

Abstract
The epidermal growth factor receptor (EGFR) kinase is activated by a variety of mutations in human cancers. R776H is one such recurrent mutation (R752H in another numbering system) in the αC-β4 loop of the tyrosine kinase domain that activates EGFR in the absence of the activating EGF ligand. However, the mechanistic details of how R776H contributes to kinase activation are not well understood. Here using cell-based cotransfection assays, we show that the R776H mutation activates EGFR in a dimerization-dependent manner by preferentially adopting the acceptor position in the asymmetric dimer. The acceptor function, but not the donor function, is enhanced for the R776H mutant, supporting the “superacceptor” hypothesis proposed for oncogenic mutations in EGFR. We also find that phosphorylation of monomeric EGFR is increased by R776H mutation, providing insights into EGFR lateral phosphorylation and oligomerization. On the basis of molecular modeling and molecular dynamics simulation, we propose a model in which loss of key autoinhibitory αC-helix capping interaction and alteration of coconserved cis regulatory interactions between the kinase domain and the flanking regulatory segments contribute to mutational activation. Since the R776 equivalent position is mutated in ErbB2 and ErbB4, our studies have implications for understanding kinase mutational activation in other ErbB family members as well.
Graphical abstract
Epidermal growth factor receptor (EGFR) associated pathways are critical for regulating cell growth, proliferation, differentiation, and survival.1–3 While EGFR signaling is tightly controlled by a diverse array of regulatory mechanisms in normal cells, in many cancer cells, the regulatory constraint on EGFR signaling is lost, resulting in abnormal cell growth and proliferation.2–5 Indeed, cancer genome sequencing studies have revealed hundreds of mutations in EGFR, many of which map to the intracellular kinase domain. The kinase domain of EGFR is also a major target for drug discovery, and many clinically approved inhibitors, such as getfitinib, erlotinib, and afatinib, target the ATP binding pocket to inhibit kinase activity.2,3 However, the sensitivity of these inhibitors varies based on mutations in the kinase domain. For example, L858R, the most commonly observed lung cancer mutation in EGFR, shows increased sensitivity toward gefitinib,6,7 whereas L861Q, a mutation only three residues C-terminal of L858, is relatively insensitive to gefitinib.7,8 The differential sensitivity of kinase domain mutations to cancer drugs opens up the exciting possibility of administering personalized drugs based on the mutational profile of the patient’s genome. To fully realize this possibility, however, a detailed mechanistic understanding of how mutations alter kinase structure, function, and regulation is needed.
Detailed structural and functional studies on wild type (WT) and mutant EGFR have provided important insights into EGFR regulation in normal and disease states. It is well established that ligand binding to the extracellular domain of WT EGFR induces activation of the intracellular kinase domain by promoting an asymmetric dimer.9 In the asymmetric dimer, the substrate binding C-lobe of one EGFR molecule (donor) docks to the ATP binding N-lobe of the other EGFR molecule (acceptor) to position the acceptor αC-helix in the active conformation.9 Stabilization of the αC-helix results in allosteric activation of the acceptor by positioning key catalytic residues in the active site.9 However, in disease states, dimerization of the kinase domain is altered by oncogenic mutations.10,11 Most oncogenic mutations are dimerization dependent,10,12,13 and based on enhanced association of mutant (L858R and the co-occurring gatekeeper T790M mutation) and WT EGFR, Brewer et al. proposed a “superacceptor” model of EGFR activation in which oncogenic mutations enhance kinase activity by preferentially adopting the acceptor position in the asymmetric dimer.13 However, it is not known whether the “superacceptor” model is specific to the L858R/T790M mutant or is more generally adopted by other oncogenic mutations.
Molecular dynamics simulations have emerged as a powerful tool to study kinase regulation in disease and normal states.11,14–18 Long time scale simulations of WT and mutant EGFR have shown that the αC-helix of WT EGFR is intrinsically disordered, and cancer mutations activate the kinase domain by quenching intrinsic disorder.11 In particular, the L858R mutation was proposed to activate the kinase domain by stabilizing the flexible αC-helix, the precise positioning of which is critical for kinase dimerization and activation. Likewise, comparisons of molecular dynamics trajectories in various conformational states have provided insights into kinase domain regulation by flanking juxtamembrane (JM) and C-terminal tail segments, and allosteric regulation by oncogenic mutations.19 Free energy landscape analysis, likewise, have provided insights into epistatic interactions altered by oncogenic mutations16 and community structures associated with kinase allosteric communication.20
Although cancer genome sequencing studies have revealed more than 800 unique mutations in EGFR, only a handful of recurrent mutations have been structurally and functionally characterized. There are several less frequently occurring mutations whose mechanisms of action are poorly understood. R776H/C/G is one such mutation in the αC-β4 loop region of EGFR kinase domain (Figure S1). A total of 22 unique samples (patients) have been reported to carry mutations at R776 position in COSMIC database, and 14 of them are R776H mutation. In one clinical report, R776H mutation is associated with lung cancer patients without smoking history and is found both in normal and tumor tissues.26 In addition, R776H/C mutations are known to co-occur with other common oncogenic mutations, such as L858R, G719A, and L861Q, and confer sensitivity to cancer drugs.26–28 R776H/C mutations have been shown to activate EGFR kianse domain in the absence of the EGF ligand.21 However, the mechanism by which R776H/C activates the kinase domain is not fully understood.
Here we employ a combination of computational and experimental approaches to characterize the R776H mutation in EGFR. First, we show that R776H activates EGFR in a ligand-independent manner; however, the mutant still relies on the asymmetric dimer for its activity. When coexpressed with WT EGFR, R776H mutant preferentially adopts the acceptor position to better exert its activity;i.e., it functions as a “superacceptor”. We further demonstrate that the enzymatic activity of R776H is not restricted to the dimer itself in that the activated EGFR dimer can phosphorylate inactive monomeric EGFR in vivo. Lastly, we performed molecular dynamics to investigate the activation mechanism of R776H mutant. Our study suggests a model in which the R776H mutation activates EGFR by relieving autoinhibitory interactions with the αC-helix as well as the autoinhibitory C-terminal tail. Our results provide an emerging scheme of how mutations activate EGFR by regulation of the critical αC-helix and provide clues for designing mutant specific kinase inhibitors.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Anti-GFP, anti-p-Y1197, HRP conjugated mouse monoclonal and rabbit polyclonal antibodies were purchased from Cell signaling (Danvers, MA). Anti-FLAG and human recombinant EGF was purchased from Sigma (St. Louis, MO). Lipofectamine was obtained from Invitrogen (Carlsbad, CA). Protease inhibitor cocktail and G418 were purchased from Calbiochem. Quick-Change site-directed mutagenesis kit was bought from Statagene.
DNA Constructs
pEGFP-N1-EGFR plasmid was a kind gift from Dr. Graham Carpenter (Vanderbilt University, Nashville, TN). Mutagenesis was performed using the QuikChange II kit and confirmed via DNA sequencing.
Cell Culture and Transfections
CHO cells were grown in high-glucose Dubeccos modified Eagle medium (DMEM) (Cellgro, Manassas, VA, USA) with 10% fetal bovine serum (Bioexpress, UT, USA) without antibiotics. Transfection in CHO cells was performed using lipfectamine-2000 according to the manufacturer’s protocol with GFP-pEGFP-N1-WT-EGFR (WT), GFP-pEGFP-N1-R776H-EGFR (R776H), FLAG-pEGFP-N1-L704N-EGFR (L704N), FLAG-pEGFP-N1-R776H/L704N-EGFR (R776H/L704N), GFP-pEGFP-N1-V948R-EGFR (V948R), GFP-pEGFP-N1-R776H/V948R-EGFR (R776H/V948R), GFP-pEGFP-N1-Y1197F-EGFR (Y1197F), GFP-pEGFP-N1-R776H/Y1197F-EGFR (R776H/Y1197F), FLAG-pEGFP-N1-L704N/R776H/Y1197F-EGFR (L704N/R776H/Y1197F), FLAG-pEGFP-N1-L704N/V948R-EGFR (L704N/V948R), GFP-pEGFP-N1-D855G-EGFR (D855G), GFP-pEGFP-N1-R776H/D855G-EGFR (R776H/D855G) DNA constructs. Transiently transfected cell population was pooled, and protein expression was analyzed under a fluorescent microscope for GFP tagged WT/mutant EGFR and on Western blot using anti-GFP/FLAG antibody.
EGF Stimulation, Cell Lysis, and Immunoblotting
CHO cells stably transfected with WT and mutant EGFR plasmids were cultured in DMEM containing 10% FBS on 60 mm plate. To detect autophosphorylation of WT and mutant EGFR, 30% confluent cells were serum-starved in Hams F-12 media for 18 h. EGF stimulation was carried out using 50 ng/mL human EGF for 5 min. Cells were washed and immediately lysed with lysis buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 10% Triton X-100, 1 mM PMSF, and 1× protease inhibitor cocktail Set V, EDTA-free). Total cell lysate was spun at 15 000 rpm at 4 °C for 5 min. Protein concentration of cell lysates was determined by Bradford assay. Samples for SDS-PAGE gel were prepared in 2× Laemelli buffer (25 µg total protein). Proteins were resolved on 10% SDS-PAGE and transferred onto polyvinylidenedifluoride (PVDF) membrane using Trans-Blot SD semidry transfer cell (Bio-Rad). Western blotting was done using anti-GFP, anti-pY1197, and anti-FLAG antibodies. Proteins were detected by using chemiluminescent substrate (Western Blotting ECL substrate, Pierce, Rockford, IL).
Modeling and Molecular Dynamics Simulations
The PDB structures of EGFR in active conformation (2GS6) were used to model the active state.9 The two disordered regions (β3-αC loop and part of C-terminal tail) were modeled using MODELLER.22 R776H mutation was then introduced in the modeled WT structure using the loop refine module. The backbone was nearly identical for both mutant and wild-type structures, and no steric clashes were observed in the final structures.
Molecular dynamics (MD) simulations were done using GROMACS version 4.6.1.23 All-atom modified AMBER ff99SB force field24 was used with TIP3P water in a box that was at least 1 nm bigger than the protein on all sides. Steepest descent and conjugate-gradient energy minimization was performed on the solvated protein for 10000 steps until the Fmax was less than 50 kcal/mol. NVT simulations were carried out by heating from 0 to 298.15 K by coupling it to a Berendsen thermostat for 200 ps. The restraints on the protein backbone atoms over multiple stages of equilibration under NPT ensemble (P = 1 atm, T = 298.15 K) were released to obtain a relaxed protein, and a Parinello–Rahman barostat was used to maintain pressure and density. The unrestrained MD productions were run for 1 µs using a time step of 3 fs and the NPT ensemble. The particle-mesh ewald (PME) method was used to calculate electrostatics with a cutoff distance of 2 nm. Root mean square deviation was checked to be stable before further analysis (Figure S2). Analysis of MD simulations was carried out using programs in the GROMACS suite. All protein visualization was done using PyMOL.25
RESULTS
R776H Mutation Activates EGFR in a Dimerization-Dependent Manner
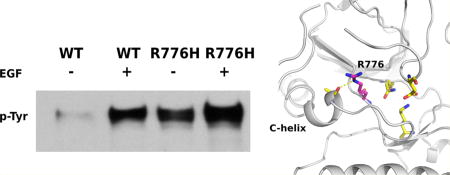
We previously demonstrated that the R776H mutant is constitutively active and displays catalytic activity in the absence of the activating EGF ligand21 (Figure 1a, lanes 3–4); however, the role of dimerization in R776H mediated EGFR activation was not studied. To test the dimerization dependency of R776H, we introduced an N-lobe dimerization deficient mutation L704N (L680N in another numbering system) and a C-lobe dimerization-deficient mutation V948R (V924R in another numbering system) in the R776H background.9 Western blot analysis indicates that dimerization-deficient mutants, R776H/L704N and R776H/V948R, are inactive (Figure 1a, lanes 5–8). However, Y1197 (Y1173 in another numbering system) phosphorylation can be restored when both constructs are coexpressed (Figure 1a, lanes 9–10), indicating that the activation of R776H relies on the intact dimerization interface. Thus, the asymmetric dimer is required for R776H mediated activation (Figure 1b). Ligand-independent phosphorylation of R776H also depends on the asymmetric dimer (Figure 1a, lanes 3, 9) because autophosphorylation is not detected when R776H is dimerization deficient (Figure 1a, lanes 5, 7).
Figure 1.

R776H depends on the asymmetric dimer for activation. (a) Lanes from left to right, WT EGFR (−), WT EGFR (+), R776H (−), R776H (+), R776H/V948R (−), R776H/V948R (+), R776H/L704N (−), R776H/L704N (+), R776H/V948R and R776H/L704N (−), R776H/V948R and R776H/L704N (+). + and − indicate the presence and absence of EGF ligand, respectively. WT EGFR, R776H, R776H/V948R are GFP tagged, whereas R776H/L704N is FLAG tagged. (b) Cartoon representation of the asymmetric dimer that is formed between R776H/V948R and R776H/L704N.
R776H Mutant Preferentially Adopts the Acceptor Position in the Asymmetric Dimer
To test whether R776H mutant preferentially adopts the acceptor position in the asymmetric dimer, we used the complementation assay, as described in a recent study.13 Specifically, we reconstituted the asymmetric dimer by coexpressing designated acceptor (V948R and R776H/V948R) (Figure 2, lanes 4, 6, 14, 16) and donor (L704N and R776H/L704N) (Figure 2, lanes 3, 5, 13, 15) constructs and probed for C-terminal tail auto-phosphorylation (Y1197). Y1197 phosphorylation is greatly enhanced when R776H mutant is in the acceptor position (Figure 2, lane 7 vs 8, 9 vs 10 and Figure 2, lane 17 vs 18, lane 19 vs 20), suggesting that the R776H mutant has a higher intrinsic kinase activity compared to WT. Cotransfection of enforced donor and acceptor constructs shows that phosphorylation of Y1197 decreases when R776H is in the donor position (Figure 2, lane 7 vs 9, 8 vs 10, 17 vs 19, and 18 vs 20). Interestingly, the highest tyrosine phosphorylation is observed when the WT donor (L704N) is reconstituted with R776H acceptor (R776H/V948R) (Figure 2, lane 8, 18). Taken together, these results indicate that the R776H mutant preferentially adopts the acceptor position when paired with WT EGFR, providing support for the “superacceptor” hypothesis proposed for other lung cancer mutations.13
Figure 2.

R776H is a “superacceptor”. + and − indicate the presence and absence of EGF ligand, respectively. Lanes from left to right, WT EGFR (−), R776H (−), L704N (−), V948R (−), R776H/L704N (−), R776H/V948R (−), L704N and V948R (−), L704N and R776H/V948R (−), R776H/L704N and V948R (−), R776H/L704N and R776H/V948R (−), WT EGFR (+), R776H (+), L704N (+), V948R (+), R776H/L704N (+), R776H/V948R (+), L704N and V948R (+), L704N and R776H/V948R (+), R776H/L704N and V948R (+), R776H/L704N and R776H/V948R (+). + and − indicate the presence and absence of EGF ligand, respectively. WT EGFR, R776H, V948R, R776H/V948R are GFP tagged, whereas L704N and R776H/L704N are FLAG tagged.
R776H Mutant Enhances Lateral Phosphorylation of Monomeric EGFR
In the traditional view of EGFR activation, only the acceptor kinase is activated upon dimerization.9 Once activated, the acceptor kinase phosphorylates tyrosine residues in the C-terminal tail of the donor in a trans manner, although recent coarse-grained MD simulation suggest an alternative hypothesis in which the acceptor kinase is phosphorylated in cis.29 To test whether the C-terminal tail of the acceptor kinase is phosphorylated in the asymmetric dimer in vivo, we mutated Y1197 in the donor to a phenylalanine (Y1197F,L704N/Y1197F) (Figure 3, lanes 3–6). When we coexpressed L704N/Y1197F with the enforced acceptor (V948R), Y1197 phosphorylation is detected in the presence of EGF (Figure 3, lanes 9, 10). We also performed the same set of experiments in the background of R776H mutant (Figure 3, lanes 11–20) and noticed that Y1197 phosphorylation is observed even in the absence of EGF (Figure 3, lanes 19, 20) suggesting that the acceptor kinase gets phosphorylated in the asymmetric dimer and the R776H mutant enhances acceptor kinase phosphorylation.
Figure 3.

Receiver kinase phosphorylation. + and − indicate the presence and absence of EGF ligand, respectively. Cartoon scheme below shows the position of the mutation introduced to EGFR. Lanes from left to right, WT EGFR (−), WT EGFR (+), Y1197 (−), Y1197 (+), L704N/Y1197F (−), L704N/Y1197F (+), V948R (−), V948R (+), L704N/Y1197F and V948R (−), L704N/Y1197F and V948R (+), R776H (−), R776H (+), R776H/Y1197F (−), R776H/Y1197F (+), L704N/R776H/Y1197F (−), L704N/R776H/Y1197F (+), R776H/V948R and L704N/R776H/Y1197F (−), R776H/V948R and L704N/R776H/Y1197F (+). L704N/Y1197F and L704N/R776H/Y1197F are FLAG tagged. All other constructs are GFP tagged.
Phosphorylation of the acceptor kinase in the asymmetric dimer can be explained by two competing hypotheses/models: (1) The acceptor kinase phosphorylates itself (cis phosphorylation) within the asymmetric dimer, because only the acceptor kinase is active when EGFR dimerizes.9 (2) The acceptor kinase is phosphorylated by other EGFR dimers. To test the second hypothesis, we generated a monomeric form of EGFR that contains both the N-lobe dimer deficient mutation (L704N) and C-lobe dimer deficient mutation (V948R).9 As expected, the enforced kinase monomer is not active (Figure 4a, lanes 7, 8; Figure 4c). However, when the enforced kinase monomer is cotransfected with WT EGFR, with Y1197 mutated to phenylalanine (Figure 4a, lanes 3, 4, 4b), phosphorylation of Y1197 is detected in the presence of EGF (Figure 4a, lanes 5, 6; Figure 4d). In addition, coexpression of R776H/Y1197F with the enforced kinase monomer reveals that Y1197 phosphorylation is enhanced even in the absence of EGF (Figure 4a, 13, 14; Figure 4d). Because Y1197 is mutated to phenylalanine in the asymmetric dimer, the observed Y1197 phosphorylation is from the enforced kinase monomer (Figure 4d). CHO cells express low levels of endogenous ErbB2,30,31 which can potentially contribute to Y1197 phosphorylation by forming heterodimers with EGFR in our experiment. To rule out this possibility, we made a kinase dead construct, in which the catalytic DFG-Asp is mutated to a glycine (D855G). Coexpression of D855G or R776H/D855G with enforced kinase monomer (L704N/V948R) shows no Y1197 phosphorylation (Figure 5, lanes 9–12 vs 13–16). Thus, the enforced kinase monomer (L704N/V948R) phosphorylation is due to EGFR but not due to endogenous ErbB2. Although our experiments cannot rule out the possibility of cis phosphorylation, lateral phosphorylation of the enforced kinase monomer by EGFR dimers has implications for understanding substrate phosphorylation and EGFR signaling in the oligomeric state (see Discussion). In the R776H background, this lateral phosphorylation is enhanced even in the absence of EGF (Figure 4a, lane 13).
Figure 4.

Lateral phosphorylation of EGFR. + and − indicate the presence and absence of EGF ligand, respectively. (a) Lanes from left to right, WT EGFR (−), WT EGFR (+), Y1197F (−), Y1197F (+), Y1197F and L704N/V948R (−), Y1197F and L704N/V948R (+), L704N/V948R (−), L704N/V948R (+), R776H (−), R776H (+), R776H/Y1197F (−), R776H/Y1197F (+), R776H/Y1197F and L704N/V948R (−), R776H/Y1197F and L704N/V948R (+). L704N/V948R is FLAG tagged. All other constructs are GFP tagged. (b) Cartoon scheme for lanes 3, 4 and lanes 11, 12. (c) Cartoon scheme for enforced kinase monomer, lane 7, 8. (d) Cartoon scheme for lanes 5, 6 and lane 13, 14.
Figure 5.

Kinase activity of EGFR is responsible for lateral phosphorylation. + and − indicate the presence and absence of EGF ligand. Lanes from left to right, WT EGFR (−), WT (+), R776H (−), R776H (+), D855G (−), D855G (+), R776H/D855G (−), R776H/D855G (+), D855G and L704N/V948R (−), D855G and L704N/V948R (+), R776H/D855G and L704N/V948R (−), R776H/D855G and L704N/V948R (+), Y1197F and R776H/V948R (−), Y1197F and L704N/V948R (+), R776H/Y1197F and R776H/V948R (−), R766H/Y1197F and L704N/V948R (+) L704N/V948R is FLAG tagged. All other constructs are GFP tagged.
αC-Helix Conformational Transition Is Correlated with a Capping Interaction between R776 and A767
To investigate the atomic details of how R776H activates EGFR, we performed 1 µs atomic molecular dynamics simulation of WT and mutant EGFR in the active state. Previous long time scale molecular dynamics simulation of EGFR showed that the regulatory αC-helix is intrinsically disordered, and the salt bridge interaction between the conserved αC-helix glutamate (E762) and the ATP coordinating lysine (K745) breaks within a short period of time (less than 200 ns) during the simulation.11 Consistent with these studies, we observe a state shift from active to inactive state during 240 ns to 480 ns in our simulation, in which the K745-E762 salt bridge is lost and the αC-helix breaks/cracks into two parts (Figure 6a,d). However, the K745-E762 salt bridge and αC-helix are stable in the R776H mutant (Figure 6a,c,d), indicating that the R776H mutant favors the active form. Furthermore, αC-helix cracking is strongly correlated with a capping interaction between R776 and A767 in WT simulations. Figure 6b plots the shortest distance between the R776/(NH1,NH2,NE) group and A767/O (Figure 6a,b). At around 240 ns to 480 ns, R776 hydrogen bonds to the backbone oxygen of A767, which correlates with the loss of K745-E762 salt bridge and αC-helix breaking (Figure 6a,c). The breaking point of αC-helix is another alanine (A763), which is four residues N-terminus of A767 and forms the canonical i-i+4 hydrogen bond in the intact αC-helix conformation (Figure 6d). However, upon αC-helix breaking the canonical i-i+4 interaction between A763 and A767 is lost, and the unsatisfied backbone hydrogen bonds are partially stabilized by the capping interactions between R776 and A767 (Figure 6d). Notably, the hydrogen bond frequency between R776 and A767 is reduced in the R776H mutant (26.9% vs 9.5%), suggesting a loss of inhibitory capping interaction, which correlates with a stable αC-helix conformation (Figure 6d). However, it should be noted that the αC-helix capping interaction in itself does not fully explain αC-helix conformational transitions because at around 600 ns the K745-E762 salt bridge is formed even though the αC-helix capping interaction is maintained (Figure 6a,b).
Figure 6.

Molecular dynamics simulation of EGFR. (a) K745-E762 salt bridge distance across 1 µs active monomer simulation of WT and R776H. (b) Distance plot between the side chain nitrogen of R767 and the backbone oxygen of A767 in WT simulation. (c) Secondary structure assignment of αC-helix residues during the 1 µs simulation. Coil: ~, Bend: S, Turn: T, A-Helix: H, 3-Helix: G. Y-axis spans residues 752–767 which correspond to αC-helix in EGFR. (d) Representative snapshot of WT and R776H simulation at 0, 300, and 900 ns.
cis Regulatory Interactions between the Kinase Domain and the Flanking JM and C-Terminal Tail Contribute to Kinase Conformational Transitions and R776H Mediated Activation
We previously demonstrated that the C-terminal tail and juxtamembrane (JM) segment are distinguishing features of the EGFR family that have coevolved with the kinase core to uniquely regulate catalytic activity.32 R776 associates with these conserved flanking segments in the crystal structures and MD trajectories. In particular, a segment of the C-tail (1011–1018) tethered to the kinase hinge forms extensive interaction with R776 during our simulation (Figure S3a,3b). However, these interactions are not observed in the R776H mutant (Figure S3a,b). Per-residue interaction energy profiles reveal that C-tail residues: D1012, A1013, D1014, I1018, and P1019 form favorable interactions with R776 but not with the mutant (R776H) (Figures S4 and S5). D1014 is one of the conserved C-tail residues that hydrogen bonds to the interlobe salt bridge (Q791 and K852) associated with interlobe movement.32 The association of R776 with the C-tail and JM segment suggests that these cis regulatory interactions may also be altered in the R776H mutant in addition to the αC-helix capping interaction described above.
DISCUSSION
The allosteric activation of EGFR kinase domain involves conformational transitions from an inactive αC-helix “out” conformation to an active αC-helix “in” conformation.14,33 Our studies are consistent with a model in which the R776H mutant enhances kinase activity by altering αC-helix conformational transition. In particular, the R776H mutant increases affinity for dimerization by stabilizing the acceptor αC-helix in an “in” conformation, thereby priming the N-lobe interface for dimerization.11,13 The increased affinity for dimerization is the biochemical basis for “superacceptor” activity, and analogous to the recently described L858R/T790M mutation, R776H also appears to display “superacceptor” activity. Notably, both L858R/T790M and R776H display impaired donor activity compared to WT EGFR,13,34 indicating that only acceptor functions are selectively enhanced by oncogenic mutations. Furthermore, the co-occurrence of R776H with L858R, L861Q, and G719A suggests that the double mutants (R776H/L858R, R776H/L861Q, and R776H/G719A) may have a synergistic effect on EGFR activation.
Free energy landscape theory suggests that activating allosteric mutations shift the conformational ensemble of proteins toward an active state by destabilizing inactive conformations.35 Our MD studies suggest a possible transition state intermediate in which the αC-helix is held in an inactive “broken” conformation by a capping interaction between R776 and A767. Additional support for the αC-helix capping interaction is provided by crystal structures of inactive symmetric dimer in which the R776 mediated capping interaction is stabilized through interactions with the juxtamembrane segment.36 In fact, comparisons of all available crystal structures of EGFR indicates that the R776 to A767 capping interaction is correlated with αC-helix “out” conformation (p-value 6.077e-10) (Figure S6, Table S1). Thus, disruption of autoinhibitory αC-helix capping interaction and C-terminal tail interaction appears to be the most likely allosteric mechanism by which R776H mutation activates the kinase domain.
Because the αC-helix “in” conformation is critical for kinase activity, key autoinhibitory mechanisms have evolved to prevent inadvertent EGFR activation.33 We identify the αC-helix capping interaction mediated by R776 as a critical autoinhibitory interaction that prevents αC-helix from adopting an active conformation in the monomeric form. The αC-helix cap may work in conjunction with other regions such as the β3-αC loop and the activation loop that are also associated with αC-helix movement. Oncogenic mutations appear to activate the kinase domain by overcoming these autoinhibitory interactions (Figure 7). L858 and L861 in the activation loop, for example, pack against the αC-helix in the inactive conformation, and oncogenic mutations at these positions (L858R and L861Q) potentially relieve autoinhibitory interactions between the activation loop and αC-helix to activate the kinase domain.5,11 The β3-αC loop is rich in deletion mutation (account for 15% of EGFR cancer mutations in COSMIC database), and although these deletion mutants have not been structurally characterized, they are likely to favor an αC-helix “in” conformation by restricting αC-helix conformational flexibility.11,33 The αC-β4 loop, on the other hand, is an insertion hotspot, and insertions in the loop also favor αC-helix “in” conformation.33,37 Indeed, in one crystal structure of αC-β4 loop insertion mutant (D770_N771insNPG, PDBID: 4LRM), the inserted residues form another turn of the αC-helix that prohibits R776 from forming the αC-helix capping interaction.37
Figure 7.

Emerging acvitation scheme of oncogenic mutations in EGFR. Right: autoinhibitory regions/interactions are highlighted rendering EGFR inactive. Left: oncogenic mutations overcome autoinhibitory mechanisms and actives EGFR.
R776 is a mutation hotspot in the kinase domain as the residue equivalent to R776 is mutated in multiple cancers and congenital disorders.21 A total of 68 missense mutations have been found at the R776 equivalent position in 44 different kinases (Table S2). Other than R776H, R776C/S/G are also observed in the COSMIC database. These mutants are likely to be activating as well, since cysteine, serine, and glycine are also predicted to destabilize the autoinhibitory αC-helix capping interaction. Indeed, we previously showed that R776C also activates EGFR in a ligand-independent manner.21 Notably, ErbB2 (R784C/L) and ErbB4 (R782Q) also harbor the arginine mutation at the equivalent position, but not ErbB3 (Table S2). Although the intrinsic disorder of the αC-helix is a unique feature of EGFR and not observed in closely related kinases such as ErbB2 and ErbB4, they may still share some aspect of EGFR conformational transition.11,14 Thus, R776H equivalent mutations in ErbB2 and ErbB4 are predicted to be activating as well.
EGFR auto-phosphorylation is believed to occur in trans through the formation of the asymmetric dimer.9 Our experimental results show that EGFR kinase activity is not restricted to the asymmetric dimer itself. Upon ligand stimulation, the activated asymmetric dimer is able to phosphorylate monomeric inactive EGFR. This is an extension of our traditional view of EGFR signal transduction. The result also indirectly supports the observed oligmers reported by several studies.38,39 One previous report showed that EGFR lacking the extracellular domain is able to phosphorylate ErbB3 in the absence of ligand stimulation,40 similar to the lateral phosphorylation suggested by our studies. These data support the model that dimeric EGFR functions as a holoenzyme to phosphorylate other monomeric EGFR and members of the ErbB family. An activating mutation in EGFR, such as R776H, increases such lateral phosphorylation even in the absence of ligand stimulation, leading to constitutive activation and downstream signaling. A logical extension of this model is that other members of the ErbB family (ErbB2, ErbB3, and ErbB4) can potentially be phosphorylated by the R776H dimer via lateral phosphorylation. This hypothesis, however, needs to be tested in future studies. The concept of lateral phosphorylation was first proposed by Verveer et al. in 2000 when lateral propagation of EGFR signals in the plasma membrane was observed using fluorescence imaging.41 Our studies on the enforced kinase monomer are consistent with previous studies linking EGFR dimerization and lateral phosphorylation.
Supplementary Material
Acknowledgments
The authors thank Dr. Samiksha Katiyar for her help with Western blot analysis. This study was supported in part by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia’s Office of the Vice President for Research and Office of the Vice President for Information Technology.
Funding
Funding for N.K. from the American Cancer Society (RSG-10-188-01-TBE), Georgia Cancer Coalition (GCC) and University of Georgia is acknowledged.
Footnotes
ASSOCIATED CONTENT
Figure S1: Structure of EGFR showing the position of R776 and associated interactions in active (2GS6) and inactive (3W32) states. Figure S2: RMSD plot of active monomer simulation of WT and R776H. Figure S3: R776H loses the interaction with C-terminal tail of EGFR. Figure S4: Coulombic interaction between R776/H776 to residues in C-terminal tail tether. Figure S5: Van Der Waals interaction between R776/H776 to residues in C-terminal tail tether. Figure S6: Histogram plot showing the shortest distances between R776(NE,NH1,NH2) to A767(O) between C-helix “in” crystal structures and C-helix “out” crystal structures. Table S1: List of crystal structures considered in Figure S6. Table S2: Cancer mutations in the equivalent position of R776 in EGFR of other kinases. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00444.
The authors declare no competing financial interest.
References
- 1.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Yewale C, et al. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials. 2013;34:8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- 4.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 5.Nussinov R, Tsai CJ. Allostery in disease and in drug discovery. Cell. 2013;153:293–305. doi: 10.1016/j.cell.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Yun CH, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kancha RK, Peschel C, Duyster J. The Epidermal Growth Factor Receptor-L861Q Mutation Increases Kinase Activity without Leading to Enhanced Sensitivity Toward Epidermal Growth Factor Receptor. J. Thorac. Oncol. 2011;6:387–392. doi: 10.1097/JTO.0b013e3182021f3e. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe S, et al. Effectiveness of gefitinib against non-small-cell lung cancer with the uncommon EGFR mutations G719X and L861Q. J. Thorac. Oncol. 2014;9:189–194. doi: 10.1097/JTO.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Cho J, et al. Cetuximab Response of Lung Cancer-Derived EGF Receptor Mutants Is Associated with Asymmetric Dimerization. Cancer Res. 2013;73:6770–6779. doi: 10.1158/0008-5472.CAN-13-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shan Y, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;11:860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 12.Wang ZH, et al. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011;18:1388–1393. doi: 10.1038/nsmb.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brewer MR, et al. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E3595–E3604. doi: 10.1073/pnas.1220050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan Y, et al. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. U. S. A. 2013;110:7270–7275. doi: 10.1073/pnas.1220843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arkhipov A, et al. Membrane Interaction of Bound Ligands Contributes to the Negative Binding Cooperativity of the EGF Receptor. PLoS Comput. Biol. 2014;10:e1003742. doi: 10.1371/journal.pcbi.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutto L, Gervasio FL. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10616–10621. doi: 10.1073/pnas.1221953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arkhipov A, et al. Architecture and Membrane Interactions of the EGF Receptor. Cell. 2013;152:557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih AJ, et al. Molecular dynamics analysis of conserved hydrophobic and hydrophilic bond-interaction networks in ErbB family kinases. Biochem. J. 2011;436:241–251. doi: 10.1042/BJ20101791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mustafa M, Mirza A, Kannan N. Conformational regulation of the EGFR kinase core by the juxtamembrane and C-terminal tail: A molecular dynamics study. Proteins. 2011;79:99–114. doi: 10.1002/prot.22862. [DOI] [PubMed] [Google Scholar]
- 20.James KA, Verkhivker GM. Structure-Based Network Analysis of Activation Mechanisms in the ErbB Family of Receptor Tyrosine Kinases: The Regulatory Spine Residues Are Global Mediators of Structural Stability and Allosteric Interactions. PLoS One. 2014;9:e113488. doi: 10.1371/journal.pone.0113488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McSkimming DI, et al. ProKinO: A Unified Resource for Mining the Cancer Kinome. Hum. Mutat. 2015;36:175–186. doi: 10.1002/humu.22726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The PyMOL Molecular Graphics System, Version 1.3r1. Schrödinger LLC; New York: 2010. [Google Scholar]
- 26.Noesel JV, et al. Activating Germline R776H Mutation in the Epidermal Growth Factor Receptor Associated With Lung Cancer With Squamous Differentiation. J. Clin. Oncol. 2013;31:e161–e164. doi: 10.1200/JCO.2012.42.1586. [DOI] [PubMed] [Google Scholar]
- 27.Yang CH, et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J. Clin. Oncol. 2008;26:2745–2753. doi: 10.1200/JCO.2007.15.6695. [DOI] [PubMed] [Google Scholar]
- 28.Wu JY, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin. Cancer Res. 2008;14:4877–4882. doi: 10.1158/1078-0432.CCR-07-5123. [DOI] [PubMed] [Google Scholar]
- 29.Koland JG. Coarse-grained molecular simulation of epidermal growth factor receptor protein tyrosine kinase multi-site self-phosphorylation. PLoS Comput. Biol. 2014;10:e1003435. doi: 10.1371/journal.pcbi.1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzahar E, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996;16:5276–87. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colagar AH, et al. Minimal HER1 and HER2 expressions in CHO and HEK-293 cells cause them appropriate negative cells for HERs-related studies. Res. Mol. Med. 2013;1:6–12. [Google Scholar]
- 32.Mirza A, Mustafa M, Talevich E, Kannan N. Co-conserved features associated with cis regulation of ErbB tyrosine kinases. PLoS One. 2010;5:e14310. doi: 10.1371/journal.pone.0014310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai CJ, Nussinov R. The molecular basis of targeting protein kinases in cancer therapeutics. Semin. Cancer Biol. 2013;23:235–242. doi: 10.1016/j.semcancer.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Littlefielda P, Jura N. EGFR lung cancer mutants get specialized. Proc. Natl. Acad. Sci. U. S. A. 2013;110:15169–15170. doi: 10.1073/pnas.1314719110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai CJ, Nussinov R. The free energy landscape in translational science: how can somatic mutations result in constitutive oncogenic activation? Phys. Chem. Chem. Phys. 2014;16:6332–6341. doi: 10.1039/c3cp54253j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jura N, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yasuda H, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci. Transl Med. 2013;5 doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofman EG, et al. Ligand-induced EGF Receptor Oligomerization Is Kinase-dependent and Enhances Internalization. J. Biol. Chem. 2010;285:39481–39489. doi: 10.1074/jbc.M110.164731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clayton AHA, et al. Ligand-induced Dimer-Tetramer Transition during the Activation of the Cell Surface Epidermal Growth Factor Receptor-A Multidimensional Microscopy Analysis. J. Biol. Chem. 2005;280:30392–30399. doi: 10.1074/jbc.M504770200. [DOI] [PubMed] [Google Scholar]
- 40.Kancha RK, Bubnoff NV, Duyster J. Asymmetric kinase dimer formation is crucial for the activation of oncogenic EGFRvIII but not for ERBB3 phosphorylation. Cell Commun. Signal. 2013;11 doi: 10.1186/1478-811X-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verveer PJ, et al. Quantitative Imaging of Lateral ErbB1 Receptor Signal Propagation in the Plasma Membrane. Science. 2000;24:1567–1570. doi: 10.1126/science.290.5496.1567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.