

Abstract
Herein we report a highly efficient method for nickel-catalyzed C–N bond formation between sulfonamides and aryl electrophiles. This technology provides generic access to a broad range of N-aryl and N-heteroaryl sulfonamide motifs, which are widely represented in drug discovery. Initial mechanistic studies suggest an energy-transfer mechanism wherein C–N bond reductive elimination occurs from a triplet excited NiII complex. Late-stage sulfonamidation in the synthesis of a pharmacologically relevant structure is also demonstrated.
Keywords: energy transfer, heterocycles, nickel, photocatalysis, sulfonamides
Graphical Abstract
A method for C–N bond formation between sulfonamides and aryl electrophiles is reported. This method provides generic access to a broad range of N-aryl and N-heteroaryl sulfonamide motifs, which are widely represented in drug discovery. Initial mechanistic studies suggest an energy-transfer mechanism wherein C–N bond reductive elimination occurs from a triplet excited NiII complex.
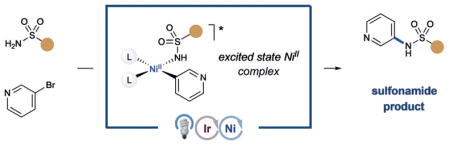
Aniline synthesis by means of C–N bond formation is a widely used transformation in medicinal chemistry and pharmaceutical synthesis.[1] Key structural motifs within this important class of compounds are N-aryl and N-heteroaryl sulfonamides, which are represented in a significant portion of widely prescribed pharmaceuticals (Scheme 1).[2] Indeed, the sulfonamide motif has been known to exhibit high levels of bioactivity for almost a century, forming the basis of a series of antibacterial drugs, some of which are still in use to this day.[3] In addition to a range of valuable biochemical properties, secondary sulfonamides can be readily exploited as carboxylic acid isosteres, wherein the inherent N–H pKa can be readily modulated by pendent aryl and heteroaryl groups.[4] Access to sulfonamides traditionally involves amine nucleophiles in combination with sulfonyl chlorides, reagents that are often toxic and non-trivial to prepare. While the development of technologies to directly cross-couple amines with aryl halides has proceeded rapidly in recent years, the attenuated nucleophilicity of sulfonamides relative to alkyl amines presents an additional challenge.[5]
Scheme 1.

Nickel-catalyzed synthesis of aryl sulfonamides.
The utility and scope of C–N bond-forming reactions have increased dramatically over the past several decades, mainly arising from the advent of Buchwald–Hartwig aryl amination cross-coupling.[6] A central feature enabling the success of this technology is the now ubiquitous family of dialkyl biaryl phosphine ligands carefully designed to prevent Pd dimer formation and accelerate reductive elimination steps.[7] In contrast to the large body of literature describing C–N bond formation through Pd and Cu catalysis, the use of Ni catalysis remains largely underdeveloped, an unfortunate circumstance given the relative abundance and economic advantages afforded by nickel.[8] The lack of success for nickel in this area has long been attributed to the fact that NiII C-NR2 reductive elimination is a high-barrier step, a mechanistic feature that has curtailed broad application.[9]
Recently, however, it has been demonstrated that a number of fundamental steps in nickel cross-coupling processes can be generically “switched on” through the modulation of oxidation states or electronic energy levels using photocatalysis. In this context, our group has described several different transformations that employ photocatalyst excited states with nickel to enable otherwise disfavored reductive elimination steps involving C–O and C–N bonds (Figure 1).[10] Moreover, recent reports from the groups of Fu and Peters, Nocera, Molander, and Doyle describing direct photoexcitation of transition-metal catalysts provide elegant demonstrations of this paradigm.[11] Our group has also demonstrated that energy-transfer photosensitization can be used to access triplet excited states of NiII complexes, specifically in the context of C–O bond formation between carboxylic acids and aryl halides.[10b] A key benefit of this methodology is the separation of light-harvesting and cross-coupling roles between two different transition-metal complexes. Indeed, utilization of a discrete energy-transfer catalyst bypasses the question of whether an organometallic complex can efficiently undergo direct visible-light excitation, the capacity for which often varies on a case-by-case basis.
Figure 1.
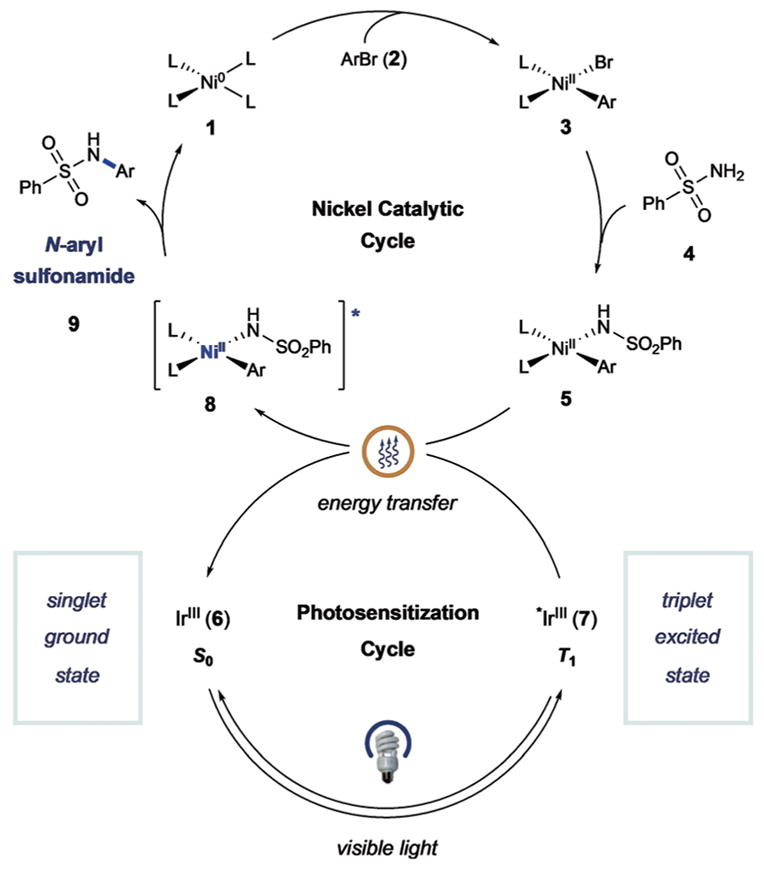
Proposed mechanism of sulfonamidation.
Recently, we hypothesized that photosensitized organometallic catalysis might enable C–N bond formation between aryl halides and sulfonamides in a general sense. The proposed mechanism for this sulfonamidation method is outlined in Figure 1. This dual catalysis process begins with oxidative addition of Ni0 complex 1 into aryl halide electro-phile 2 to generate NiII-aryl complex 3. At this stage, ligand exchange with benzenesulfonamide (4) and deprotonation would form NiII-aryl amido complex 5. At the same time, irradiation of iridium(III) photocatalyst Ir(ppy)2(bpy)PF6 (6; ppy =2-phenylpyridine; bpy =bipyridine) with visible light would produce the long-lived triplet photoexcited state *IrIII 7 (τ =0.3 μs).[12] Based on our previous studies, we hypothesized that nickel complex 5 and excited-state Ir system 7 would undergo triplet–triplet energy transfer to form excited nickel complex 8. The resulting triplet-excited-state species 8 should readily undergo a reductive elimination step to deliver N-aryl sulfonamide product 9 and regenerate Ni0 catalyst 1.
To our delight, initial studies revealed that the proposed C–N bond-forming reaction was indeed possible when using of Ir(ppy)2(bpy)PF6 (1), NiCl2·glyme, tetramethylguanadine (TMG) as base, and MeCN as solvent (see the Supporting Information for optimization studies). At this juncture, we sought to evaluate our hypothesis that an energy-transfer pathway was operative, as outlined in our mechanistic scheme (Figure 1). Indeed, control experiments revealed that the presence of both nickel and visible light were critical to product formation (Table 1).[13] However, in the absence of photocatalyst, a significant amount of product was still obtained through direct irradiation with blue LED light, and UV/Vis studies support the formation of a nickel complex capable of visible-light absorption (see the Supporting Information). The efficiency of this process is increased in the presence of benzophenone, an organic sensitizer. This observation demonstrates that formation of a NiII excited-state species can induce reductive elimination, which is consistent with an energy-transfer mechanism being operative in the presence of the photocatalyst.[14] Moreover, the improved efficiency observed under dual catalytic conditions (cf. light-only direct excitation) highlights the important role of light harvesting by the photocatalyst. As further evidence for photosensitization of the nickel catalyst, additional experiments revealed that the efficiency of product formation is correlated with the triplet energy of the photocatalyst (see the Supporting Information).
Table 1.
C–N sulfonamidation control experiments.[a]
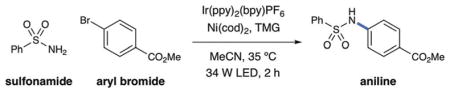
| ||
|---|---|---|
| Entry | Conditions | Yield [%] |
| 1 | as shown | 99 |
| 2 | no light | 0 |
| 3 | no nickel | 0 |
| 4 | no photocatalyst | 11 |
| 5 | benzophenone (0.5%) as photocatalyst | 18 |
| 6 | no photocatalyst, no light | 0 |
| ||
Performed with Ir(ppy)2(bpy)PF6 (0.05 mol%), Ni(cod)2 (5 mol%), TMG (1.5 equiv), aryl halide (1.0 equiv), and benzenesulfonamide (1.5 equiv).
Yields were obtained by 1H NMR analysis.
Having determined optimal conditions for C–N bond formation, we set out to determine the substrate scope of this photocatalytic reaction (Table 2). As a general design principle, we sought to examine the proposed transformation in the presence and absence of bipyridine ligands, given that our recent studies demonstrated the possibility of C–N coupling using “ligand-free” conditions. Notably, the inclusion of dtbbpy (1 mol%) in DMSO as solvent enabled the reaction to be conducted at ambient temperature and displayed high efficiency for electron-rich aryl halide substrates. In the absence of ligand, the amination reaction was also successful at elevated reaction temperatures (68°C), presumably to facilitate oxidative addition, and the reaction could be conducted with reduced photocatalyst loading (0.05 mol%). Given the inherent advantages of each system, we have elected to report the results for both methods (Table 2).
Table 2.
Scope of photosensitized nickel-catalyzed cross-coupling.[a]

|
All yields are of isolated product. Performed with Ir photocatalyst (0.5 or 0.05 mol%), NiCl2·glyme (5 mol%), aryl bromide (1.0 equiv), sulfonamide (1.5 equiv), and tetramethylguanadine (1.5 equiv). For detailed experimental procedures, see the Supporting Information.
A large number of electron-neutral and electron-rich halides provided excellent yields, despite a potentially challenging nickel oxidative addition step in these cases (10–17, 41–86% yield without ligand, 66–98% yield with ligand). As expected, the ligand-added conditions were generally more effective for electron-rich arenes. Notably, ortho-substituted aryl rings were readily tolerated (16, 19, and 24, 50–83% yield). Furthermore, a number of electron-deficient aryl halides containing fluoro, amido, trifluoromethyl, and cyano functionalities were successful coupling partners (18–24, 50–88% yield without ligand, 72–96% yield with ligand). It is important to note that good to excellent yields were observed with a range of heteroaryl halide electrophiles, including pyridine, pyrimidine, and pyrazine (25–31, 49–90% yield without ligand, 5–73% yield with ligand). Importantly, 5-membered ring heterocyclic aryl halides, which are notoriously difficult cross-coupling partners in general, also provided good yields of the desired C–N-coupled products (32 and 33, 62% and 55% yield, respectively). Among this class of substrates, “ligand-free” conditions were uniformly more effective. We next turned our attention to the scope of the sulfonamide nucleophile. Gratifyingly, a number of aryl and heteroaryl sulfonamides were tolerated (34–39, 19–99% yield). It should also be noted that complete selectivity for bond formation at the primary sulfonamide moiety versus alternative N–H sites was observed (39, 91% and 99% yields). Moreover, efficient coupling was achieved with a range of alkyl sulfonamide examples (40–43, 41–99% yield).
Finally, as a demonstration of this new C–N bond-forming method and its potential application to the preparation of drug-like molecules, we undertook a synthesis of dabrafenib, a selective B-Raf kinase inhibitor.[15] As shown in Figure 2, the drug precursor 44 incorporates both an aryl halide and an unprotected anilinic nitrogen on a pyrimidine ring. By implementing high-throughput evaluation (96-well-plate format) we were able to find optimal conditions for the desired C–N bond formation (see the Supporting Information). Indeed, photosensitized nickel cross-coupling between 2,6-difluorobenzenesulfonamide and aryl bromide 44 using ligated nickel provided dabrafenib (45) in a useful level of efficiency (57% yield) without the requirement for protection/deprotection sequences.
Figure 2.
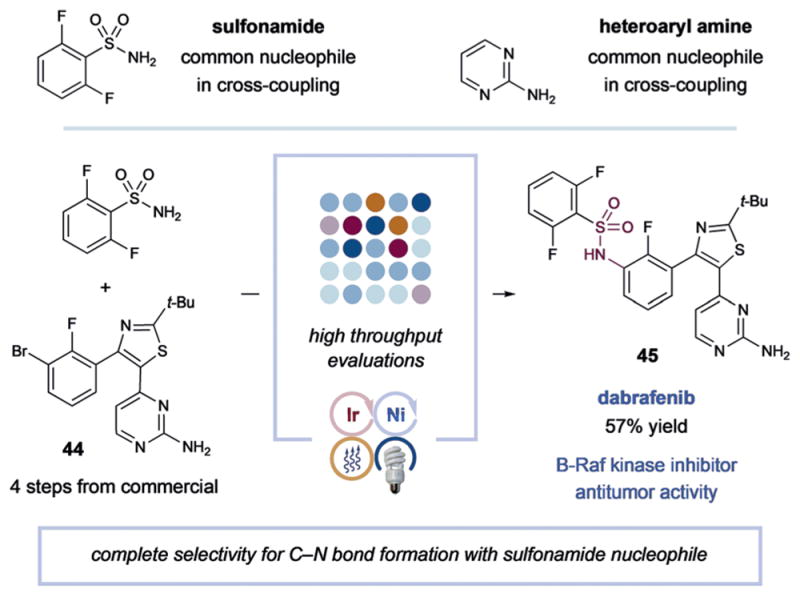
Synthesis of dabrafenib.
Supplementary Material
Acknowledgments
The authors are grateful for financial support provided by the NIH General Medical Sciences (Grant NIHGMS (R01 GM103558-05), Seoul National University, the NRF (2017 R1A2B3002869) of Korea, and kind gifts from Merck, BMS, Janssen, and Eli Lilly. The authors thank Professor Stephen Buchwald for helpful discussions.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201800699.
Contributor Information
Taehoon Kim, Department of Chemistry, Seoul National University Seoul 08826 (South Korea).
Stefan J. McCarver, Merck Center for Catalysis at Princeton University, Washington Road, Princeton, NJ 08544 (USA).
Prof. Dr. Chulbom Lee, Department of Chemistry, Seoul National University Seoul 08826 (South Korea)
Prof. Dr. David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Washington Road, Princeton, NJ 08544 (USA)
References
- 1.Fischer C, Koenig B. Beilstein J Org Chem. 2011;7:59–74. doi: 10.3762/bjoc.7.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Feng M, Tang B, Liang SH, Jiang X. Curr Top Med Chem. 2016;16:1200–1216. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr Med Chem. 2003;10:925–953. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- 3.a) Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]; b) Kalgutkar AS, Jones RA. Sawant in Metabolism, Pharmacokinetics and Toxicity of Functional Groups. In: Smith DA, editor. Sulfonamide as an Essential Functional Group in Drug Design. Royal Society of Chemistry; Cambridge: 2010. pp. 210–274. [Google Scholar]
- 4.a) Ballatore C, Huryn DM, Smith AB., III ChemMedChem. 2013;8:385–395. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Şanli N, Şanli S, Özkan G, Denizli A. J Braz Chem Soc. 2010;21:1952–1960. [Google Scholar]
- 5.Reports on aryl halide sulfonamidation with Cu and Pd: Wolfe JP, Rennels RA, Buchwald SL. Tetrahedron. 1996;52:7525–7546.Baffoe J, Hoe YM, Touré BB. Org Lett. 2010;12:1532–1535. doi: 10.1021/ol100263r.He H, Wu YJ. Tetrahedron Lett. 2003;44:3385–3386.Nasrollahzadeh M, Ehsani A, Maham M. Synlett. 2014:505–508.Yin J, Buchwald SL. Org Lett. 2000;2:1101–1104. doi: 10.1021/ol005654r.Burton G, Cao P, Li G, Rivero R. Org Lett. 2003;5:4373–4376. doi: 10.1021/ol035655u.
- 6.a) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805–818. [Google Scholar]; b) Hartwig JF. Acc Chem Res. 1998;31:852–860. [Google Scholar]
- 7.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Wolfe JP, Buchwald SL. J Am Chem Soc. 1997;119:6054–6058. [Google Scholar]; b) Corcoran EB, Pirnot MT, Lin S, Dreher SD, DiRocco DA, Davies IW, Buchwald SL, MacMillan DWC. Science. 2016;353:279–283. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ge S, Green R, Hartwig JF. J Am Chem Soc. 2014;136:1617–1627. doi: 10.1021/ja411911s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lavoie CM, MacQueen PM, Rotta-Loria NL, Sawatzky RS, Borzenko A, Chisholm AJ, Hargreaves BKV, McDonald R, Ferguson MJ, Stradiotto M. Nat Commun. 2016;7:11073. doi: 10.1038/ncomms11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koo K, Hillhouse GL. Organometallics. 1995;14:4421–4423. [Google Scholar]
- 10.a) Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Welin ER, Le CC, Arias-Rotondo DM, McCusker JK, MacMillan DWC. Science. 2017;355:380–385. doi: 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647–651. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]; b) Kainz QM, Matier CD, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hwang SJ, Powers DC, Maher AG, Anderson BL, Hadt RG, Zheng SL, Chen YS, Nocera DG. J Am Chem Soc. 2015;137:6472–6475. doi: 10.1021/jacs.5b03192. [DOI] [PubMed] [Google Scholar]; d) Heitz DR, Tellis JC, Molander GA. J Am Chem Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shields BJ, Doyle AG. J Am Chem Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochola JR, Wolf MO. Org Biomol Chem. 2016;14:9088–9092. doi: 10.1039/c6ob01717g. [DOI] [PubMed] [Google Scholar]
- 13.The use of Ni(cod)2 for these mechanistic experiments is critical in that it circumvents the necessary initial reduction of the NiII precatalyst to Ni0.
- 14.Stern–Volmer experiments rule out the involvement of N-centered sulfonamidyl radicals generated by a proton-coupled electrontransfer mechanism (see the Supporting Information).
- 15.Rheault TR, Stellwagen JC, Adjabeng GM, Hornberger KR, Petrov KG, Waterson AG, Dickerson SH, Mook RA, Jr, Laquerre SG, King AJ, Rossanese OW, Arnone MR, Smitheman KN, Carson Kane LS, Han C, Moorthy GS, Moss KG, Uehling DE. ACS Med Chem Lett. 2013;4:358–362. doi: 10.1021/ml4000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.