

Abstract
Membrane-engineered cells displaying antigen-targeting ligands are useful as both scientific tools and clinical therapeutics. While genetically-encoded artificial receptors have proven efficacious, their scope remains limited as this approach is not amenable to all cell types and the modification is often permanent. Our group has developed a non-genetic method to rapidly, stably, and reversibly modify any cell membrane with a chemically self-assembled nanoring (CSAN) that can function as a prosthetic receptor. Bifunctional CSANs displaying epithelial cell adhesion molecule (EpCAM)-targeted fibronectin domains were installed on the cell membrane through hydrophobic insertion and remained stably bound for ≥72 h in vitro. These CSAN-labeled cells were capable of recognizing EpCAM-expressing target cells, forming intercellular interactions that were subsequently reversed by disassembling the nanoring with the FDA-approved antibiotic, trimethoprim. This study demonstrates the use of this system to engineer cell surfaces with prosthetic receptors capable of directing specific and reversible cell-cell interactions.
TOC image
INTRODUCTION
The ability to direct cell-cell interactions has tremendous value across numerous fields – including tissue engineering,1, 2 regenerative medicine,3, 4 and adoptive immunotherapy5, 6 – and as a tool for elucidating fundamental biology.7, 8 To this end, several approaches for modifying cell surfaces have been developed, perhaps the most notable being that of chimeric antigen receptor (CAR) T cells.9 Though clinically efficacious, the genetic engineering underlying the CAR T cell platform makes it irreversible and yields several limitations that hinder its use for alternative applications.10 Specifically, not all cell types – such as regenerative stem cells11 – are readily amenable to such modification, and the permanence of the genetically encoded receptor has led to significant adverse events in the clinic, including B cell aplasia,12 solid organ damage,13 and neurotoxicity.14
To address these limitations and expand the use of cell-directing therapies, many groups have sought non-genetic approaches to introduce artificial receptors and targeting elements to the cell surface. Liposome fusion has been used to integrate bioorthogonal functional groups into the cell membrane, which can either be paired with complementarily modified cells or reacted with appropriately-conjugated targeting ligands.15, 16 Reactive groups can be introduced to the cell surface through metabolic engineering, wherein cells are grown in media supplemented with chemically-functionalized sugar analogs that get incorporated into membrane glycoproteins; similarly, these functional groups can then be conjugated to antigen targeting elements.1, 5 Others have taken advantage of naturally existing cell-surface amines (primarily lysine side chains) to bind activated esters tethered to a variety of species to the cell surface nonspecifically.4, 17 Finally, various alkyl-, lipid-, and glycophosphatidylinositol (GPI)-tagged species have been hydrophobically inserted and anchored into the cell membrane.18–21
While many of these non-genetic approaches have demonstrated the ability to direct specific cell-cell interactions, relatively few do so in a reversible fashion. Additionally, reversal mechanisms employed thus far – including irradiation with ultraviolet (UV) light,5, 22 changes in electrochemical redox potential,23 alterations in temperature,24, 25 and enzymatic cleavage of the tethering species26 – are largely unfit for in vivo applications, especially when surface-modified cells are distributed throughout an organism.
Expanding upon this body of prior work, we designed a cell membrane engineering methodology that would be broadly applicable to a variety of cell types and possess a reversal mechanism suitable for in vivo use. To accomplish this, we utilized a protein scaffold developed by our lab called the chemically self-assembled nanoring (CSAN; Figure 1A).27 CSANs are formed when bivalent dihydrofolate reductase (DHFR2) fusion proteins are spontaneously oligomerized by a chemical dimerizer, bis-methotrexate (bisMTX).27 CSANs can be further functionalized by fusing various binding entities to the DHFR2 subunits28, 29 – in this case, either a monovalent streptavidin (mSA30) unit or a fibronectin (Fn3) domain with engineered specificity for epithelial cell adhesion molecule (EpCAM) was fused.31 Similarly, the bisMTX moiety can be chemically modified to incorporate a bioorthogonal ligation handle, such as an azide group.29, 32 Using stoichiometric combinations of the fusion proteins and the bisMTX, one can form multivalent, heterobifunctional CSANs capable of targeting multiple distinct antigens.33 Importantly, the CSAN scaffold can be disassembled through exposure to the FDA-approved antibiotic trimethoprim, providing a pharmacologic mechanism for removing the targeting ligands from the cell surface.6, 32, 33
Figure 1. Cell Surface Engineering with Chemically Self-Assembled Nanorings (CSANs).
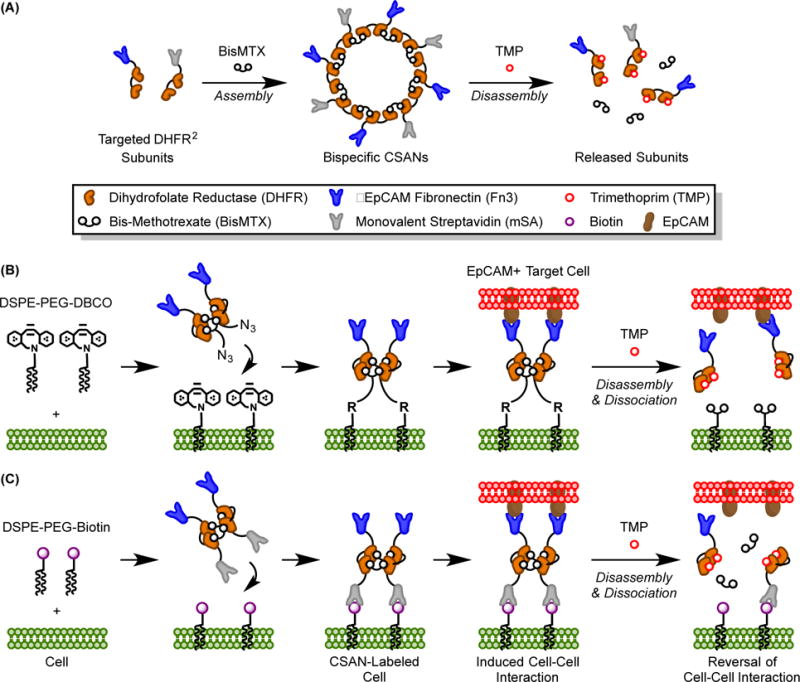
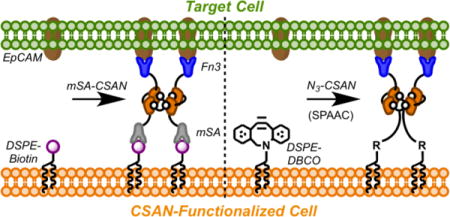
(A) CSANs are composed of targeted-DHFR2 fusion proteins that are spontaneously oligomerized by the chemical dimerizer, bisMTX; they can be pharmacologically disassembled by the FDA-approved antibiotic trimethoprim. (B) DSPE-PEG2000-DBCO moieties spontaneously insert into cell membranes and are stabilized in the lipid bilayer by the hydrophobic effect.(19) EpCAM-targeted Fn3 CSANs oligomerized with an azide-bisMTX dimerizer are then installed on the cell surface through a copper-free, strain-promoted alkyne/azide cycloaddition. The CSAN-functionalized cells can then form targeted interactions with EpCAM+ cells, and these interactions can be reversed with trimethoprim. (C) Similarly, cells modified with DSPE-PEG2000-biotin moieties can be functionalized with bispecific mSA/Fn3 CSANs, enabling recognition of EpCAM+ target cells. Trimethoprim-induced disassembly of the CSAN reverses the intercellular interactions.
Consistent with the aim to develop a surface engineering approach that would be applicable to multiple cell types, we devised a system based upon the spontaneous hydrophobic insertion of commercially available phospholipid conjugates (Figure 1B-C). Using either 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-{biotinyl(polyethylene glycol)-2000} (DSPE-PEG2000-biotin) or 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-{dibenzocyclooctyl(polyethylene glycol)-2000} (DSPE-PEG2000-DBCO), cell surfaces can be decorated with biotin and DBCO moieties, respectively. Targeted CSANs are then attached to the lipid-modified cells via a non-covalent biotin/mSA interaction or a copper-free, strain-promoted alkyne/azide cycloaddition (SPAAC) involving the DBCO/azide groups, thereby functionalizing the cell with the EpCAM-binding domains. As demonstrated herein, the CSAN-functionalized cells are capable of interacting with EpCAM+ target cells, and these intercellular interactions are readily reversed with trimethoprim.
As such, this study details a non-genetic, two-component strategy to functionalize cells with antigen-binding ligands capable of directing targeted cell-cell interactions in a pharmacologically reversible fashion.
RESULTS AND DISCUSSION
Functionalized Phospholipids Hydrophobically Insert into Cell Membranes
The spontaneous membrane insertion of hydrophobic species – including alkyl chains, phospholipids, and GPI-conjugated proteins – has been demonstrated in numerous cell types,34–36 including mesenchymal stem cells (MSCs).3, 18, 37 These results have shown that this insertion is innocuous to the modified cell, having no effect on cell viability, proliferation, or differentiation. Furthermore, this approach is facile, requiring no specialized reagents or techniques, and is universally applicable to essentially any cell type. Therefore, we decided to use hydrophobic insertion to tether our CSANs to the cell surface (Figure 1B-C).
The commercially available phospholipid conjugates DSPE-PEG2000-biotin and DSPE-PEG2000-DBCO were selected for this study. These species were chosen because we hypothesized that the hydrophobic lipid would enable membrane insertion while the long, flexible PEG linker would improve the accessibility of the biotin and DBCO groups. We also envisioned two approaches to labeling the cells with the phospholipids: (1) resuspending the cells ex vitro in buffer supplemented with the phospholipids; and (2) actively culturing the cells in vitro in phospholipid-supplemented media. Importantly, cell viability was not affected by either lipid-modification approach, even when concentrations of up to 100 μM of DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO were used (Figure S1). This was true for both of the model cell lines, adherent MCF-7 and suspensive Raji cells.
To simultaneously assess the membrane insertion of phospholipids and ensure that the biotin and DBCO groups were accessible, cells were analyzed via flow cytometry using streptavidin- and azide-conjugated fluorophores, respectively. Both MCF-7 and Raji cells were modified with increasing concentrations of DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO through both the buffer (ex vitro) and culture (in vitro) methods. In all instances, the biotin and DBCO moieties were readily detected on the cell surface after lipid modification, indicating both successful membrane insertion and availability of the functional groups for subsequent labeling (Figure 2 and S2). Furthermore, the extent of the modification could be easily modulated by varying the concentration of the phospholipid conjugate that was used.
Figure 2. Phospholipid Conjugates Hydrophobically Insert into Cell Membranes.
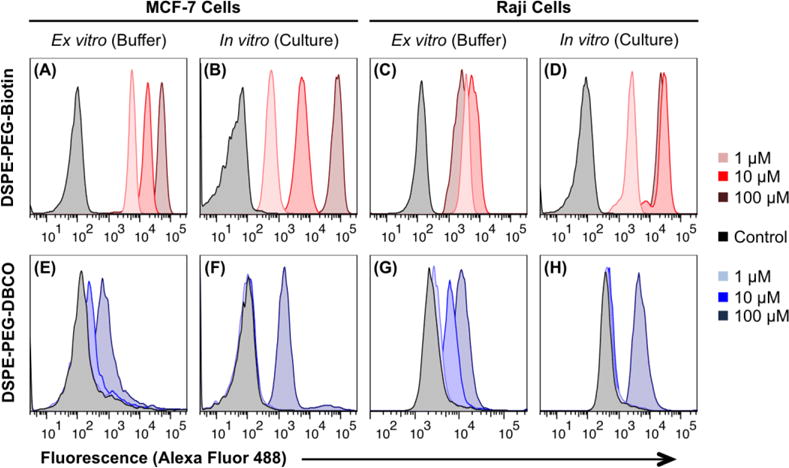
Cells were modified with increasing concentrations of either DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO through one of two methods: (1) resuspension in phospholipid-containing buffer (ex vitro), or (2) active culture in phospholipid-containing media (in vitro). Cells were subsequently analyzed by flow cytometry using streptavidin- or azide-conjugated Alexa Fluor 488 to assess the presence of biotin and DBCO moieties, respectively, on the cell surface. Both adherent MCF-7 cells and suspensive Raji cells can be successfully modified with DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO through both the ex vitro and in vitro approaches. While insertion experiments were performed in triplicate, a representative trial of each condition is shown here. A quantitative analysis of the triplicate data is presented in Figure S2 of the Supporting Information.
Seemingly, the DSPE-PEG2000-biotin species affords a more tunable modification than DSPE-PEG2000-DBCO (Figure 2). However, this observation is likely an artifact of the relatively short time (1 hour) and low temperature (4 °C) for which the lipid-modified cells were incubated with the secondary reagent, as the biotin/streptavidin interaction forms more rapidly38 than the slower azide/alkyne ligation39 necessary to detect the DBCO species. Indeed, extending this incubation time to ≥3 h and raising the temperature to 37 °C enhances conjugation to surface DBCO groups (Figure S3). Therefore, it is possible that both DSPE-PEG2000-biotin and DSPE-PEG2000-DBCO insert into the cell membrane to a similar extent, and that the discrepancies between the labeling observed in Figures 2 and S2 are due to the inherent differences between the subsequent binding and ligation efficiencies. Additionally, the hydrophobicity of the DBCO group itself may enable it to interact with hydrophobic membrane components, further slowing the azide ligation reaction.
Lastly, the Raji cells appear to become saturated with DSPE-PEG2000-biotin following incubation with 10 μM of the phospholipid, as incubation with 100 μM does not afford an increase in the fluorescent signal. Across the concentration range tested, no such saturation was observed for the MCF-7 cells. This observation is most likely explained by the difference in size between the two cell types – the Raji cells are smaller and thus their membranes cannot support the same quantity of the DSPE-PEG2000-biotin as the larger MCF-7 cells.
Collectively, this data shows that a variety of cell types can be effectively modified with phospholipid conjugates via hydrophobic insertion into the cell membrane without effecting cell viability. These results are consistent with those obtained by others performing similar hydrophobic insertions and further validates this approach as a universal method for cell surface modification.3, 4, 18, 34–37
Production and Characterization of Cell-Binding CSANs
EpCAM is a cell surface antigen that is overexpressed by numerous carcinomas and several cancer stem cells.40 We previously reported the development of EpCAM-binding Fn3 ligands, based upon the human tenth type III fibronectin domain.31 To impart EpCAM-targeting capabilities to our CSANs, we fused Fn3 clone C5 (Kd = 17 ± 1 nM) to the C-terminus of our DHFR2 fusion proteins. When these DHFR2-Fn3 monomers were exposed to a molar excess of the chemical dimerizer, bisMTX, they rapidly and completely oligomerized into Fn3 CSANs, as demonstrated by size exclusion chromatography (Figure S4A-B). Importantly, the Fn3 CSANs continued to bind to EpCAM-expressing MCF-7 cells with high affinity (apparent Kd = 21 ± 6 nM and selectivity (Figures S5A-B).
We next sought to develop methods for binding these Fn3 CSANs to cells that had been modified with DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO. To recognize the biotin-decorated cells, we fused a monovalent streptavidin domain (mSA) to the N-terminus of the DHFR2 fusion proteins. In the presence of bisMTX, these mSA-DHFR2 monomers readily oligomerized into biotin-binding CSANs (Figure S4C). Furthermore, equimolar mixtures of the mSA- and Fn3-fused monomers could be co-assembled into CSANs with bispecificty for both biotin and cellular EpCAM (Figure S4D). Importantly, these bispecific mSA/Fn3 CSANs retained their apparent affinity for EpCAM+ cells (apparent Kd = 24 ± 6 nM; Figure S5A).
Our group has previously reported the synthesis and use of a bisMTX analog that incorporates a free amine suitable for further conjugation and additional functionalization.29, 32 To produce CSANs capable of binding to the DBCO-decorated cells, we coupled a PEG4-azide moiety to this amine via N-hydroxysuccinimide (NHS) chemistry, generating an azide-bisMTX analog that contains a free azide group (Figure S6). As with the parent bisMTX dimerizer, DHFR2-Fn3 fusion proteins exposed to azide-bisMTX oligomerized into azide/Fn3-CSANs (Figure S4B).
We further verified the formation of the mSA, Fn3, and mSA/Fn3 bispecific CSAN species via cryo-electron microscopy (cryo-EM). Nanoring structures were readily visualized for all three species (Figures 3A-C, respectively), and analysis of multiple samples indicated similar sizes for the mSA (18 ± 3 nm), Fn3 (19 ± 4 nm), and mSA/Fn3 bispecific (19 ± 4 nm) CSANs (Figure 3D). These diameters are in close agreement to dynamic light scattering (DLS) measurements of the hydrodynamic radii of these species (Figure S7).
Figure 3. Cryo-EM Characterization of CSAN Species.
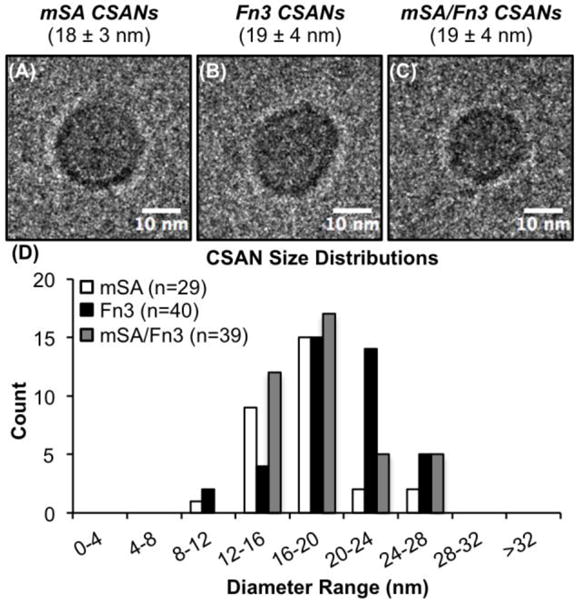
The formation of (A) mSA CSANs, (B) Fn3 CSANs, and (C) bispecific mSA/Fn3 CSANs was demonstrated by cryo-EM. The values in parentheses represent the mean diameter ± standard deviation of n=29, 40, and 39 nanorings, respectively. (D) The size distribution of mSA, Fn3, and mSA/Fn3 CSANs as assessed by cryo-EM.
CSANs are Readily Installed on Cells Modified with Phospholipid Conjugates
After confirming the membrane insertion of the phospholipid conjugates, we sought to use the associated functional groups as handles for the attachment of our nanoring platform. Cells were first modified with DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO ex vitro. They were subsequently incubated with CSANs of various functionalities at 4 °C for 1 h, or in the case of the Fn3 CSANs oligomerized with azide-bisMTX, 37 °C for 3 h. Specifically: (1) mSA CSANs were successfully bound to biotin-modified MCF-7 cells (Figure 4A); (2) Fn3 CSANs oligomerized with azide-bisMTX were conjugated to DBCO-modified Raji cells (Figure 4B); and (3) Fn3 CSANs were bound to EpCAM-expressing MCF-7 cells (Figure 4C). Additionally, mSA/Fn3 bispecific CSANs could be installed on both biotin-modified Raji cells (Figure 4D) and unmodified MCF-7 cells (Figure 4E), demonstrating the retained bifunctionality of these co-assembled CSANs. These experiments also verified the presence of both the mSA-DHFR2 and DHFR2-Fn3 subunits within a single CSAN, as the analyzed events were positive for both the FLAG and MYC epitope tags present on the respective fusion proteins (Figure S8A). Finally, in preparation for future cell-targeting experiments, the optimal labeling concentration of mSA/Fn3 CSANs on Raji cells modified with DSPE-PEG2000-biotin was assayed by flow cytometry and found to be 100 nM (Figure S8B).
Figure 4. CSANs are Readily Installed on Cells Modified with Phospholipid Conjugates.
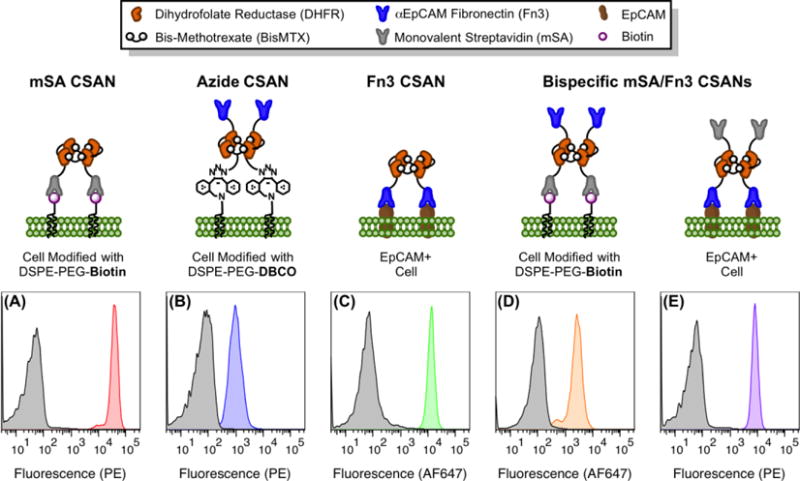
For all panels, cells were labeled with CSANs through one of several methods and then analyzed by flow cytometry with staining for surface-bound nanorings via either an anti-FLAG phycoerythrin or anti-MYC Alexa Fluor 647 conjugate. The non-specific binding of the antibody-fluorophore conjugate is shown in grey, while the specific detection of the indicated CSAN is shown in color. (A) mSA CSANs bind to MCF-7 cells modified with DSPE-PEG2000-biotin. (B) Fn3 CSANs formed with azide-bisMTX are conjugated to Raji cells modified with DSPE-PEG2000-DBCO. (C) Fn3 CSANs bind to unmodified, EpCAM+ MCF-7 cells. (D) Bispecific mSA/Fn3 CSANs bind to Raji cells modified with DSPE-PEG2000-biotin. (E) Bispecific mSA/Fn3 CSANs bind to unmodified, EpCAM+ MCF-7 cells. All experiments were performed in triplicate, with a representative histogram shown for each scenario.
In Vitro Stability of Phospholipid-Anchored CSANs
While the insertion of hydrophobic anchors into the lipid bilayer is an energetically favorable process,19 it is typically a transient modification with a half-life on the order of hours for cells in active culture.3, 35 Additionally, because the lipids can insert into essentially any cell membrane, it was conceivable that a lipid-anchored species could dissociate from the membrane into which it was principally installed and subsequently label a neighboring cell, essentially “hopping” from the intended cell to a bystander cell. However, we hypothesized that by engaging multiple lipid anchors per nanoring, the multivalency of the CSAN would afford an improved surface stability relative to single lipid species and keep the CSANs localized to the principally modified cell.
To test both the surface stability of lipid-anchored CSANs and their potential to transfer amongst cells, two populations of Raji cells were differentially labeled. The first population was labeled only with CellTrace Violet (CTV) dye. The second population was modified with DSPE-PEG2000-biotin in vitro and then labeled with “reduced valency” mSA CSANs. To more accurately recapitulate the valency of mSA domains that would be present in a bifunctional mSA/targeted CSAN, the CSANs used in this study were co-assembled with an equal ratio of mSA-DHFR2 monomers and non-targeted DHFR2 monomers. In this manner, the reduced valency CSANs used in this experiment serve as a surrogate for any bispecific mSA/targeted CSAN, including the mSA/Fn3 CSANs previously introduced.
The CTV-labeled and CSAN-labeled Raji cell populations were combined and co-cultured for 72 h; every 24 h, the culture media was refreshed (to partially simulate the effect of clearance) and a sample of the pooled population was analyzed for CTV and CSAN presence by flow cytometry. For comparison, the same analysis was performed for a mixed population of CTV-labeled Raji cells and Raji cells only modified with the DSPE-PEG2000-biotin (no CSANs). As shown in Figure 5A, the lipid-anchored CSANs remained stably bound to the cell surface for ≥72 h. In contrast, significant loss of the monomeric phospholipid conjugates was observed over this same time frame (p<0.0025). This indicates that, through the engagement of multiple phospholipid conjugates, the multivalent CSANs possess an increased avidity for the cell surface and thus an enhanced surface stability relative to species that are anchored by only a single lipid. Furthermore, the CSANs exhibited a surface half-life of approximately 24 h when incubated in mouse plasma (Figure S9), making them considerably more stable than previously-reported phospholipid-anchored constructs and thus potentially useful for future in vivo applications. Furthermore, Figure 5C demonstrates that there is minimal migration of a lipid-anchored CSAN from one cell to another. Specifically, the percentage of CTV+/CSAN+ Raji cells in the population increases only marginally over the course of three days, from 0.9 ± 0.3% of the population on day zero to 2.9 ± 0.9% on day three; this correlates to a decrease in the number of CTV+/CSAN− Raji cells from 27.1 ± 0.9% to 24.5 ± 0.4% over the same time period. A similar effect is observed for the monomeric DSPE-PEG2000-biotin moieties (Figure 5D), with an increase in the number of CTV+/lipid+ cells from 0.6 ± 0.6% to 4.1 ± 0.8% and a corresponding decrease in the number of CTV+/lipid− cells from 26.9 ± 0.5% to 23.3 ± 0.5% over three days. This data suggests that, while the phospholipid conjugates and their tethered cargo can dissociate from the cell surface, very few of the dissociated species re-insert themselves into the membranes of neighboring cells. This is likely due to the low concentration of the dissociated species in the media and the frequent refreshing of the cell media (every 24 h), thus reducing the accumulation of free phospholipid conjugates.
Figure 5. Membrane Stability and Controlled Dissociation of Phospholipid-Anchored CSANs.
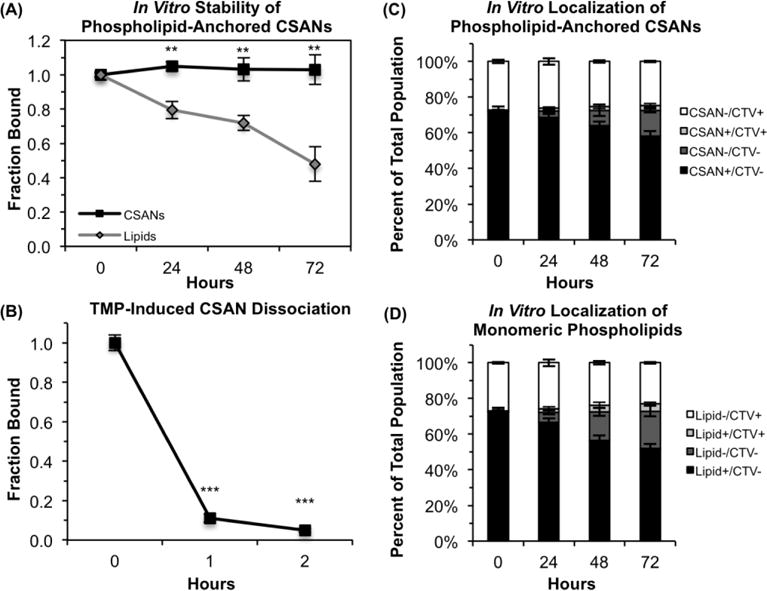
(A) Biotin-modified Raji cells were labeled with reduced-avidity mSA CSANs and analyzed by flow cytometry every 24 h, staining for either the CSANs (black) or, in the case of the lipid-only control (grey), for biotin. For this analysis, the mean fluorescence intensity (MFI) values are corrected for the number of cell divisions (as determined by CellTrace Violet labeling) and scaled relative to the MFI values obtained at t = 0 h. (B) Biotin-modified Raji cells were labeled with mSA/Fn3 CSANs and then resuspended in culture media with or without 2 μM trimethoprim for 1-2 h at 37 °C. Cells were then analyzed by flow cytometry to detect the surface-bound CSANs. (C) Biotin-modified Raji cells labeled with reduced-avidity mSA CSANs were pooled with CTV-labeled Raji cells at a 7:3 ratio and co-cultured for 72 h. Cells were analyzed by flow cytometry every 24 h to ascertain whether the lipid-anchored CSANs had migrated onto the membranes of the CTV+ Raji cells. (D) Raji cells modified with only DSPE-PEG2000-biotin (no CSANs) were pooled with CTV-labeled Raji cells at a 7:3 ratio and co-cultured for 72 h. Cells were analyzed by flow cytometry every 24 h to ascertain whether the phospholipid conjugates had migrated onto the membranes of the CTV+ Raji cells. For all panels, data is presented as the mean ± standard deviation of at least three trials.
Trimethoprim Removes Targeting Elements from the Cell Surface
To date, relatively few cell surface engineering approaches – either genetic or non-genetic in origin – possess mechanisms for removing the artificial receptors from the cell surface. Furthermore, many of those reversal stimuli are not currently suited for in vivo applications.22–26 Accordingly, we sought to utilize the trimethoprim-induced disassembly of the CSAN scaffold as a pharmacologic mechanism for removing the targeting ligands from the surface of a CSAN-functionalized cell. To demonstrate this, Raji cells were sequentially modified with DSPE-PEG2000-biotin in vitro and labeled with mSA/Fn3 bispecific CSANs. The CSAN-functionalized Raji cells were then resuspended in culture media supplemented with a clinically-relevant concentration of trimethoprim (2 μM; serum concentrations of trimethoprim have been shown to reach peak concentrations of ~6-15 μM within 2 h of oral dosing41, 42) and incubated at 37 °C for up to 2 h. An aliquot of cells was analyzed by flow cytometry at 0, 1, and 2 h. As shown in Figure 5B, the targeting ligands were dissociated from the cell surface in a time-dependent manner, with 95% of the EpCAM-targeted Fn3 domains removed within 2 h.
CSANs Direct Reversible Cell-Cell Interactions In Vitro
The ability of CSANs to direct reversible intercellular interactions in vitro was assessed by fluorescence microscopy (Figure 6A-C). CFSE-labeled Raji cells were sequentially modified with DSPE-PEG2000-biotin, labeled with mSA/Fn3 bispecific CSANs, and then incubated with a monolayer of EpCAM-expressing target cells (MCF-7) adhered to glass coverslips. The CSAN-functionalized Raji cells readily bound to the monolayer of target cells (Figure 6B), and these cell-cell interactions were readily reversed via a brief (1 h) exposure to trimethoprim (Figure 6C). Importantly, phospholipid-modified cells that were not functionalized with the mSA/Fn3 CSANs were not able to interact with the target cells (Figure 6A), indicating that the observed interactions were induced by the CSANs and not non-specific adherence.
Figure 6. CSANs Direct Reversible Cell-Cell Interactions.
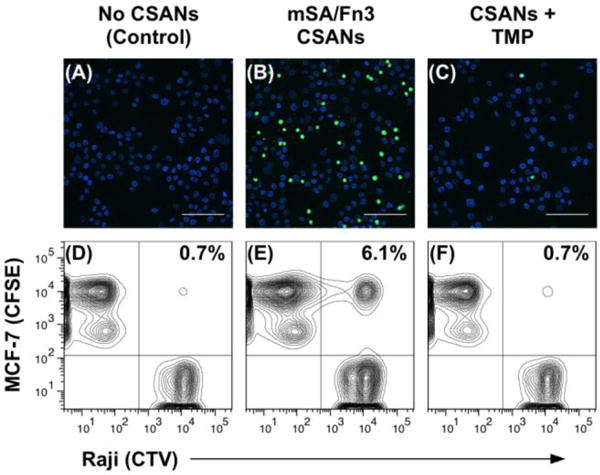
For the fluorescence microscopy experiment (top row), Raji cells were sequentially labeled with CFSE, DSPE-PEG2000-biotin and with or without mSA/Fn3 CSANs; they were then incubated with a monolayer of EpCAM+ MCF-7 cells. (A) In the absence of CSANs, the phospholipid-modified Raji cells are unable to interact with the MCF-7 cells. (B) When functionalized with the mSA/Fn3 CSANs, the EpCAM-targeted Raji cells adhere to the MCF-7 cell monolayer. (C) The EpCAM-targeted Raji cells can be dissociated from the MCF-7 cell monolayer by disassembling the CSAN with trimethoprim (TMP). Scale bars in (a-c) represent 100 μm. For the flow cytometry experiment (bottom row), the target MCF-7 cells were labeled with CFSE while the Raji cells were labeled with CTV. Raji cells were again modified with DSPE-PEG2000-biotin and labeled with or without mSA/Fn3 CSANs. (D) In the absence of CSANs, the phospholipid-modified Raji cells are unable to interact with the MCF-7 cells. (E) When functionalized with the mSA/Fn3 CSANs, the EpCAM-targeted Raji cells formed stable clusters with the MCF-7 cells. (F) The Raji/MCF-7 cell clusters were readily dissociated with trimethoprim. Data are representative of replicate (n = 3) experiments.
A similar experiment was conducted via flow cytometry. In this case, CTV-labeled Raji cells were again modified with DSPE-PEG2000-biotin and labeled with mSA/Fn3 bispecific CSANs. They were then combined with detached, CFSE-labeled MCF-7 cells and incubated together on a rotating platform for 1 h. Samples were subsequently resuspended in either standard culture media or media supplemented with 2 μM trimethoprim. After another 1 h incubation with rotation, samples were thoroughly washed and analyzed on a flow cytometer. Similar to the microscopy results, very few non-specific cell-cell interactions are observed in the absence of the CSANs (Figure 6D). However, the CSAN-functionalized Raji cells were able to form targeted cell clusters with the MCF-7 cells (Figure 6E); again, nearly all of the targeted interactions were dissociated with trimethoprim treatment (Figure 6F). Even under the high-flow conditions of the cytometer (an instrument designed for single cell analyses), the CSAN-functionalized Raji cells were able to form significantly more interactions with the MCF-7 cells relative to the non-functionalized Raji cells (7.3 ± 1.1% vs. 0.8 ± 0.1%; p<0.001). Brief exposure to trimethoprim drove significant dissociation of these clusters (p<0.01), returning nearly the baseline number of non-specific interactions (1.9 ± 0.3%). As demonstrated above, the trimethoprim-induced dissociation of the CSAN scaffold is time-dependent; therefore, it is conceivable that prolonging the trimethoprim incubation in this experiment beyond 1 h would drive further reversal of the cell-cell interactions.
Bioorthogonal CSANs Enable Formation of Multicellular Interactions
Taking advantage of the modular nature of the CSAN platform, we sought to use a combination of multifunctional CSANs to induce controlled interactions between three different model cell populations (Figure 7). MCF-7 cells were again adhered to glass coverslips to form a monolayer of EpCAM-positive target cells. Then, CFSE-labeled Raji cells that had been sequentially modified with DSPE-PEG2000-biotin and mSA/Fn3 CSANs were bound to the MCF-7 cells, as before. To introduce a third cell population, a separate aliquot of Raji cells was labeled with CellTrace Far Red, modified with DSPE-PEG2000-DBCO, and functionalized with azide/mSA bispecific CSANs capable of targeting the unoccupied biotin moieties on the surface of the preceding CFSE-labeled Raji population. After washing the cell layer to remove unbound CFSE-Raji cells, the functionalized FarRed-Raji cell population was added and incubated in an analogous fashion. After washing, fixation, and mounting on glass slides using DAPI-containing mountant, the coverslips were analyzed by fluorescence microscopy. In the absence of CSANs, we again observed that the phospholipid-modified Raji cells were unable to interact with the MCF-7 cells (Figure 7A). However, the CSAN-functionalized Raji cells were able to form targeted cell arrangements, adhering to the monolayer of EpCAM-expressing cells and to each other (Figure 7B). Many of these interactions were reversed after a 1 h incubation in trimethoprim-containing media (Figure 7C). Finally, when the FarRed-Raji cells that had been functionalized with the azide/mSA CSANs were incubated with just the primary MCF-7 cell layer, they were not able to adhere as these cells did not express the target “antigen”, in this case, biotin (Figure 7D). These results indicate that by exchanging the various targeting domains utilized in the CSAN platform, diverse cell populations can be driven to interact with one another in a controlled and pharmacologically reversible fashion.
Figure 7. Bioorthogonal CSANs Enable Formation of Multicellular Interactions.
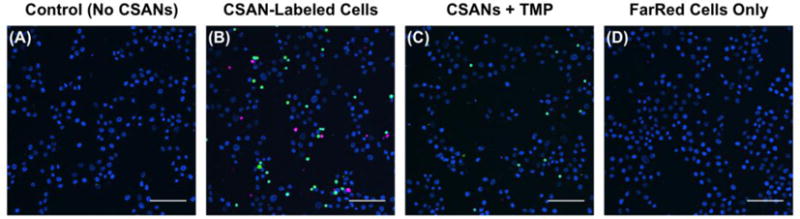
For this experiment, three populations of cells were used: (1) EpCAM+ MCF-7 cells adhered to glass coverslips; (2) CFSE-labeled Raji cells sequentially modified with DSPE-PEG2000-biotin and mSA/Fn3 CSANs; and (3) a second population of Raji cells labeled with CellTrace Far Red, DSPE-PEG2000-DBCO, and mSA CSANs oligomerized with azide-bisMTX, granting them the ability to target unoccupied biotin moieties on the CFSE-labeled Raji cells. (A) In the absence of CSANs, neither Raji cell population is able to adhere to the MCF-7 cell monolayer. (B) When functionalized with the CSANs, the two Raji cell populations are able to interact with each other and with the MCF-7 cell monolayer. (C) The cell-cell interactions are largely reversed when the CSAN scaffold is disassembled with trimethoprim. (D) As the Far Red-labeled Raji cells only have the capability of targeting biotin, they are unable to adhere to the MCF-7 cell monolayer in the absence of the CFSE-labeled Raji cells that were modified with DSPE-PEG2000-biotin. Scale bars represent 100 μm.
CONCLUSIONS
In conclusion, the CSAN platform offers a modular approach for reversibly functionalizing cell membranes with targeting ligands. Through the spontaneous membrane insertion of phospholipids conjugated to biotin and DBCO groups, CSANs can be installed on essentially any cell surface and function as prosthetic receptors. In contrast to genetic engineering approaches, which require both manipulation of the target cell’s genome and extensive culturing to expand the modified cells, the method described here is rapid, scalable to large cell numbers, and broadly applicable to diverse cell types.
Once installed on cell surfaces, EpCAM-targeting CSANs were capable of inducing specific intercellular interactions between the CSAN-functionalized cells and EpCAM-expressing target cells in vitro. Due to the modularity of the CSAN platform, this approach was further expanded to direct targeted interactions between three different cell populations. Additionally, these interactions were rapidly reversed through exposure to trimethoprim.
The capacity to remove CSAN-based surface modifications pharmacologically with trimethoprim makes them distinct from other reversal approaches developed thus far. While photoirradiation, enzymatic degradation, and changes in redox potential or temperature have all been demonstrated, none of these mechanisms are currently applicable in an in vivo setting, especially when the modified cells are broadly distributed throughout an organism (as would be the case for immunotherapy and some regenerative medicine applications). Because trimethoprim is an FDA-approved antibiotic that is used systemically, CSAN-directed cell-cell interactions could conceivably be readily reversed in vivo via trimethoprim administration, providing a safe mechanism for deactivating the targeted cells in the case of adverse events or initiating processes dependent on the loss of intercellular interactions. Ultimately, this work demonstrates that lipid-anchored prosthetic receptors provide a broadly applicable approach to cell surface engineering that could be used to expand the formation of targeted, reversible cell-cell interactions across diverse fields.
MATERIALS & METHODS
Cells and Cell Culture
The MCF-7, U-87 MG, and Raji cell lines were previously purchased from the American Type Culture Collection (ATCC). MCF-7 and U-87 MG cells were grown at 37 °C in a humidified atmosphere with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose, L-glutamine, and supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Raji cells were grown at 37 °C in a humidified atmosphere with 5% CO2 in Roswell Park Memorial Institute (RPMI) media with L-glutamine and supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. When needed for passaging or harvesting, adherent cell lines MCF-7 and U-87 MG were detached via trypsin. Cell count and viability was determined via trypan blue staining/exclusion using a Bio Rad TC20 automated cell counter (Bio Rad Laboratories, Inc.).
Expression Plasmids
gBlock Gene Fragments coding for the DHFR2-Fn3 and mSA-DHFR2 fusion proteins were ordered from Integrated DNA Technologies (IDT) and cloned into the Novagen pET28a(+) vector (EMD Millipore, Cat: 69864-3) via NcoI and XhoI restriction sites. Notably, the DHFR2-Fn3 fusion proteins contain an N-terminal MYC epitope tag and C-terminal polyhistidine tag to facilitate detection via flow cytometry and purification via immobilized metal affinity chromatography (IMAC), respectively. Similarly, the mSA-DHFR2 fusion proteins contain a C-terminal FLAG epitope tag to enable flow cytometric detection.
Protein Expression and Purification
The DHFR2-Fn3 fusion proteins were produced in Escherichia coli (E. coli) and purified from the soluble fraction of the cell lysates by IMAC according to the methods previously reported for the parent Fn3 clones.31 The mSA-DHFR2 fusion proteins produced in E. coli and purified from the insoluble inclusion bodies of the cell lysates according to our previously reported refolding methods.6, 43 Purified proteins were analyzed by SEC on a Superdex 200 Increase 10/300 gel filtration column (GE Healthcare Life Sciences, Cat: 28990944) in phosphate buffered saline (PBS, pH 7.4) running buffer (Figure S4). Fusion protein retention times were compared to those of commercial molecular weight standards (Sigma Aldrich, Cat: MWGF1000-1KT).
CSAN Formation and Characterization
CSANs were formed by adding a 1.1-3.0 fold molar excess of the desired chemical dimerizer – either bisMTX or aizde-bisMTX – to a solution of targeted DHFR2 fusion protein monomers in PBS. Consistent with our previous studies, CSAN oligomerization occurs within minutes of adding the dimerizer.27 Cryo-EM samples were prepared using a Vitrobot Mark IV (FEI). Briefly, 3 μL of CSANs in PBS was applied to a lacey formvar/carbon grid (Ted Pella, Inc.; Cat: 01883) in a humidified chamber, blotted, and plunged into liquid ethane for vitrification. Grids were imaged on a Tecnai Spirit G2 BioTWIN (FEI) equipped with an Eagle 2k CCD camera (FEI) under a high tension of 120 kV. Images were analyzed in ImageJ and, for the size distribution analysis, only nanoparticles with ≥70% circularity were included. For DLS, 60 μL of CSANs in PBS was loaded into a cuvette and analyzed on a Punk DLS unit (Unchained Labs). Hydrodynamic diameter values represent the mean ± standard deviation of at least three measurements.
Affinity Determination of Fn3 and mSA/Fn3 CSANs
The apparent affinity and selectivity of the Fn3 and mSA/Fn3 CSANs was determined by flow cytometry, as previously described.31 Briefly, EpCAM-expressing MCF-7 cells and EpCAM-negative U-87 MG cells were cultured to approximately 80% confluency, detached, and counted, as described above. Aliquots of 5×104 cells were washed with PBSA (PBS with 0.1% w/v bovine serum albumin) and labeled with varying concentrations of Fn3 or mSA/Fn3 CSANs for ≥90 min at 4 °C. Cells were then pelleted (500g, 5 min, 4 °C) and resuspended in 50 μL anti-MYC (clone 9E10) Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Cat: MA1-980-A647; 5 μg/mL in PBSA). After incubating at 4 °C for ≥30 min in the dark, cells were again pelleted and washed thrice with 1 mL cold PBSA before the fluorescence was analyzed on an LSR II flow cytometer (BD Biosciences).
Hydrophobic Insertion of Phospholipid Conjugates
DSPE-PEG2000-biotin and DSPE-PEG2000-DBCO were purchased from Avanti Polar Lipids (Cat: 880129P and 880229P, respectively) and resuspended in PBS at pH 7.4. Cells were modified with DSPE-PEG2000-biotin and DSPE-PEG2000-DBCO via one of two methods: (1) through resuspension in phospholipid-containing PBS (ex vitro), or (2) through active culture in media supplemented with the phospholipid (in vitro).
For the ex vitro (buffer) method, cells were harvested from culture, pelleted at 300g for 5 min, and washed with 1 mL PBS. Cells were then resuspended in PBS containing the desired concentration of phospholipid (0-100 μM) at a ratio of 2.5×106 cells/mL. The cell suspension was then placed on a rotating platform and incubated at room temperature for 1 h. Cells were then pelleted at 300g for 5 min, and washed thrice with 1 mL cold PBS to remove uninserted DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO. Cells were then used directly for subsequent applications.
For the in vitro (culture) method, cells were grown in culture media (DMEM or RPMI, as above) supplemented with the desired concentration (0-100 μM) of DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO for 24-48 h. Cells were then harvested from culture (adherent cells were detached with trypsin), pelleted at 300g for 5 min, and washed thrice with 1 mL cold PBS to remove uninserted DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO. Cells were then used directly for subsequent applications.
Following each modification, flow cytometry was used to determine whether the DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO conjugates had inserted into the cell membrane. To probe for the biotin and DBCO moieties on the cell surface, the phospholipid-modified cells were washed as above and resuspended in 50 μL of either streptavidin Alexa Fluor 488 conjugate (Thermo Fisher Scientific, Cat: S32354; 10 μg/mL in PBS) or azide Alexa Fluor 488 conjugate (Thermo Fisher Scientific, Cat: A10266; 5 μM in PBS), respectively. After incubating at 4 °C for 1 h, the cells were pelleted (500g, 5 min, 4 °C) and washed thrice with 1 mL cold PBS before the fluorescence was analyzed on an LSR II flow cytometer (BD Biosciences). For data analysis, the maximum mean fluorescence intensity (MFI) obtained within each experimental series is normalized to 1.0, with the other samples in that series scaled relative to this value.
Functionalizing Phospholipid-Modified Cells with CSANs
Cells were cultured, harvested, and modified with 10 μM of either DSPE-PEG2000-biotin or DSPE-PEG2000-DBCO ex vitro, as described above. Generally, 0.5×106 cells were then labeled with 500 μL of 100 nM CSANs of the desired functionality (Figure 5) in PBS at 4°C for 1 h. However, to install the Fn3 CSANs formed with azide-bisMTX onto DBCO-modified Raji cells, 100 μL of 500 nM CSANs in PBS was used and the cells were incubated at 37 °C for 3 h. After the primary incubation, cells were washed once with 1 mL cold PBS to remove unbound CSANs. The cells were then resuspended in 50 μL of either anti-MYC (clone 9E10) Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Cat: MA1-980-A647; 5 μg/mL in PBS) or anti-FLAG PE conjugate (Biolegend, Cat: 637309; 1 μg/mL in PBS) to probe for the MYC epitope tag present on the DHFR2-Fn3 subunits or the FLAG epitope tag present on the mSA-DHFR2 subunits, respectively. After incubating at 4 °C for 1 h, the cells were pelleted (500g, 5 min, 4 °C) and washed thrice with 1 mL cold PBS before the fluorescence was analyzed on an LSR II flow cytometer.
Stability Studies
The in vitro longevity of the phospholipid-anchored CSANs on the cell surface was assessed by flow cytometry. Briefly, Raji cells were modified with 10 μM DSPE-PEG2000-biotin in vitro, labeled with 100 nM “reduced-avidity” mSA CSANs (CSANs formed with a 1:1 ratio of mSA-DHFR2 subunits and non-targeted DHFR2 subunits), and then returned to culture for 0-72 h. At 24 h intervals, an aliquot of 0.5×106cells was taken, labeled with an anti-FLAG PE conjugate (1 μg/mL in PBS) to detect cell surface CSANs, and analyzed on an LSR II flow cytometer, as described above. To compare the surface longevity of the CSANs to that of the individual DSPE-PEG2000-biotin moieties themselves, a separate population of Raji cells was modified with only 10 μM DSPE-PEG2000-biotin in vitro (no CSANs) and returned to culture for 0-72 h. An aliquot of these cells was taken, labeled with streptavidin Alexa Fluor 488 conjugate (10 μg/mL in PBS) to detect cell surface biotin moieties, and analyzed on an LSR II flow cytometer in parallel with the CSAN-labeled samples. To determine the number of cell divisions over the course of the experiment, a third aliquot of Raji cells was labeled with CellTrace Violet (CTV; Thermo Fisher Scientific, Cat: C34571) according to the manufacturer’s instructions and cultured/analyzed in parallel with the CSAN and phospholipid samples. For data analysis, the MFI of the samples at t=0 was normalized to 1.0, representing maximum labeling, and the MFI on subsequent days was scaled relative to this value. Because cell division reduces the MFI value through dilution of the CSANs/phospholipids across daughter cell membranes and not due to loss of the constructs, the MFI values of subsequent analyses were corrected for the number of cell divisions, as determined by the CTV labeling.
To ascertain whether the phospholipid-anchored CSANs could “migrate” from the principally modified cell to an unmodified neighbor cell, two populations of Raji cells were prepared. The first population was labeled only with CTV. The second population was modified with 10 μM DSPE-PEG2000-biotin in vitro and then labeled with “reduced valency” mSA CSANs (see above). The CTV-labeled and CSAN-labeled Raji cell populations were combined at a 3:7 ratio and co-cultured in RPMI for 72 h; every 24 h, the culture media was refreshed (to partially simulate the effect of clearance) and a 0.5×106 cell sample of the pooled population was analyzed for CTV and CSAN presence by flow cytometry. CSANs were detected by labeling the cells with anti-FLAG PE conjugate (1 μg/mL in PBS), as above. At each time point, the percentage of CTV+/CSAN− (original CTV-modified population), CTV+/CSAN+ (CTV cells that had acquired a “migrating” CSAN), CTV-/CSAN+ (original CSAN-functionalized population), and CTV-/CSAN− (cell that has lost their CSAN functionalization) cells was quantified by flow cytometry. For comparison, the same analysis was performed for a mixed population of CTV-labeled Raji cells and Raji cells modified with only the 10 μM DSPE-PEG2000-biotin (no CSANs).
Trimethoprim-Induced CSAN Dissociation
Raji cells (0.5×106) were modified with 10 μM DSPE-PEG2000-biotin ex vitro and then labeled with 100 nM mSA/Fn3 CSANs, as above. The CSAN-labeled cells were then divided into two equal aliquots, one of which was resuspended in 200 μL of RPMI and the other in 200 μL of RPMI supplemented with 2 μM trimethoprim (Fisher Scientific, Cat: AAJ66646MD). Cells were then incubated at 37 °C for 1-2 h, labeled with anti-FLAG PE conjugate (1 μg/mL in PBS) to detect cell surface CSANs, and analyzed on an LSR II flow cytometer, as described above. For data analysis, the MFI of the samples in plain RPMI was normalized to 1.0, representing maximum labeling, and the MFI of the samples in RPMI with was scaled relative to this value.
Formation of Intercellular Interactions
To form intercellular interactions between two cell types (Figure 6), a monolayer of MCF-7 cells was adhered to glass coverslips (Thermo Scientific, Cat: 12-541-B) via overnight culture in a 6-well plate. Separately, a population of Raji cells was labeled with CFSE (Biolegend, Cat: 423801) according to the manufacturer’s protocol and returned to culture overnight. The following day, the CFSE-labeled Raji cells were sequentially modified with 10 μM DSPE-PEG2000-biotin ex vitro and 100 nM mSA/Fn3 CSANs, as above. The CSAN-functionalized, and thus EpCAM-targeted, Raji cells were then washed once in 1 mL PBS, resuspended in 1 mL DMEM, and added to the wells containing the MCF-7 cells on the coverslips. The two cell populations were then incubated static at 4 °C for 1 h. Media and unbound cells were then removed via aspiration, and the cell layers were washed thrice with 1 mL PBS. Then, 1 mL of DMEM with or without 2 μM trimethoprim was added to the wells and the cell layers were incubated for another 1 h at 4 °C. Media and unbound cells were again removed via aspiration, and the cell layers were washed thrice with 1 mL PBS. The cell layers were then fixed in 4% paraformaldehyde in PBS at room temperature for 15 min before washing thrice with 1 mL PBS. Coverslips were then rinsed twice in ultrapure water, blotted to remove excess liquid, and mounted on glass coverslips using ProLong Gold Antifade Reagent with DAPI (Thermo Fisher Scientific, Cat: P36935). After curing for ≥24 h in the dark, slides were imaged on an Eclipse Ti-E Wide Field Deconvolution Inverted Microscope (Nikon Instruments, Inc.).
Intercellular interactions between three different cell populations (Figure 7) were formed similarly; however, the CFSE-labeled Raji cells were modified with only 50 nM mSA/Fn3 CSANs instead of 100 nM. Then, a third cell population was prepared by sequentially labeling Raji cells with CellTrace Far Red (Thermo Fisher Scientific, Cat: 34572) according to the manufacturer’s protocols, 100 μM DSPE-PEG2000-DBCO ex vitro, and 500 nM mSA CSANs oligomerized with azide-bisMTX, as above. This population of Raji cells – now capable of targeting the unoccupied biotin moieties on the CFSE-labeled Raji cell layer – was added to the wells after the incubation with the CFSE-labeled Raji cells (but before the addition of the DMEM with trimethoprim). After incubating the cell layers with this third cell population at 4 °C for 1 h, the coverslips were washed, fixed, mounted, and imaged on an Eclipse Ti-E Wide Field Deconvolution Inverted Microscope, as above.
For the flow cytometry analysis of cell pairing, target MCF-7 cells were labeled with CFSE, cultured for 24 h, and detached, as above. Separately, a population of Raji cells were sequentially labeled with CTV, cultured for 24 h, modified with 10 μM DSPE-PEG2000-biotin, and functionalized with mSA/Fn3 CSANs, as above. The two cell populations were resuspended in PBS, combined at a 1:1 ratio, and incubated at 4 °C in the dark with rotation for 1 h. Cells were then pelleted (500 g, 5 min, 4 °C), washed once with 1 mL cold PBS, and resuspended in PBS with or without 2 μM trimethoprim. After incubating at 4 °C in the dark with rotation for 1 h, the cells were washed thrice with PBS and analyzed on an LSR II flow cytometer to ascertain the number of CTV+/CFSE+ cell clusters. As controls, unmodified Raji cells, unmodified MCF-7 cells, CTV-labeled Raji cells, CFSE-labeled MCF-7 cells, and CTV-Raji cells modified with only 10 μM DSPE-PEG2000-biotin (no CSANs) plus CFSE-MCF-7 cells were all prepared and analyzed in parallel.
Statistical Considerations
Unless otherwise stated, experiments were performed in triplicate and data is presented as the mean ± standard deviation of three independent trials. Differences between means are compared using a two-tailed Student’s t-test, and a p-value <0.05 is denoted in graphics with an (*), p<0.01 is denoted with (**), and p<0.001 is denoted with (***).
Safety Considerations
No unexpected or unusually high safety hazards were encountered.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health R21 CA185627 (CRW), F30 CA210345 (CMC), T32 GM008244 (CMC), R21 EB019518 (BJH), and the University of Minnesota. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program. The authors gratefully acknowledge Dr. Bob Hafner (Characterization Facility, University of Minnesota) for his assistance with the transmission electron microscopy experiments.
ABBREVIATIONS
- CFSE
carboxyfluorescein succinimidyl ester
- CSAN
chemically self-assembled nanoring
- CTV
CellTrace Violet
- DBCO
dibencocyclooctyne
- DHFR
dihydrofolate reductase
- DSPE
1,2-distearoyl-sn-glycero-3-phosphoethanolamine
- EpCAM
epithelial cell adhesion molecule
- Fn3
tenth type III fibronectin domain
- MFI
mean fluorescence intensity
- mSA
monovalent streptavidin domain
- MTX
methotrexate
- PEG
polyethylene glycol
- SEC
size exclusion chromatography
- TMP
trimethoprim
Footnotes
SUPPORTING INFORMATION
Cell viability data, quantitative analysis of phospholipid insertion experiments, SEC and DLS characterization of CSANs, affinity and selectivity of EpCAM-targeted CSANs, synthesis and characterization of azide-bisMTX, CSAN-labeling experiments demonstrating optimization and bispecificity, plasma stability data, and fusion protein sequences
References
- 1.Gartner ZJ, Bertozzi CR. Programmed assembly of 3-dimensional microtissues with defined cellular connectivity. Proc Natl Acad Sci USA. 2009;106:4606–10. doi: 10.1073/pnas.0900717106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogozhnikov D, O’Brien PJ, Elahipanah S, Yousaf MN. Scaffold Free Bio-orthogonal Assembly of 3-Dimensional Cardiac Tissue via Cell Surface Engineering. Sci Rep. 2016;6:39806. doi: 10.1038/srep39806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kean TJ, Duesler L, Young RG, Dadabayev A, Olenyik A, Penn M, Wagner J, Fink DJ, Caplan AI, Dennis JE. Development of a peptide-targeted, myocardial ischemia-homing, mesenchymal stem cell. J Drug Target. 2012;20:23–32. doi: 10.3109/1061186X.2011.622398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarkar D, Spencer JA, Phillips JA, Zhao W, Schafer S, Spelke DP, Mortensen LJ, Ruiz JP, Vemula PK, Sridharan R, et al. Engineered cell homing. Blood. 2011;118:e184–91. doi: 10.1182/blood-2010-10-311464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi P, Ju E, Yan Z, Gao N, Wang J, Hou J, Zhang Y, Ren J, Qu X. Spatiotemporal control of cell-cell reversible interactions using molecular engineering. Nat Commun. 2016;7:13088. doi: 10.1038/ncomms13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrielse K, Gangar A, Kumar N, Lee JC, Fegan A, Shen JJ, Li Q, Vallera D, Wagner CR. Reversible re-programing of cell-cell interactions. Angew Chem Int Ed. 2014;53:5112–6. doi: 10.1002/anie.201310645. [DOI] [PubMed] [Google Scholar]
- 7.Merzaban JS, Imitola J, Starossom SC, Zhu B, Wang Y, Lee J, Ali AJ, Olah M, Abuelela AF, Khoury SJ, et al. Cell surface glycan engineering of neural stem cells augments neurotropism and improves recovery in a murine model of multiple sclerosis. Glycobiology. 2015;25:1392–409. doi: 10.1093/glycob/cwv046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao W, Loh W, Droujinine IA, Teo W, Kumar N, Schafer S, Cui CH, Zhang L, Sarkar D, Karnik R, et al. Mimicking the inflammatory cell adhesion cascade by nucleic acid aptamer programmed cell-cell interactions. Faseb j. 2011;25:3045–56. doi: 10.1096/fj.10-178384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–81. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nowakowski A, Andrzejewska A, Janowski M, Walczak P, Lukomska B. Genetic engineering of stem cells for enhanced therapy. Acta Neurobiol Exp (Wars) 2013;73:1–18. doi: 10.55782/ane-2013-1918. [DOI] [PubMed] [Google Scholar]
- 12.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamers CHJ, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, et al. Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Mol Ther. 21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7:1–16. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutta D, Pulsipher A, Luo W, Mak H, Yousaf MN. Engineering cell surfaces via liposome fusion. Bioconjugate Chem. 2011;22:2423–33. doi: 10.1021/bc200236m. [DOI] [PubMed] [Google Scholar]
- 16.Luo W, Westcott N, Dutta D, Pulsipher A, Rogozhnikov D, Chen J, Yousaf MN. A Dual Receptor and Reporter for Multi-Modal Cell Surface Engineering. ACS Chem Biol. 2015;10:2219–26. doi: 10.1021/acschembio.5b00137. [DOI] [PubMed] [Google Scholar]
- 17.Cheng H, Byrska-Bishop M, Zhang CT, Kastrup CJ, Hwang NS, Tai AK, Lee WW, Xu X, Nahrendorf M, Langer R, et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell-selectin interaction kinetics. Biomaterials. 2012;33:5004–12. doi: 10.1016/j.biomaterials.2012.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko IK, Kean TJ, Dennis JE. Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials. 2009;30:3702–10. doi: 10.1016/j.biomaterials.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeong JH, Schmidt JJ, Kohman RE, Zill AT, DeVolder RJ, Smith CE, Lai MH, Shkumatov A, Jensen TW, Schook LG, et al. Leukocyte-mimicking stem cell delivery via in situ coating of cells with a bioactive hyperbranched polyglycerol. J Am Chem Soc. 2013;135:8770–3. doi: 10.1021/ja400636d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todhunter ME, Jee NY, Hughes AJ, Coyle MC, Cerchiari A, Farlow J, Garbe JC, LaBarge MA, Desai TA, Gartner ZJ. Programmed synthesis of three-dimensional tissues. Nat Methods. 2015;12:975–81. doi: 10.1038/nmeth.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamdy N, Goustin AS, Desaulniers JP, Li M, Chow CS, Al-Katib A. Sheep red blood cells armed with anti-CD20 single-chain variable fragments (scFvs) fused to a glycosylphosphatidylinositol (GPI) anchor: a strategy to target CD20-positive tumor cells. J Immunol Methods. 2005;297:109–24. doi: 10.1016/j.jim.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Luo W, Pulsipher A, Dutta D, Lamb BM, Yousaf MN. Remote Control of Tissue Interactions via Engineered Photo-switchable Cell Surfaces. Sci Rep. 2014;4:6313. doi: 10.1038/srep06313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pulsipher A, Dutta D, Luo W, Yousaf MN. Cell-surface engineering by a conjugation-and-release approach based on the formation and cleavage of oxime linkages upon mild electrochemical oxidation and reduction. Angew Chem Int Ed. 2014;53:9487–92. doi: 10.1002/anie.201404099. [DOI] [PubMed] [Google Scholar]
- 24.Altman MO, Chang YM, Xiong X, Tan W. Modifying cellular properties using artificial aptamer-lipid receptors. Sci Rep. 2013;3:3343. doi: 10.1038/srep03343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amaral AJ, Pasparakis G. Macromolecular cell surface engineering for accelerated and reversible cellular aggregation. Chem Commun. 2015;51:17556–9. doi: 10.1039/c5cc07001e. [DOI] [PubMed] [Google Scholar]
- 26.Xiong X, Liu H, Zhao Z, Altman MB, Lopez-Colon D, Yang CJ, Chang LJ, Liu C, Tan W. DNA Aptamer-Mediated Cell Targeting. Angew Chem Int Ed. 2013;52:1472–1476. doi: 10.1002/anie.201207063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlson JCT, Jena SS, Flenniken M, Chou T-F, Siegel RA, Wagner CR. Chemically Controlled Self-Assembly of Protein Nanorings. J Am Chem Soc. 2006;128:7630–7638. doi: 10.1021/ja060631e. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, So CR, Fegan A, Cody V, Sarikaya M, Vallera DA, Wagner CR. Chemically Self-Assembled Antibody Nanorings (CSANs): Design and Characterization of an Anti-CD3 IgM Biomimetic. J Am Chem Soc. 2010;132:17247–17257. doi: 10.1021/ja107153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah R, Petersburg J, Gangar AC, Fegan A, Wagner CR, Kumarapperuma SC. In Vivo Evaluation of Site-Specifically PEGylated Chemically Self-Assembled Protein Nanostructures. Mol Pharm. 2016;13:2193–203. doi: 10.1021/acs.molpharmaceut.6b00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim KH, Huang H, Pralle A, Park S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol Bioeng. 2013;110:57–67. doi: 10.1002/bit.24605. [DOI] [PubMed] [Google Scholar]
- 31.Stern LA, Csizmar CM, Woldring DR, Wagner CR, Hackel BJ. Titratable Avidity Reduction Enhances Affinity Discrimination in Mammalian Cellular Selections of Yeast-Displayed Ligands. ACS Combi Sci. 2017;19:315–323. doi: 10.1021/acscombsci.6b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fegan A, Kumarapperuma SC, Wagner CR. Chemically Self-Assembled Antibody Nanostructures as Potential Drug Carriers. Mol Pharm. 2012;9:3218–3227. doi: 10.1021/mp300303k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, Vallera DA, Wagner CR. Prosthetic Antigen Receptors. J Am Chem Soc. 2015;137:10108–11. doi: 10.1021/jacs.5b06166. [DOI] [PubMed] [Google Scholar]
- 34.Lim KS, Lee DY, Valencia GM, Won YW, Bull DA. Cell surface-engineering to embed targeting ligands or tracking agents on the cell membrane. Biochem Biophys Res Commun. 2017;482:1042–1047. doi: 10.1016/j.bbrc.2016.11.155. [DOI] [PubMed] [Google Scholar]
- 35.de Kruif J, Tijmensen M, Goldsein J, Logtenberg T. Recombinant lipid-tagged antibody fragments as functional cell-surface receptors. Nat Med. 2000;6:223–7. doi: 10.1038/72339. [DOI] [PubMed] [Google Scholar]
- 36.Weber RJ, Liang SI, Selden NS, Desai TA, Gartner ZJ. Efficient targeting of fatty-acid modified oligonucleotides to live cell membranes through stepwise assembly. Biomacromolecules. 2014;15:4621–6. doi: 10.1021/bm501467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo CY, Antonopoulos A, Dell A, Haslam SM, Lee T, Neelamegham S. The use of surface immobilization of P-selectin glycoprotein ligand-1 on mesenchymal stem cells to facilitate selectin mediated cell tethering and rolling. Biomaterials. 2013;34:8213–22. doi: 10.1016/j.biomaterials.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srisa-Art M, Dyson EC, deMello AJ, Edel JB. Monitoring of Real-Time Streptavidin−Biotin Binding Kinetics Using Droplet Microfluidics. Anal Chem. 2008;80:7063–7067. doi: 10.1021/ac801199k. [DOI] [PubMed] [Google Scholar]
- 39.Karver MR, Weissleder R, Hilderbrand SA. Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew Chem Int Ed. 2012;51:920–922. doi: 10.1002/anie.201104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. 2012;38:68–75. doi: 10.1016/j.ctrv.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Eatman FB, Maggio AC, Pocelinko R, Boxenbaum HG, Geitner KA, Glover W, Macasieb T, Holazo A, Weinfeld RE, Kaplan SA. Blood and salivary concentrations of sulfamethoxazole and trimethoprim in man. J Pharmacokinet Biopharm. 1977;5:615–624. doi: 10.1007/BF01059687. [DOI] [PubMed] [Google Scholar]
- 42.Watson ID, Stewart MJ. Trimethoprim: Prediction of serum concentrations from saliva measurements. Eur J Clin Pharmacol. 1986;30:457–461. doi: 10.1007/BF00607960. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Hapka D, Chen H, Vallera DA, Wagner CR. Self-assembly of antibodies by chemical induction. Angew Chem Int Ed. 2008;47:10179–82. doi: 10.1002/anie.200803507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.