 37 °C in a humidified atmosphere and 5% CO2. Cells were allowed to attach on the surfaces overnight (12 h).
Peptide Amphiphile Treatment of Cells for Confocal Analysis
HeLa cells were incubated with 2.5 μM PAs and 0.1 μM transferrin (Alexa Fluor 647 labeled, Thermo Fisher Science) for 1 h in advanced DMEM (Invitrogen) supplemented with 1% FBS. The media was then removed, and cells were washed and either fixed immediately with 4% paraformaldehyde in PBS for 10 min at room temperature or replaced with new peptide-free media; next, cells were allowed to incubate for another 1, 2, or 5 h before being fixed. The fixed cells were then washed and left in PBS before being imaged. For accumulation experiments, 10 μM PAs were incubated individually and left on the cells for 24 h, and the confocal images were taken in different time periods from live cells.
Confocal and Superresolution Microscopy
Images were taken by a Marianas Yokogawa-type spinning disk (inverted confocal microscope). The following lasers were used: (1) green: λexc = 488 nm and green filter; (2) red: λexc = 565 nm and red filter; (3) transferrin: λexc = 640 nm and far-red filter; and (4) FRET channel: λexc = 488 nm and red filter. Super-resolution images were taken on a Leica SP5 II STED-CW super-resolution laser scanning confocal microscope. All imaging was performed at the Integrated Light Microscope Core Facility at the University of Chicago. Images were analyzed by ImageJ software.
Extracellular Vesicle Analysis
HeLa cells were grown in the T25 with 10% FBS. Cells were washed twice with PBS and incubated with (1) 10 μM diC16–GGG–p53(14–29), (2) 10 μM diC16–VC–PABA–p53(14–29), and (3) media alone for 1 h in advanced DMEM supplemented with 1% FBS. This media was then removed, and cells were washed twice with PBS. New 10% FBS media was then added on the cells. Following incubation for 6 and 24 h, 1 mL of media was collected from each of the samples and analyzed using a Nanosight NS300 flow cell (Nanosight, UK) following the manufacturer protocol. Nanoscale particles (10–1000 nm) were analyzed using the NTA software for size distribution and total number of particles per frame. Particles were also tracked using red filters to detect red-laden particles. The ratio of detected red particles per milliliter to total particle per milliliter for each sample was then calculated.
Supplementary Material
Acknowledgments
We thank Eric Smith for graphics assistance. We also thank Dr. Vytas Bindokas and Dr. Christine Labno at the Integrated Light Microscope Core Facility, University of Chicago for their help during the imaging. The authors also acknowledge M. Necdet Ersoy and family for their support of this research as well as the NIH’s MSTP training grant (no. T32GM007281).
ABBREVIATIONS
- PPI
protein-protein interaction
- PA
peptide amphiphile
- FRET
Forster resonance transfer
- catB
cathepsin-B
- PABC
para-amino benzyl carbamate
- PABA
para-amino benzoic acid
- diC16
double palmitic acid
- VC
valine–citruline
- GGG
glycine–glycine–glycine
- AMC
7-amino-4-methylcoumarin
- TEM
transmission electron microscopy
- DLS
dynamic light scattering
- FAM
5-carboxyfluorescein
- Tamra
5(6)-carboxyte-tramethylrhodamine
- DMF
dimethylformamide
- TFA
trifluoroacetic acid
- CMC
critical micellar concentration
- PBS
phosphate-buffered saline
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.bioconj-chem.7b00364.
Figures showing experimental chemical structures, length between FAM and FRET fluorophores, wide-field microscope setup, relative intensities, LC–MS data, dynamic light scattering and critical micellar concentration results, and HPLC analysis. (PDF)
ORCID
James L. LaBelle: 0000-0001-6776-4695
Notes
The authors declare no competing financial interest.
References
- 1.London N, Raveh B, Schueler-Furman O. Druggable protein-protein interactions–from hot spots to hot segments. Curr Opin Chem Biol. 2013;17(6):952–9. doi: 10.1016/j.cbpa.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. A proteome-scale map of the human interactome network. Cell. 2014;159(5):1212–26. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 2005;17(6):631–6. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–70. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9(12):862–73. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- 6.Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12(4):355–66. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a non-overlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29(1):92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 9.Bond GL, Hu W, Levine A. A single nucleotide polymorphism in the MDM2 gene: from a molecular and cellular explanation to clinical effect. Cancer Res. 2005;65(13):5481–4. doi: 10.1158/0008-5472.CAN-05-0825. [DOI] [PubMed] [Google Scholar]
- 10.Ohtsubo C, Shiokawa D, Kodama M, Gaiddon C, Nakagama H, Jochemsen AG, Taya Y, Okamoto K. Cytoplasmic tethering is involved in synergistic inhibition of p53 by Mdmx and Mdm2. Cancer Sci. 2009;100(7):1291–9. doi: 10.1111/j.1349-7006.2009.01180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 12.Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, Inguilizian H, Mermel C, Currie T, Dogan A, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell. 2012;22(3):359–72. doi: 10.1016/j.ccr.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acar H, Srivastava S, Chung EJ, Schnorenberg MR, Barrett JC, LaBelle JL, Tirrell M. Self-assembling peptide-based building blocks in medical applications. Adv Drug Delivery Rev. 2017:110–111. 65–79. doi: 10.1016/j.addr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Ceil. 2010;18(5):411–22. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shangary S, Ding K, Qiu S, Nikolovska-Coleska Z, Bauer JA, Liu M, Wang G, Lu Y, McEachern D, Bernard D, et al. Reactivation of p53 by a specific MDM2 antagonist (MI-43) leads to p21-mediated cell cycle arrest and selective cell death in colon cancer. Mol Cancer Ther. 2008;7(6):1533–42. doi: 10.1158/1535-7163.MCT-08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Nati Acad Sci U S A. 2008;105(10):3933–8. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 19.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129(9):2456–7. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Missirlis D, Krogstad DV, Tirrell M. Internalization of p53(14–29) peptide amphiphiles and subsequent endosomal disruption results in SJSA-1 cell death. Mol Pharmaceutics. 2010;7(6):2173–84. doi: 10.1021/mp100193h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110(36):E3445–54. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450(7172):1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 23.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5(3):161–73. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 24.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 25.Lim YB, Lee E, Lee M. Cell-penetrating-peptide-coated nanoribbons for intracellular nanocarriers. Angew Chem, Int Ed. 2007;46(19):3475–3478. doi: 10.1002/anie.200604576. [DOI] [PubMed] [Google Scholar]
- 26.Trent A, Marullo R, Lin B, Black M, Tirrell M. Structural properties of soluble peptide amphiphile micelles. Soft Matter. 2011;7(20):9572–9582. [Google Scholar]
- 27.Missirlis D, Chworos A, Fu CJ, Khant HA, Krogstad DV, Tirrell M. Effect of the peptide secondary structure on the peptide amphiphile supramolecular structure and interactions. Langmuir. 2011;27(10):6163–70. doi: 10.1021/la200800e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–51. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Missirlis D, Teesalu T, Black M, Tirrell M. The non-peptidic part determines the internalization mechanism and intracellular trafficking of peptide amphiphiles. PLoS One. 2013;8(1):e54611. doi: 10.1371/journal.pone.0054611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan R, Richardson SC. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol Pharmaceutics. 2012;9(9):2380–402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 31.Yu B, Tai HC, Xue W, Lee LJ, Lee RJ. Receptor-targeted nanocarriers for therapeutic delivery to cancer. Mol Membr Biol. 2010;27(7):286–98. doi: 10.3109/09687688.2010.521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thanou M, Duncan R. Polymer-protein and polymer-drug conjugates in cancer therapy. Curr Opin Investig Drugs. 2003;4(6):701–9. [PubMed] [Google Scholar]
- 33.Li YC, Rodewald LW, Hoppmann C, Wong ET, Lebreton S, Safar P, Patek M, Wang L, Wertman KF, Wahl GM. A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep. 2014;9(5):1946–58. doi: 10.1016/j.celrep.2014.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, Knipe JO, Lasch SJ, Trail PA. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002;13(4):855–69. doi: 10.1021/bc025536j. [DOI] [PubMed] [Google Scholar]
- 35.Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, Loo C, et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjugate Chem. 2015;26(4):650–9. doi: 10.1021/bc5005747. [DOI] [PubMed] [Google Scholar]
- 36.Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett. 1998;8(23):3341–3346. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]
- 37.Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33(7):733–5. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 38.Carter PJ, Senter PD. Antibody-drug conjugates for cancer therapy. Cancer J. 2008;14(3):154–69. doi: 10.1097/PPO.0b013e318172d704. [DOI] [PubMed] [Google Scholar]
- 39.Staben LR, Koenig SG, Lehar SM, Vandlen R, Zhang D, Chuh J, Yu SF, Ng C, Guo J, Liu Y, et al. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody-drug conjugates. Nat Chem. 2016;8(12):1112–1119. doi: 10.1038/nchem.2635. [DOI] [PubMed] [Google Scholar]
- 40.Bouchard H, Viskov C, Garcia-Echeverria C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24(23):5357–63. doi: 10.1016/j.bmcl.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 41.Farias SE, Strop P, Delaria K, Casas MG, Dorywalska M, Shelton DL, Pons J, Rajpal A. Mass Spectrometric Characterization of Transglutaminase Based Site-Specific Antibody-Drug Conjugates. Bioconjugate Chem. 2014;25(2):240–250. doi: 10.1021/bc4003794. [DOI] [PubMed] [Google Scholar]
- 42.Maniganda S, Sankar V, Nair JB, Raghu KG, Maiti KK. A lysosome-targeted drug delivery system based on sorbitol backbone towards efficient cancer therapy. Org Biomol Chem. 2014;12(34):6564–6569. doi: 10.1039/c4ob01153h. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Delivery Rev. 2017:110–111. 112–126. doi: 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burkhart DJ, Barthel BL, Post GC, Kalet BT, Nafie JW, Shoemaker RK, Koch TH. Design, synthesis, and preliminary evaluation of doxazolidine carbamates as prodrugs activated by carboxylesterases. J Med Chem. 2006;49(24):7002–12. doi: 10.1021/jm060597e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maly DJ, Huang L, Ellman JA. Combinatorial strategies for targeting protein families: Application to the proteases. ChemBioChem. 2002;3(1):16–37. doi: 10.1002/1439-7633(20020104)3:1<16::AID-CBIC16>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 46.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Ada, Proteins Proteomics. 2012;1824(1):68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5(6):507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Law B, Weissleder R, Tung CH. Mechanism-based fluorescent reporter for protein kinase A detection. ChemBioChem. 2005;6(8):1361–1367. doi: 10.1002/cbic.200500027. [DOI] [PubMed] [Google Scholar]
- 49.Ochocki JD, Mullen DG, Wattenberg EV, Distefano MD. Evaluation of a cell penetrating prenylated peptide lacking an intrinsic fluorophore via in situ click reaction. Bioorg Med Chem Lett. 2011;21(17):4998–5001. doi: 10.1016/j.bmcl.2011.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salim M, Minamikawa H, Sugimura A, Hashim R. Amphiphilic designer nano-carriers for controlled release: from drug delivery to diagnostics. MedChemComm. 2014;5(11):1602–1618. [Google Scholar]
- 51.Missirlis D, Khant H, Tirrell M. Mechanisms of Peptide Amphiphile Internalization by SJSA-1 Cells in Vitro. Biochemistry. 2009;48(15):3304–3314. doi: 10.1021/bi802356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopkins CR. Intracellular Routing of Transferrin and Transferrin Receptors in Epidermoid Carcinoma A431-Cells. Cell. 1983;35(1):321–330. doi: 10.1016/0092-8674(83)90235-0. [DOI] [PubMed] [Google Scholar]
- 53.Hopkins CR, Trowbridge IS. Internalization and Processing of Transferrin and the Transferrin Receptor in Human Carcinoma A431-Cells. J Cell Biol. 1983;97(2):508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5(2):121–32. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 55.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 56.Jaishy B, Abel ED. Lipids, lysosomes, and autophagy. J Lipid Res. 2016;57(9):1619–35. doi: 10.1194/jlr.R067520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol CellBiol. 2008;9(2):112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dupont E, Prochiantz A, Joliot A. Penetratin Story: An Overview. Methods Mol Biol. 2015;1324:29–37. doi: 10.1007/978-1-4939-2806-4_2. [DOI] [PubMed] [Google Scholar]
- 59.Lonn P, Dowdy SF. Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin Drug Delivery. 2015;12(10):1627–36. doi: 10.1517/17425247.2015.1046431. [DOI] [PubMed] [Google Scholar]
- 60.Lonn P, Kacsinta AD, Cui XS, Hamil AS, Kaulich M, Gogoi K, Dowdy SF. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci Rep. 2016;6:32301. doi: 10.1038/srep32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122(6):2018–31. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chanphai P, Bekale L, Tajmir-Riahi HA. Effect of hydrophobicity on protein-protein interactions. Eur Polym J. 2015;67:224–231. [Google Scholar]
- 63.Dyson HJ, Wright PE, Scheraga HA. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc Natl Acad Sci U S A. 2006;103(35):13057–13061. doi: 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Young L, Jernigan RL, Covell DG. A Role for Surface Hydrophobicity in Protein-Protein Recognition. Protein Sci. 1994;3(5):717–729. doi: 10.1002/pro.5560030501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berndt P, Fields GB, Tirrell M. Synthetic Lipidation of Peptides and Amino-Acids – Monolayer Structure and Properties. J Am Chem Soc. 1995;117(37):9515–9522. [Google Scholar]
- 66.Chung EJ, Cheng Y, Morshed R, Nord K, Han Y, Wegscheid ML, Auffinger B, Wainwright DA, Lesniak MS, Tirrell MV. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials. 2014;35(4):1249–56. doi: 10.1016/j.biomaterials.2013.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trent A, Ulery BD, Black MJ, Barrett JC, Liang S, Kostenko Y, David NA, Tirrell MV. Peptide amphiphile micelles self-adjuvant group A streptococcal vaccination. AAPS J. 2015;17(2):380–8. doi: 10.1208/s12248-014-9707-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Black M, Trent A, Kostenko Y, Lee JS, Olive C, Tirrell M. Self-assembled peptide amphiphile micelles containing a cytotoxic T-cell epitope promote a protective immune response in vivo. Adv Mater. 2012;24(28):3845–9. doi: 10.1002/adma.201200209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.