

Abstract
A robust network of transcription factors and an open chromatin landscape are hallmarks of the naïve pluripotent state. Recently, the acetyllysine reader Brd4 has been implicated in stem cell maintenance, but the relative contribution of Brd4 to pluripotency remains unclear. Here we show that Brd4 is dispensable for self-renewal and pluripotency of embryonic stem cells (ESCs). When maintained in their ground state, ESCs retain transcription factor binding and chromatin accessibility independent of Brd4 function or expression. In metastable ESCs, Brd4 independence can be achieved by increased expression of pluripotency transcription factors including STAT3, Nanog or Klf4 so long as the DNA methylcytosine oxidases, Tet1 and Tet2, are present. These data reveal that Brd4 is not essential for ESC self-renewal. Rather, the levels of pluripotency transcription factor abundance and Tet1/2 function determine the extent to which bromodomain recognition of protein acetylation contributes to the maintenance of gene expression and cell identity.
The interplay between transcription factors and the chromatin landscape is a critical determinant of lineage-specific gene expression programs that define cell identity. In embryonic stem cells (ESCs), a network of transcription factors including Oct4, Sox2 and Nanog contributes to self-renewal and pluripotency1, 2. The ability of transcription factors to control gene expression can be amplified or repressed by histone and DNA modifications; in turn, transcription factors influence the expression and localization of chromatin modifying proteins3, 4. Repressive chromatin modifications, such as methylation of DNA and certain histone lysine residues, have been reported to occlude transcription factor binding and block the ability of transcription factors to maintain transcriptional networks5–7. In contrast, histone acetylation can promote the recruitment of transcription factors and bromodomain-containing proteins that are required for pluripotency8, 9.
Mouse ESCs cultured in conventional medium containing serum and leukemia inhibitory factor (LIF; hereafter S/L) exhibit heterogeneous expression of pluripotency-associated transcription factors and levels of DNA methylation comparable to that observed in somatic cells. The addition of kinase inhibitors against MEK and GSK3β (‘2i’) drives murine ESCs into a naïve “ground state” of pluripotency marked by homogenous expression of pluripotency-associated transcription factors and global DNA hypomethylation10. Whereas a fraction of S/L-cultured ESCs can be considered naïve11, the majority is “metastable” and prone to spontaneous differentiation. In contrast, 2i-cultured ESCs are homogenously naïve and continuously self-renew in culture10. Histone and DNA demethylation have been implicated in the establishment of the naïve ground state12–17, but the role of acetylation of either histones or transcription factors in maintaining naïve pluripotency has been less clear.
Histone acetylation promotes gene expression in cis, by modulating charge-driven nucleosomal contacts, and in trans, by recruiting transcriptional coactivator complexes. Bromodomain and extraterminal (BET) domain-containing proteins such as Brd4 bind to acetylated histone tails and acetylated transcription factors and recruit complexes, including the Mediator complex, that promote gene transcription18. Inhibitors against BET family members inhibit gene expression and proliferation in cancer cells and ESCs19. Brd4, in particular, has been reported to be essential for mouse post-implantation development and the culture of ESCs in vitro, suggesting that the reading of acetylation by BET family members is essential for the maintenance of embryonic pluripotency8, 20–23. However, whether Brd4 is essential for the maintenance of the naïve pluripotent state remains an open question.
Results
Brd4 is dispensable in naïve ground state ESCs
Acetylation of histone residues such as H3K9 and H3K27 decorates promoters of active genes and is associated with transcription24, 25. Analyses of chromatin immunoprecipitation (ChIP)-sequencing datasets of H3K9ac and H3K27ac in metastable ESCs26 revealed that genes marked by high promoter histone acetylation are enriched for pluripotency associated gene signatures (Fig. 1a). We asked whether these marks are modulated in the ground state of pluripotency. As serum is lipid-laden, traditional serum-free naïve culture conditions (2i/L) impose a significant burden for de novo lipid biosynthesis. To exclude possible confounding effects of serum on histone acetylation, which competes with lipid biosynthesis for cytosolic acetyl-CoA, we compared histone acetylation in ESCs cultured in S/L with or without 2i, as 2i is sufficient to drive many of the epigenetic and metabolic changes characteristic of ground state pluripotency14, 27.
Figure 1. 2i increases acetylation at key pluripotency loci.
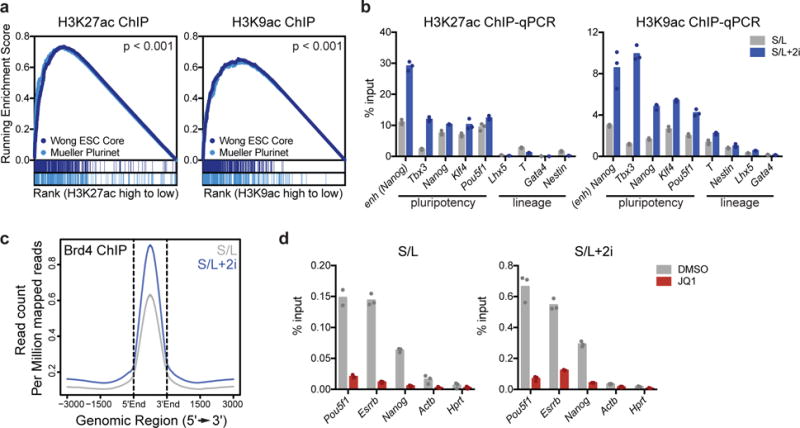
(a) Gene set enrichment plot showing that genes associated with high H3K9ac and H3K27ac are enriched for two independently defined pluripotency gene sets: Muller Plurinet (genes involved in the protein-protein network shared by diverse pluripotent cell types53) and Wong ESC Core (genes coordinately upregulated in mouse and human ESCs54). Data are derived from a single ChIP-Seq experiment26. P values are calculated based on 1000 permutations by the GSEA algorithm and was not adjusted for multiple comparisons. (b) 2i increases acetylation at key pluripotency genes. H3K27ac (left) and H3K9ac (right) at enhancer (enh) or promoters of indicated genes as assessed by ChIP-qPCR. (c) ChIP-seq meta profile for Brd4 binding in ESCs cultured in S/L or S/L+2i. The metaprofile is centered on the midpoint of all Brd4 ChIP-seq peaks. (d) Brd4 ChIP-qPCR illustrating Brd4 binding in ESCs cultured in S/L (left) or S/L+2i (right) treated with DMSO (vehicle) or 500 nM JQ1 for 24 h. (b,d) Bars represent mean of n=3 technical replicates from one IP.
The addition of 2i increased both H3K9ac and H3K27ac at key cis-regulatory loci including enhancers and promoters of pluripotency genes (Fig. 1b), prompting us to examine the role of Brd4 in the ground state of pluripotency. Genome-wide analyses of Brd4 binding confirmed that Brd4-binding correlated with H3K9ac and H3K27ac and revealed that 2i increased Brd4 binding to chromatin genome-wide (Fig. 1c and Supplementary Fig. 1a,b). The genes associated with the largest Brd4 peaks were enriched for pluripotency-associated gene signatures (Supplementary Fig. 1c) and included key pluripotency loci such as the Nanog enhancer and Pou5f1 promoter (Fig. 1d).
Treating ESCs with small molecule inhibitors of Brd4 such as JQ1 has been reported to block stem cell self-renewal by displacing Brd4 from key stem cell genes and reducing their expression8, 23. Consistent with these findings, JQ1 treatment inhibited Brd4 binding to pluripotency genes in cells cultured in either S/L or S/L+2i (Fig. 1d). Surprisingly, while S/L-cultured cells ceased proliferation when treated with JQ1 (Fig. 2a), S/L+2i-grown cells were resistant to the antiproliferative effects of JQ1 (Fig. 2b). Whereas ESCs cultured in S/L rapidly lost pluripotency in the presence of JQ1, acquiring a flattened morphology marked by increased cell volume, lost reactivity to alkaline phosphatase (AP) and downregulated Nanog expression, 2i-cultured ESCs appeared unaffected by JQ1 treatment (Fig. 2c-f and Supplementary Fig. 2a). Metastable ESCs exhibited dose-dependent sensitivity to JQ1, but naïve ESCs resisted JQ1 treatment across a wide range of doses (Supplementary Fig. 2b). Moreover, 2i-cultured ESCs retained proliferation and ESC-like morphology even upon prolonged passage with JQ1 (Fig. 2g, Supplementary Fig. 2c). An alternative BET inhibitor, iBET28, similarly caused metastable ESCs to arrest and enlarge while naïve ESCs remained resistant (Supplementary Fig. 2d,e). 2i-induced resistance to BET inhibitors did not require an embryonic cell of origin: pro-B cells reprogrammed to pluripotency maintained proliferation, volume and AP staining when treated with JQ1 (Fig. 2h, Supplementary Fig. 2f,g).
Figure 2. Naïve ESCs are resistant to Brd4 inhibitors.
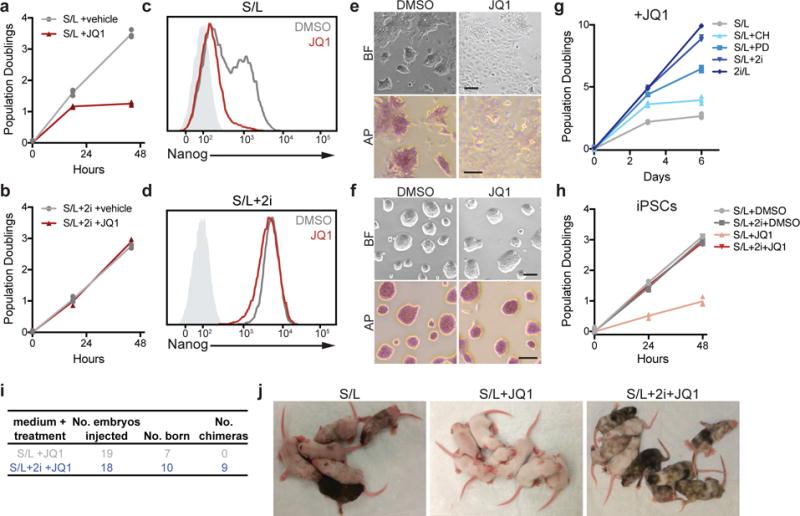
(a,b) Growth curve of ESCs cultured in S/L (a) or S/L+2i (b) with DMSO (vehicle) or 500 nM JQ1. (c,d) GFP intensity of Nanog-GFP reporter ESCs cultured in S/L (c) or S/L+2i (d) treated with DMSO or 500 nM JQ1 for 24 h as measured by FACS. Light grey shaded peak represents negative control. (e,f) Representative images of ESCs grown in S/L (e) or S/L+2i (f) treated with DMSO (vehicle) or 500 nM JQ1 for 24 h. Top, brightfield (BF); bottom, alkaline phosphatase staining (AP). Scale bar, 100 μm. (g) Population doublings of ESCs grown in indicated medium and cultured with 500 nM JQ1. CH, GSK3β inhibitor; PD, MEK inhibitor. (h) Population doublings of induced pluripotent stem cells (iPSCs) generated from hematopoietic CD19+ pro-B cells grown in S/L or S/L+2i with DMSO or 500 nM JQ1. (i,j) Chimeras generated from injection of ESCs cultured in S/L or S/L+2i with or without 500 nM JQ1 for 48 h. Black coat color indicates ESC contribution to live offspring. For growth curves (a,b,g,h), n=3 independent samples are shown and the connecting line joins the mean values of each time point (a,b,c,h); all experiments (a-h) were repeated independently at least two times with similar results.
While each inhibitor (CHIR, PD) provided partial resistance to JQ1, the combined effect of both inhibitors (2i) was required for full JQ1 resistance (Fig. 2g, Supplementary Fig. 2h-j). The presence of serum did not appear to influence susceptibility to bromodomain inhibition: ESCs cultured in 2i/L were resistant to JQ1 (Fig. 2g, Supplementary Fig. 2h-j). These results suggest that the ground state of pluripotency, rather than MEK or GSK3β inhibition individually, induces JQ1 resistance. Naïve ESCs, but not metastable ESCs, were capable of giving rise to AP+ colonies even in the ongoing presence of JQ1 (Supplementary Fig. 2k). To determine whether ESCs remain bona fide pluripotent cells after JQ1 treatment, ESCs exposed to JQ1 were injected into blastocysts to assay for differentiation competence in vivo. After treatment with JQ1, only ESCs cultured in S/L+2i contributed to live born chimeras (Fig. 2i,j).
Although BET inhibitors such as JQ1 and iBET target the bromodomain of BET family proteins, inhibition of Brd4 drives the majority of the published effects of BET inhibitors8, 19, 21. To probe the role of Brd4 specifically, we performed CRISPR/Cas9-mediated editing of Brd4 using sgRNA against the BD1 domain of Brd429. Expression of sgRNA against Brd4 severely compromised the number of viable colonies formed in S/L-cultured ESCs but had no effect on colony formation in S/L+2i-cultured ESCs (Fig. 3a and Supplementary Fig. 3a,b). Just as 2i preserved Nanog expression in response to JQ1 treatment, genetic editing of Brd4 did not alter Nanog expression in S/L+2i-cultured ESCs (Supplementary Fig. 3a). Accordingly, each Brd4-edited clone retained normal morphology and proliferation when cultured in the presence of 2i (Supplementary Fig. 3c,d). This resistance to the effect of Brd4 editing required 2i: within 48 h of 2i removal, Brd4-deficient clones exhibited severe growth defects (Supplementary Fig. 3d). When seeded at single cell density, only clones cultured with 2i were able to form AP+ colonies (Fig. 3b,c).
Figure 3. Brd4 is dispensable in naïve ESCs.
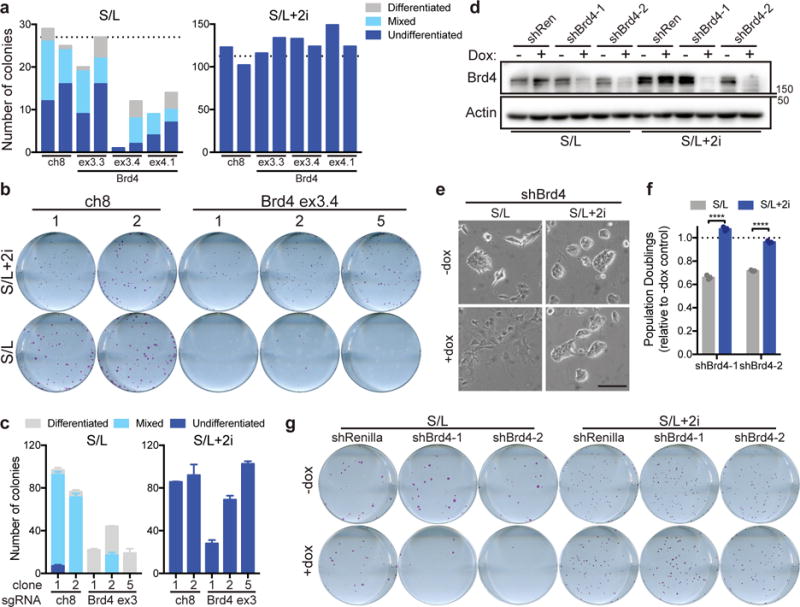
(a) Quantification of colony formation assay of cells cultured in S/L (left) or S/L+2i (right) transfected with Cas9 and the indicated sgRNA against a nongenic region of chromosome 8 (ch8, control) or exon 3 (ex3) or exon 4 (ex4) of Brd4. Dotted line represents average of ch8 controls. Each bar represents quantification of a single well of a six-well plate; transfected samples were seeded in duplicate. (b) Alkaline phosphatase staining of colonies formed from single cells of clonal ESC lines edited with sgRNA against chromosome 8 (ch8) or Brd4 exon 3 and cultured in S/L or S/L+2i. One representative well of a six-well plate is shown. (c) Quantification of colony formation assay shown in (b). (d) Western blot depicting Brd4 levels in ESCs expressing doxycycline (dox)-inducible hairpins against Renilla (shRen) or Brd4 (shBrd4). Cells were cultured with or without dox for 48 h prior to harvest. Actin is used as a loading control. Western blot was performed two independent times. (e) Brightfield images of ESCs expressing shBrd4-2 cultured for 48 h with or without doxycycline (dox). (f) Population doublings of cells cultured for 72 h in doxycycline relative to controls grown without dox. (g) Alkaline phosphatase staining of colonies formed from single cells expressing the indicated hairpins grown in the presence or absence of doxycycline (dox) and cultured in S/L or S/L+2i. One representative well of a six-well plate is shown. All bars represent mean ±SEM (c) or ±SD (f) of n=3 independent samples. ****, P < 0.0001 by 2-way ANOVA with Sidak’s multiple comparisons post test (shBrd4-1, P = 7.2e-10; shBrd4-2, P = 4.6e-8). Scale bar, 100 μm.
We next manipulated Brd4 expression using inducible shRNA against Brd4 (Fig. 3d). Brd4 knockdown recapitulated the effects of BET inhibition, flattening colonies and blocking proliferation of S/L-cultured ESCs while having no obvious effect on the morphology or proliferation of S/L+2i-cultured ESCs (Fig. 3e,f). Although Brd4 was absolutely required for the self-renewal of metastable ESCs, naïve ESCs were capable of forming AP+ colonies even in the presence of continuous Brd4 depletion (Fig. 3g). Together, these results suggested that ground state ESCs tolerate pharmacologic and genetic inhibition of Brd4.
Naïve ESCs maintain transcriptional networks independently of Brd4 function
The ability of ESCs grown in 2i to maintain pluripotency in the presence of BET inhibitors suggests that these cells are capable of maintaining pluripotency-associated transcription networks independently of BET protein recognition of acetylated lysines. Indeed, while expression of pluripotency transcription factors required Brd4 activity in S/L-cultured ESCs as previously reported8, 21, 23, 2i-cultured ESCs maintained high levels of transcription factor expression even in the presence of pharmacologic or genetic inhibition of Brd4 activity (Fig. 4a,b). To determine how ESCs sustain transcriptional networks without the help of BET protein coactivators, we analyzed gene expression in ESCs treated with JQ1 for 72 h. In metastable ESCs, most genes comprising the pluripotency-associated gene network30 lost Brd4 binding and were dramatically downregulated by prolonged exposure to JQ1 (Fig. 4c and Supplementary Fig. 4a). In contrast, expression of these genes remained high in naïve ESCs regardless of JQ1 treatment (Fig. 4c), suggesting that these cells can sustain transcription in the absence of Brd4 bromodomain function.
Figure 4. Naïve ESCs maintain transcriptional networks in the presence of BET inhibition.
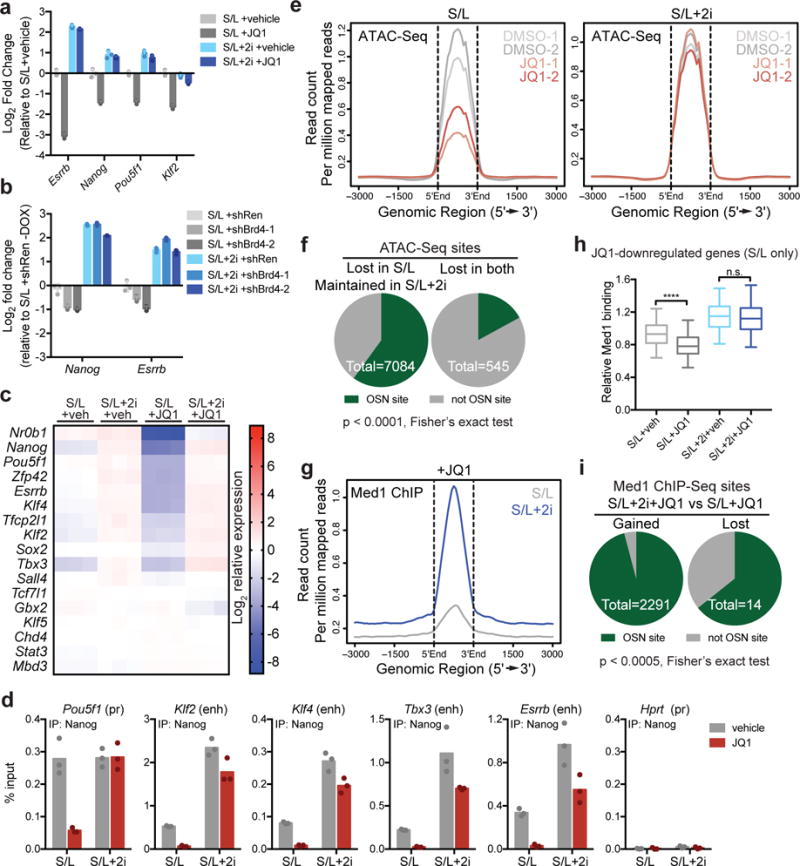
(a) qRT-PCR on key pluripotency genes in ESCs cultured in S/L or S/L+2i treated with DMSO (veh) or 500 nM JQ1 for 24h. (b) qRT-PCR on key pluripotency genes in S/L or S/L+2i-cultured ESCs expressing dox-inducible hairpins against Renilla (shRen) or Brd4 (shBrd4) and treated with dox for 48 h. Shown are levels of Nanog and Esrrb in cells with dox relative to control S/L+shRen cells without dox. (c) Heat map shows expression of pluripotency associated genes measured by RNA-Seq in ESCs cultured in S/L or S/L+2i with vehicle (veh, DMSO) or 500 nM JQ1 for 72 h. Color scale represents Log2 relative to mean expression level. (d) Nanog binding to key pluripotency loci in ESCs cultured in S/L or S/L+2i treated with DMSO or 500 nM JQ1 for 24h, assessed by ChIP-qPCR. Bar represents mean of n=3 technical replicates from one IP. (e) ATAC-Seq meta profile of chromatin accessibility in ESCs cultured in S/L (left) or S/L+2i (right) with DMSO or 500 nM JQ1. Data from 2 independent replicates are shown. (f) At baseline, 82% of Oct4, Sox2, Nanog (OSN) binding sites43 have an ATAC-Seq peak above background. OSN sites make up 60% (4,250/7,084) of the peaks that maintain accessibility despite JQ1 treatment in S/L+2i cultured ESCs but only 17% (93/545) of peaks lost in both conditions. (g) ChIP-seq meta profile for Med1 binding in ESCs cultured in S/L or S/L+2i treated 500 nM JQ1. (h) Box plot shows relative Med1 binding at n=635 genes that are downregulated > 2 fold, FDR < 5% by JQ1 in ESCs cultured in S/L but not in ESCs cultured in S/L+2i. Values from ChIP-Seq experiment shown in (g) with 1 ChIP per condition. Box, 25-75th percentile; bar, median; whiskers, 5-95th percentile. ****, P < 1e-15; ns, P = 0.27 by 2-way ANOVA with Tukey’s multiple comparisons post test. See Supplementary Table 5. (i) Sites that gain Med1 binding in S/L+2i+JQ1 relative to S/L+JQ1 are mostly OSN binding sites. Data presented as mean ± SD of n=3 independent samples (a, b).
To assess the activity of pluripotency-associated transcriptional networks, we looked at expression of genes identified as direct targets of Oct4, Sox2 and Nanog (OSN)31. In the absence of Brd4 function, OSN targets were preferentially downregulated in cells cultured in S/L medium (Supplementary Fig. 4b). Similarly, genes directly bound by pluripotency factors such as Oct4, Sox2, Nanog, Klf4 and Rex132 were repressed by JQ1 treatment of cells growing in S/L medium (Supplementary Fig. 4c). In both cases, addition of 2i preserved expression of these gene sets during JQ1 treatment (Supplementary Fig. 4d,e). Thus, while JQ1 abrogated pluripotency-associated transcription factor binding to key regulatory loci in metastable ESCs, transcription factor binding to these same loci was higher and relatively resistant to JQ1 in naïve ESCs (Fig. 4d).
Chromatin accessibility can be used to infer recruitment of transcriptional activators and co-activators to regulatory elements associated with gene expression33. The above results suggested that naïve ESCs grown in 2i-containing medium might maintain chromatin accessibility independently of Brd4. Consistent with this hypothesis, ATAC-Seq analyses revealed that while JQ1 induced profound loss of chromatin accessibility in ESCs grown in S/L medium, ESCs cultured in medium containing 2i exhibited minimal changes in chromatin accessibility in the presence of JQ1 (Fig. 4e and Supplementary Fig. 4f). Over 60% of the ATAC-Seq peaks lost in S/L-cultured cells as a result of JQ1 treatment and preserved in S/L+2i-cultured cells were OSN sites (Fig. 4f).
In addition to maintaining chromatin accessibility34, Brd4 can potentiate gene expression by associating with the Mediator complex that bridges transcription factor binding and RNA polymerase II recruitment to regulatory elements18, 35. Displacement of the Med1 subunit of the Mediator complex following JQ1 treatment correlates with loss of gene expression8, 18. Consistent with these prior findings, S/L-grown cells exhibited a profound reduction in genome-wide Med1 recruitment when treated with JQ1. In contrast, ESCs grown in S/L+2i medium retained high genome-wide Med1 recruitment despite JQ1 treatment (Fig. 4g and Supplementary Fig. 4g). While JQ1 reduced both chromatin accessibility and Med1 binding at pluripotency genes in S/L-cultured ESCs, it had no consistent effect on either parameter at these genes in S/L+2i-cultured ESCs (Supplementary Fig. 4h,i).
The above results suggested that persistent Mediator recruitment might sustain Brd4-independent gene expression in S/L+2i-cultured ESCs. In support of this model, genes that were sensitive to JQ1 in metastable cells, but not in naïve cells, lost Med1 binding under S/L culture conditions but preserved Med1 binding in the presence of 2i (Fig. 4h). Mediator facilitates transcription by promoting Pol II C-terminal domain phosphorylation by TFIIH and P-TEFb36. To test the contribution of transcriptional activation by these factors, we cultured cells with inhibitors against CDK737 and CDK938, the active subunits of TFIIH and P-TEFb, respectively39, 40. In contrast to Brd4 inhibition, both CDK7 and CDK9 inhibition triggered loss of Nanog expression and impaired proliferation, regardless of the presence of 2i (Supplementary Fig. 4j,k).
Pluripotency-associated transcription factors enable resistance to BET inhibitors
Sites associated with high Med1 binding in S/L+2i relative to S/L were highly enriched (96%) in known OSN binding sites (Fig. 4i), suggesting that the pluripotency transcription factor network enabled resistance to BET inhibition by promoting Brd4-independent Mediator recruitment. To further examine the hypothesis that a strengthened pluripotency network bestows Brd4 independence, we generated ESCs expressing a granulocyte colony-stimulating factor (G-CSF)-inducible LIF receptor transgene harboring a mutation at tyrosine 118 that results in hyperactivation of the JAK/STAT3 signaling pathway (Supplementary Fig. 5a) and drives ESCs into the ground state of pluripotency41. Induction of STAT3 signaling rendered cells resistant to JQ1: G-CSF-treated ESCs were able to maintain proliferation, cell volume and expression of pluripotency-associated genes even in the presence of JQ1 (Fig. 5a, 5b, and Supplementary Fig. 5b).
Figure 5. Increased transcription factor activity provides resistance to BET inhibition in the absence of 2i.
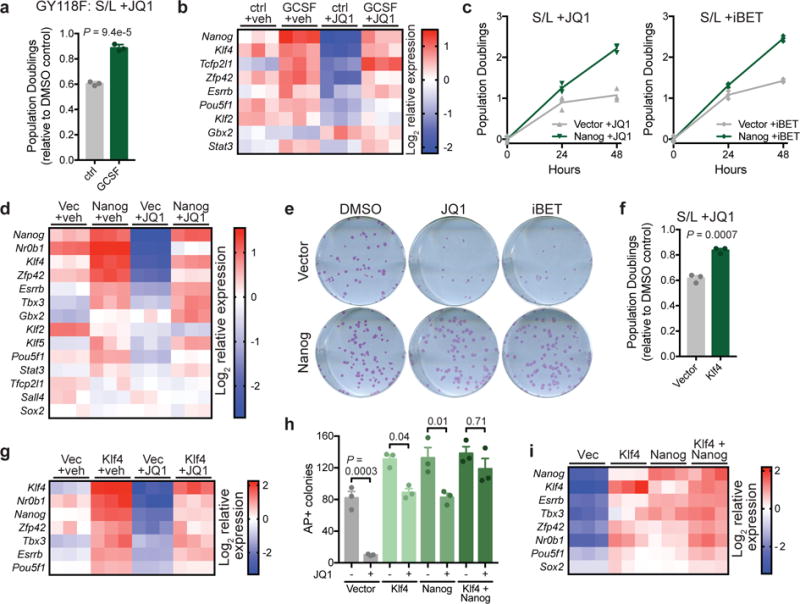
(a) Population doublings of cells expressing GCSF-activated LIFR transgene (GY118F) cultured with or without GCSF and treated for 48 h with 500 nM JQ1 relative to vehicle (DMSO)-treated controls. (b) Heat map depicting relative gene expression in cells expressing GCSF-activated LIFR transgene (GY118F) treated for 24 h with vehicle (DMSO) or 500 nM JQ1. (c) Growth curve of ESCs expressing empty vector or Nanog and cultured with 500 nM JQ1 (left) or 500 nM iBET (right). n=3 independent samples are shown and the connecting line joins the mean values of each time point. (d) Heat map depicting relative gene expression in cells expressing empty vector or Nanog and treated with vehicle (DMSO) or 500 nM JQ1 for 24 h. (e) Alkaline phosphatase staining of colony formation assays in which vector or Nanog-expressing cells are treated for the final 48 h with vehicle (DMSO), JQ1 or iBET. Experiment was performed at least two times with similar results. (f) Population doublings of cells expressing empty vector or Klf4 treated for 48 h with 500 nM JQ1 relative to vehicle (DMSO)-treated controls. (g) Heat map depicting relative gene expression in cells expressing empty vector or Klf4 and treated with vehicle (DMSO) or 500 nM JQ1 for 24 h. (h) Quantification of number of alkaline phosphatase (AP)-positive colonies formed in the presence of continuous JQ1 in cells expressing empty vector, Klf4, Nanog or Klf4+Nanog. (i) Heat map depicting relative gene expression in cells shown in (h) treated with 500 nM JQ1 for 24h. Data are presented as mean ± SD (a) or SEM (f,h) of n=3 independent samples. Statistics calculated by unpaired, two-tailed Student’s t-test (a,f) or 1-way ANOVA with Tukey’s post-test (h).
These results suggested that direct reinforcement of the pluripotency transcription network might provide JQ1 resistance. Brd4 is recruited to many of the same sites as key pluripotency transcription factors such as Nanog, Oct4, Sox2, Klf4 and Esrrb42 (Supplementary Fig. 5c). Brd4-bound sites form three major clusters: Nanog-bound, Oct4/Sox2/Klf4-bound and Esrrb-bound (Supplementary Fig. 5d). Therefore, we tested whether increased expression of transcription factors representing distinct clusters might be sufficient to enable resistance to BET inhibition. ESCs with transgenic Nanog expression (Supplementary Fig. 5e) maintained proliferation, cell volume and AP reactivity in the presence of either JQ1 or iBET (Fig. 5c,e and Supplementary Fig. 5f,g). Moreover, Nanog overexpression sustained expression of pluripotency genes in JQ1-treated cells (Fig. 5d). Similarly, transgenic expression of Klf4, which binds a different set of Brd4 targets than Nanog (Supplementary Fig. 5d,h), improved cell proliferation, prevented increases in cell volume and blunted decreases in pluripotent gene expression in response to JQ1 treatment (Fig. 5f,g and Supplementary Fig. 5i). Dual expression of both Nanog and Klf4 further enhanced resistance to JQ1: expression of pluripotency-associated genes was highest in JQ1-treated Nanog+Klf4 expressing cells (Fig. 5i), and continuous JQ1 had no significant effect on formation of AP+ colonies in Nanog+Klf4 lines (Fig. 5h and Supplementary Fig. 5j). Thus, resistance to BET inhibition correlates with the strength of the pluripotency transcriptional network.
Tet1/2 cooperate with transcription factors to maintain Brd4-independent pluripotency
To determine the mechanism through which enhanced transcription factor expression rendered metastable ESCs resistant to BET inhibitors, we performed ATAC-Seq on Nanog transgenic cells in the presence and absence of JQ1. At baseline, Nanog overexpression induced chromatin opening at loci associated with key pluripotency genes (Fig. 6a). Moreover, Nanog overexpression blunted the JQ1-induced loss of chromatin accessibility at these regions (Fig. 6a). More generally, Nanog overexpression increased accessibility across a wide range of direct targets of OSN pluripotency factors in JQ1-treated cells (Fig. 6b).
Figure 6. JQ1 affects Nanog and Tet target genes.
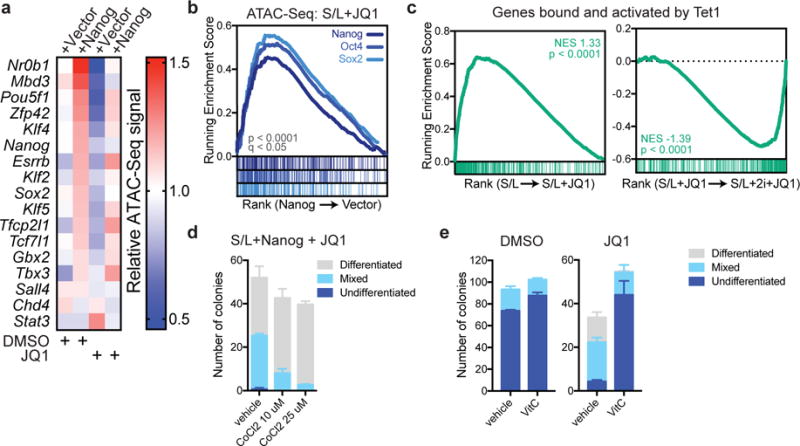
(a) Relative chromatin accessibility at peaks associated with key pluripotency genes in ESCs expressing empty vector or Nanog and treated with DMSO or 500 nM JQ1 for 72 h. See also Supplementary Table 5. (b) Gene set enrichment analysis (GSEA) showing genes ranked by fold change in total accessibility with Nanog overexpression as measured by ATAC-Seq of duplicate samples. Genes identified as direct targets of Nanog, Oct4 and Sox2 are enriched among genes whose accessibility is increased by Nanog overexpression. (c) Gene set enrichment analyses of RNA-Seq data from triplicate samples showing enrichment of genes identified as bound and activated by Tet1. For GSEA, P values are calculated based on 1000 permutations by the GSEA algorithm and was not adjusted for multiple comparisons. (d) Colony formation assay of Nanog-overexpressing cells cultured in the presence of 500 nM JQ1 and indicated dose of CoCl2 for 6 days. (e) Colony formation assay of Nanog-overexpressing cells cultured in the presence of DMSO or 200 nM JQ1 and 100 μg/mL Vitamin C for 6 days. Data are presented as mean ± SEM of n=3 independent samples.
Binding by OSN transcription factors during the transition to the naïve ground state of pluripotency is associated with local loss of DNA methylation43. Tet1 and Tet2, methylcytosine oxidases that hydroxylate DNA 5-methylcytosine, are reported to contribute to DNA hypomethylation at a subset of regulatory elements in naïve ESCs12–14, 44. Additionally, during reprogramming, Nanog cooperates with Tet1 and Tet2 to activate expression of key reprogramming genes45. Tet1 binds to 70% of Brd4-bound sites46 (Supplementary Fig. 6a,b), and 73.3% of these sites are also bound by OSN factors (p < 2.2e-16, Fisher’s exact test).
Consequently, we tested whether the ability of transcription factors to maintain gene expression in JQ1-treated cells might involve Tet1/2. As preliminary evidence, we found that genes reported to be both bound and activated by Tet1 in mouse ESCs46 are significantly enriched among genes downregulated by JQ1 treatment in S/L-cultured ESCs (Fig. 6c). However, in 2i-cultured ESCs, expression of Tet1-target genes was maintained despite JQ1 treatment (Fig. 6c). Furthermore, treatment of Nanog-overexpressing ESCs with cobalt chloride, which inhibits iron-dependent dioxygenases including Tet1/2, induced a dose-dependent loss of resistance to JQ1 (Fig. 6d). Conversely, Vitamin C, a cofactor that promotes Tet1/2 activity in ESCs47, significantly enhanced the ability of Nanog-overexpressing cells, but not control cells, to resist JQ1 treatment (Fig. 6e and Supplementary Fig. 6c). These results suggested that transcription factors, in cooperation with Tet1/2 and potentially other iron-dependent demethylases, drive JQ1 resistance.
As both cobalt chloride and Vitamin C can affect multiple cellular processes, we generated ESCs with CRISPR/Cas9 derived mutations in both Tet1 and Tet2 (Supplementary Fig. 7a). Consistent with previous studies demonstrating that Tet1 and Tet2 are dispensable for pluripotency48 Tet1/2 double mutant ESCs had no obvious defect in chromatin accessibility, Brd4 expression, growth or morphology under basal conditions (Supplementary Fig. 7b-d). However, loss of Tet1/2 function clearly sensitized cells to JQ1, indicating that in the absence of Tet1/2, cells were more dependent on Brd4 to maintain gene expression (Supplementary Fig. 7e). We next tested whether Tet enzymes contributed to the BET inhibitor resistance induced by Nanog or 2i. Overexpression of Nanog in Tet1/2 double mutant ESCs (Supplementary Fig. 7f) revealed that Tet1/2 mutation severely compromised the ability of Nanog to maintain proliferation and generate AP+ colonies in the presence of JQ1 (Fig. 7a,b and Supplementary Fig. 7g). Similarly, 2i-treated ESCs required Tet1 and Tet2 to sustain JQ1 resistance (Fig. 7c-f).
Figure 7. Tet1/2 are required for resistance to BET inhibitors.
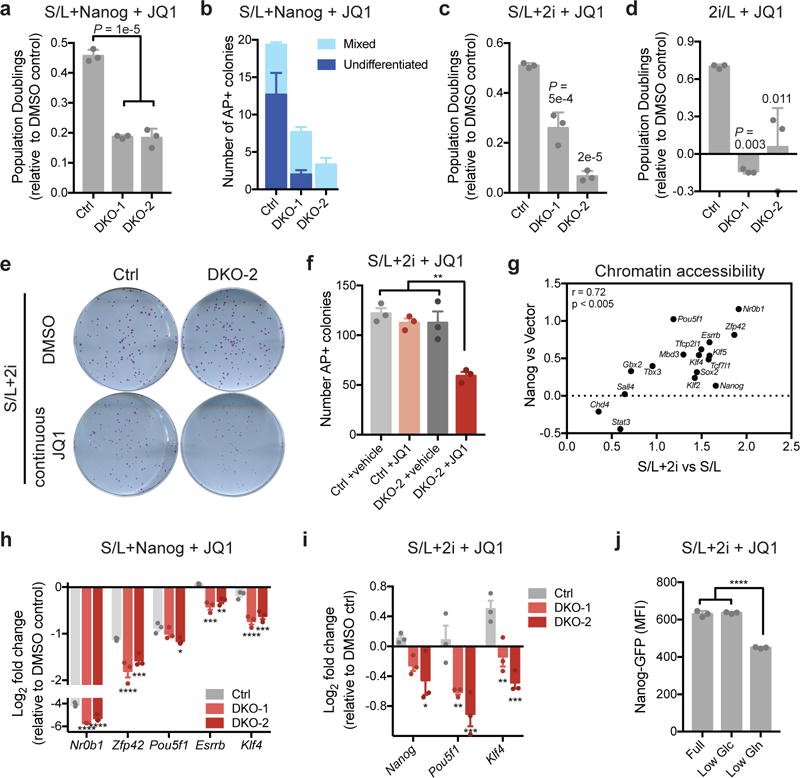
(a) Population doublings of JQ1-treated ESCs relative to DMSO controls during day 4-6 of treatment with 500 nM JQ1. All cells express Nanog and are cultured in S/L. (b) Quantification of number of alkaline phosphatase (AP)-positive colonies with mixed or undifferentiated morphology formed in the presence of continuous JQ1 treatment in control or Tet1/2 double mutant (DKO) ESCs expressing Nanog. (c and d) Population doublings of JQ1-treated ESCs relative to DMSO controls during day 4-6 of treatment with 500 nM JQ1. Cells are cultured in S/L+2i (c) or 2i/L (d). (e and f) Images (e) and quantification (f) of alkaline phosphatase (AP)-positive colonies formed in the presence of continuous JQ1 treatment in control or Tet1/2 DKO ESCs cultured in S/L+2i. (g) Relative chromatin accessibility in JQ1-treated ESCs assessed by ATAC-seq. All peaks associated with each gene were summed and plotted as the log2 fold change of accessibility with each treatment relative to control (Nanog overexpression or 2i culture). (h and i) Expression of pluripotency genes in Nanog expressing (h) or S/L+2i cultured (i) ESCs treated with 500 nM JQ1 for 72 h relative to DMSO controls. (j) Nanog-GFP expression in JQ1-treated ESCs cultured in S/L+2i in full medium (20 mM glucose, 2 mM glutamine) or medium with reduced glucose (included as a control; Low Glu, 2 mM) or reduced glutamine (Low Gln, 200 μM). Data are presented as mean ± SEM (b, f, h, i) or ± SD (a, c, d, j) of n=3 independent samples. *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.0001 by 1-way ANOVA (a, c, d, f, j) or 2-way ANOVA (h, i) with Tukey’s multiple comparison post-test.
Both 2i treatment and Nanog overexpression promoted chromatin accessibility at pluripotency genes in JQ1-treated ESCs (Fig. 7g). Tet1/2 were required for Nanog-induced increases in accessibility at peaks associated with pluripotency genes (Supplementary Fig. 7h). While Nanog expression was sufficient to preserve accessibility at a subset of JQ1-sensitive peaks, this ability was compromised absence of Tet1/2 (Supplementary Fig. 7i). In particular, a subset of the peaks whose accessibility was lost with JQ1 treatment but preserved by Nanog overexpression required Tet1/2 in order to be maintained by Nanog (Supplementary Fig. 7i). Motif analyses revealed that pluripotency-associated motifs are strongly enriched in all clusters, including Tet1/2-dependent sites (Supplementary Fig. 7i). These results suggested that Tet1/2 are required for maintenance of accessibility at a subset of pluripotency-associated transcription factor targets and loss of Tet1/2 therefore weakens the pluripotency transcriptional network and compromises JQ1 resistance. Indeed, the ability of both Nanog overexpression and 2i treatment to maintain expression of pluripotency-associated transcription factors during JQ1 treatment was blunted in Tet1/2 double mutant ESCs (Fig. 7h,i).
We next asked whether the reported ability27 of 2i-treatment to enhance glutamine-dependent production of αKG, a co-substrate for DNA and histone modifying enzymes including Tet1/2, contributed to the maintenance of pluripotent gene expression during JQ1 treatment. Culture in low glutamine did not affect Brd4 expression (Supplementary Fig. 7j). However, low glutamine impaired the ability of 2i to maintain Nanog expression in the presence of JQ1 (Fig. 7j). While glutamine withdrawal likely has pleiotropic effects, these data suggest that 2i-induced production of Tet substrates may also contribute to the resistance to BET inhibitors exhibited by naïve ESCs.
Discussion
BET inhibitors have gained wide attention for their ability to block proliferation and self-renewal of stem cells and cancer cells. The efficacy of these inhibitors is tied to their ability to inhibit expression of key stem cell maintenance genes and oncogenic transcription factors like Myc8, 19, 49, 50. Here we demonstrate that naïve ESCs exhibit inherent resistance to BET inhibition. When grown in 2i-containing medium or provided enhanced JAK/STAT3 signaling, ESCs maintain proliferation, self-renewal and pluripotency in the presence of BET inhibitors. The ability of naïve ESCs to maintain Brd4-independent proliferation and self-renewal is mediated by both a strengthened transcription factor network and Tet1/2 DNA methylcytosine oxidases which converge to maintain a transcriptionally permissive chromatin environment. Naïve ESCs represent a fully optimized cell state; under less optimized conditions, such as with metastable ESCs, the reading of histone acetylation remains essential to the maintenance of cellular identity.
The present data suggest that the dependency upon reading of histone acetylation to consolidate gene networks is most critical when transcription factor binding and chromatin accessibility is limiting. However, the data do not preclude contributions from additional chromatin modifications, transcriptional activators or co-activators to Brd4-independent maintenance of cell identity and future work may uncover additional proteins that compensate for BET family inhibition. We favor the general view that when transcription factor networks and chromatin methylation states are collectively optimized, the recognition of lysine acetylation by bromodomain containing proteins plays a relatively minor role in modulating gene expression. In support of this model, loss of DNMT3A and PRC2, which deposit repressive DNA methylation and H3K27me3, can lead to BET inhibitor resistance in AML cell lines51. Similarly, AML cells expressing mutant forms of isocitrate dehydrogenase 2 exhibit hypermethylation and enhanced sensitivity to Brd4 inhibition52. Taken together, these data reveal that a combination of transcription factor networks and permissive chromatin modifications can render Brd4 dispensable to the maintenance of cell identity and illustrate that chromatin modification-based transcriptional regulation is context-dependent.
Methods
Cell Culture
Mouse ESC lines (ESC1) were generated from C57BL/6 × 129S4/SvJae F1 male embryos as previously described27. D34 ESCs were previously described55. Nanog-GFP reporter ESCs and iPSCs generated from hematopoietic CD19+ pro-B cells were a gift from R. Jaenisch (MIT). ESC1 cells were used for all experiments unless otherwise noted. ESCs were maintained on gelatin-coated plates in serum/LIF (S/L) medium containing Knockout DMEM (Gibco) supplemented with 10% ESC-qualified FBS (Gemini), penicillin/streptomycin (Life Technologies), 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine (Life Technologies) and 1000 U/mL LIF (Millipore). For culture in 2i (S/L+2i), S/L medium was supplemented with 2i: 3 μM CHIR99021 (Stemgent) and 1 μM PD0325901 (Stemgent). Cells were adapted to S/L+2i for at least three passages prior to experiments and were maintained in S/L+2i for no more than 9 passages. Serum-free 2i/L consisted of a 1:1 mix of DMEM/F12 (Life Technologies 11302-033) and Neurobasal medium (Life Technologies 21103-049) containing N2 and B27 supplements (Life Technologies 17502-048 and 17504-044, 1:100 dilutions), 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine, LIF, CHIR99021 at 3 μM (Stemgent) and PD0325901 at 1 μM (Stemgent). For glucose and glutamine deprivation experiments, cells were cultured in medium containing a 1:1 mix of glutamine- and glucose-free DMEM (Invitrogen A14430-01) and glutamine- and glucose-free Neurobasal medium (Invitrogen 0050128DJ) including 10% dialyzed FBS and all supplements as described above and containing glucose (Sigma) at either 20 mM or 2 mM or glutamine at either 2 mM or 0.2 mM. JQ1 and iBET (Sigma) were dissolved in DMSO and used at indicated doses. Cobalt (II) Chloride (CoCl2) (0.1M) and Vitamin C (50 mg/mL) were dissolved in water and used at indicated doses. Leukocyte Alkaline Phosphatase Kit (Sigma) was used to stain cells with alkaline phosphatase reagent according to manufacturer instructions. Images of cells were taken using a Leica microscope.
Generation of ESC lines
Generation of transgenic cell lines
Nanog and Klf4 mouse cDNA were cloned into piggybac (pCAGGS-IRES-Neo, kind gift of H. Niwa, IMEG, Japan) and pEF1a-IRES-GFP (System Biosciences) plasmids. Approximately 5 × 106 ESC-1 cells cultured in S/L medium were electroporated with piggybac plus transposase (pBase) in 3:1 ratio using Amaxa ES cell nucleofector kit (VPH-1001, program A-023) and plated onto gelatin-coated plates. 36-48 h after electroporation, medium was changed to S/L+G418 at 300 μg/mL for at least 2-3 passages. Nanog+Klf4 double transgenic lines were generated by electroporating Nanog expressing lines with pEF1a-Klf4-GFP and then sorting top 10% GFP+. Hyperactivated STAT3 ESCs were generated in Nanog-GFP ESCs using GY118F chimeric LIF-receptor piggybac plasmid56 as described above and selected using 200 μg/mL hygromycin for at least three passages.
Generation of Tet1/2 knockout cell lines
Tet1/2 double mutant ESCs were obtained using CRISPR/Cas9-derived mutation of Tet1 and Tet2. Tet1 and Tet 2 gRNAs (Tet1, 5′-tggctgctgtcagggagctca-3′; Tet2, 5′-tgaaagtgccaacagatatcc-3′)57 were cloned into px459 v1 (Addgene plasmid number Plasmid #48139) containing 2A-PURO selection and electroporated (Amaxa, Lonza) into ESCs. After electroporation, cells were plated at clonal density onto feeder mouse embryonic fibroblasts (MEFs) and subjected to puromycin selection (2 μg/mL) after 36-48 h. Single colonies were picked and tested for loss of Tet1 and Tet2 by Western blot analysis and clones were confirmed by sequencing of PCR amplicons generated using the following primer pairs: Tet1 forward, 5′-CCTTCCACAGATGGAGGTACAC-3′, reverse, 5′-CTGACCGTCTTCATTCGCCT-3′; Tet2 forward, 5′-AAGCAGTTTCAGCTCAAGTCAC-3′, reverse, 5′-GCATCCTTCACATTTGCCATTCT-3′. Sequencing was performed by the CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA).
Generation of Brd4 BD1 mutant cell lines
ESCs harboring inactivating mutations at Brd4 bromodomain 1 (BD1) were obtained using CRISPR/Cas9-derived mutation of Brd4. Previously described guide RNA (gRNA) sequences targeting Brd429 or a nongenic region on mouse chromosome 8 (ch8)58 were cloned into the pCas9 (BB)2A-GFP (pX458, Addgene plasmid number Plasmid #48138), as previously described59. See Supplementary Table 1 for gRNA sequences. ESCs were passaged three times over the course of six days to adapt to S/L or S/L+2i medium. 0.4 × 106 cells, grown under either SL or S/L+2i conditions, were electroporated using the Nucleofector 4D (Amaxa, Lonza) with 5 μg pX458 plasmid encoding Cas9, EGFP and gRNA targeting Brd4 or ch8 (control), or with empty pX458 vector (control). After electroporation, cells were plated on a feeder layer of on irradiated mouse embryonic fibroblasts (MEFs) in S/L or S/L+2i medium. After 48 hours, cells were dissociated with Accutase (Invitrogen) and sorted using the BD FACSAria III sorter (BD Biosciences) to enrich for GFP-positive cells. 10,000 FACS sorted GFP-positive cells per experimental condition were seeded on 10 cm plates (on feeders) to enable clonal growth. After 7 days, individual clones (~10 per condition) were picked and split for DNA extraction and expanded (on feeders) to make frozen cell stocks as well as whole cell lysates using RIPA Buffer (Sigma). For preparation of lysates, binding to gelatin-coated dishes for 15 min two consecutive times depleted cells of feeders. Biallelic disruption of Brd4 was assessed by Western blot. Clones were confirmed by sequencing PCR amplicons generated using the following primers: forward, 5′-CCAGTAATGGGGGATGGACT-3′; reverse. 5′- CCCTGTCCAGATGGCTACTC-3′. Amplicons were cloned (Zero Blunt™ TOPO™ PCR Cloning Kit, Thermo Fisher Scientific, 450031) and sequenced using the T3 sequencing primer (Eton Biosciences). Sequencing results were confirmed by sending the same PCR amplicons for deep sequencing at the MSKCC IGO core facility.
Generation of inducible Brd4-shRNA ESC lines
Tetracycline-inducible shRNAs linked to GFP were targeted into D34 ESCs by recombinase-mediated cassette exchange (RMCE), as previously described60. See Supplementary Table 1 for shRNA sequences. D34 ESCs harbor i) a Cre-activatable tet transactivator, CAGs-LSL-rtTA3-IRES-mKate2 (CAGs-LSL-RIK), which enables doxycycline (dox)-mediated induction of tetracycline-regulated element (TRE)-controlled elements and expression of fluorescent mKate2 upon Cre-mediated recombination, and ii) a recombinase-mediated cassette exchange (RMCE)-“homing cassette”, which allows for direct single-copy transgenic integration downstream of the Collagen type I gene (Col1a1), as previously described55. Following hygromycin selection, targeted D34 clones were treated with adenoviral Cre recombinase (University of Iowa) and plated at low density for isolation of individual Cre-recombined clones, as identified by positive mKate2 fluorescence. Ten individual Cre-recombined clones were picked for each different shRNA, and consistent GFP induction was confirmed by fluorescence microscopy upon dox-induction (1 μg/mL). Adenoviral Cre-mediated LoxP recombination was also confirmed by genotyping using specific primers (Supplementary Table 2).
Chromatin immunoprecipitation
Native ChIP assays (histones) were performed with approximately 6 × 106 ESCs per experiment. Cells were subjected to hypotonic lysis and treated with micrococcal nuclease to recover mono- to tri-nucleosomes. Nuclei were lysed by brief sonication and dialysed into N-ChIP buffer (10 mM Tris pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100) for 2 h at 4 °C. Soluble material was incubated overnight at 4 °C after addition of 0.5–1 μg of H3K9ac antibody (Cell Signaling Technologies, 9649) or H3K27 antibody (Active Motif, 39133) bound to 25 μL protein A Dynal magnetic beads (Invitrogen), with 5% kept as input DNA. Magnetic beads were washed, chromatin was eluted and ChIP DNA was dissolved in 10 mM Tris pH 8 for quantitative PCR reactions (see Supplementary Table 3 for primer pairs). Three independent ChIP experiments were performed. The data shown are averaged qRT–PCR values of technical replicates (n = 3) from one representative IP.
Brd4, Nanog, and Med1 ChIP was carried out as previously described43, 61. 1 × 107 ESCs treated with DMSO or 500 nM JQ1 for 24 h were used per IP. Cells were chemically cross-linked by the addition of formaldehyde (final concentration, 1%) for 10 min at room temperature. After quenching with glycine, cells were rinsed twice with PBS, flash frozen in liquid nitrogen and stored at −80 °C prior to use.
For Med1 and Nanog IP, cells were resuspended in 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 0.1% SDS and 1% Triton X-100 and sonicated for approximately 8 min using a Covaris E220 ultrasonicator. The resulting whole cell extract was centrifuged and the supernatant was incubated for 1 h at 4 °C with 50 μL of protein G magnetic beads that had been pre-incubated with 5 μg of control rabbit IgG. The supernatant was then incubated overnight at 4 °C with 50 μL of protein G magnetic beads that had been pre-incubated with an anti-Med1 antibody (Bethyl Laboratories) or anti-Nanog antibody (Cosmo Bio). Beads were then washed 1X with sonication buffer, 1X with 20 mM Tris (pH 8.0), 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton-X100, 1X with 10 mM Tris, 250 mM lithium chloride, 2 mM EDTA, 1% NP-40 and 1X with TE containing 50 mM NaCl. Bound complexes were eluted from the beads in 10 mM Tris, 1% SDS, 10 mM EDTA by heating at 65 °C for 30 min with continuous shaking. Crosslinking was reversed by overnight incubation at 65 °C. Input DNA from the sonication step was also treated for crosslink reversal.
For Brd4 IP, cells were resuspended in 50 mM Tris (pH 8.0), 10 mM EDTA and 1% SDS and sonicated for approximately 12 min using a Diagenode bioruptor. The resulting whole cell extract was centrifuged and the supernatant was immediately diluted with 4 volumes of ChIP dilution buffer (20 mM Tris pH 8.0, 1 mM EDTA, 167 mM NaCl, 1% Triton X-100, 0.01% SDS). The supernatant was then incubated for 1 h at 4 °C with 50 μL of protein G magnetic beads that had been pre-incubated with 5 μg of control rabbit IgG and then incubated overnight at 4 °C with 50 μL of protein G magnetic beads that had been pre-incubated with an anti-Brd4 antibody (Bethyl laboratories). Beads were then washed 3X with 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100, 1X with 50 mM Tris, 500 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 1% Triton X-100; 1X with 20 mM Tris, 250 mM lithium chloride, 1 mM EDTA, 0.5% sodium deoxycholate, 0.5% NP-40, and 1X with TE. Bound complexes were eluted from the beads using 0.1 M sodium bicarbonate with 1% SDS by heating at 65 °C for 30 min with continuous shaking and crosslinking was reversed by overnight incubation at 65 °C. Input DNA from the sonication step was also treated for crosslink reversal.
Purified ChIP DNA was used for qRT-PCR (see Supplementary Table 3 for primers) or used to prepare Illumina multiplexed sequencing libraries. For both Nanog and Brd4 ChIP-qPCR, values shown are from technical replicates (n = 3) of a single IP. Each IP was performed at least two independent times. For both Brd4 and Med1 ChIP-Seq, libraries for Illumina sequencing were prepared using Nextflex Barcoding Adapters (Bioo Scientific) and a KAPA library amplification kit according to the manufacturer’s instructions. Samples were sequenced using Illumina HiSeq generating 50-bp paired-end reads. Reads were trimmed for quality and Illumina adapter sequences using ‘trim_galore’ before aligning to mouse assembly mm9 with bowtie2 using the default parameters. Aligned reads with the same start site and orientation were removed using the Picard tool MarkDuplicates (http://broadinstitute.github.io/picard/). Density profiles were created by extending each read to the average library fragment size and then computing density using the BEDTools suite (http://bedtools.readthedocs.io). Enriched regions were discovered using MACS 1.4 with default parameters and scored against matched input libraries. All genome browser tracks and read density tables were normalized to sequencing depth. Promoter-based comparisons used the normalized read density within +/−2 kb of the transcription start site, whereas non-promoter signals were additionally normalized to peak length. Dynamic regions between two conditions were scored using MACS with the second ChIP library replacing input, and only sites with an absolute fold change greater than two were scored as a gain/loss. For gene-based analyses, all peaks within 30 kb of a gene were assigned to the relevant gene(s). For ChIP-seq scatter plots, read density was calculated using featureCounts (http://subread.sourceforge.net/) in paired-end mode with a minimum quality threshold of ten. Read density was subsequently normalized to a sequencing depth of ten million mapped reads.
ATAC-Seq
ESCs were seeded at 150,000 cells per well of a six-well plate and treated the following day with DMSO or 500 nM JQ1. Cells were passaged into fresh six-well plates after 24 hours of treatment and harvested after a total of 72 hours of treatment. Duplicate samples from replicate wells were prepped for each condition. For each sample 100,000 cells were harvested and washed with ATAC buffer (Tris 10 mM pH 7.4, 10 mM NaCl, 3 mM MgCl2). Samples were spun at 1000g for 5 minutes at 4 °C. Pellets were resuspended in 50 μL of ATAC lysis buffer (Tris 10 mM pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 or Igepal-Ca630). Nuclei were collected after centrifugation at 1000g for 10 minutes at 4 °C. After discarding supernatant, tagmentation was performed at 37 °C for 30 minutes (Illumina Nextera DNA Sample Preparation Kit, FC-121-1030). DNA was collected using Qiaquick PCR purification columns according to manufacturers instructions and eluted in 10 μl of Elution buffer (10 mM Tris, pH 8). Eluted DNA was mixed with NEBNext Q5 Hot Start HiFi PCR Master Mix (NEB, M0543L) and primer mix (Nextera). DNA was amplified by PCR (65 °C, 5 min; 98 °C, 30 sec; 98 °C, 10s, 65 °C, 1m15s (12×)) and purified using AMPure XP beads (ratio of 1.5:1). Library QC was performed using a Quant-iT Picogreen dsDNA assay kit (Life Technologies, P7589) and the median fragment size using an Agilent D1000 screentape (5067-5582). Samples were pooled for multiplexing and sequenced using paired-end sequencing technology on Illumina NextSeq 500 platform. Sequencing and data processing were identical to ChIP-seq, except in the density profile creation step, in which reads were not extended. Enriched regions were discovered using MACS and scored against matched input libraries (fold change > 2 and p-value < 0.005), and peaks within 500 bp were merged. All genome browser tracks and read density tables were normalized to sequencing depth. For gene-based analyses, each peak was assigned to the most proximal transcription start site based on linear genomic distance. K-means clustering of ATAC-seq data was performed on all peaks whose accessibility was maintained and/or increased with Nanog overexpression and whose accessibility also decreased with JQ1 treatment in control cells. Dynamic peaks were annotated with Homer v4.5 (http://homer.salk.edu/homer/ngs/annotation.html) using a distance threshold of 50 kb. Unless otherwise noted, values shown are the average of duplicate samples.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA)62 was performed using ranked lists or normalized read counts. Mueller Plurinet and Wong ESC Core gene lists are found in the MSigDB62. GSEAPreranked version 4 was used with default parameters and data were exported and graphed in GraphPad Prism version 7.
Western blotting
Protein lysates were extracted in RIPA buffer (Cell Signaling) or Laemmli buffer, separated by SDS-PAGE and transferred to nitrocellulose membranes (Biorad) or PVDF membranes (Millipore). Membranes were blocked in 5% milk in Tris-buffered saline with 0.1% Tween-20 (TBST) and incubated at 4 °C with primary antibodies overnight. After TBST washes the next day, membranes were incubated with horseradish-peroxidase conjugated secondary antibodies for 1 h, incubated with ECL (Pierce or GE Healthcare) and imaged using Ecomax X-Ray Film Processor (Protec) or FluorChem M System (ProteinSimple). Antibodies used (at 1:1,000 unless otherwise noted) were pSTAT3 (Cell Signaling Technologies (CST), 9138), STAT3 (CST, 9139) pERK (CST, 4377), ERK (CST, 9102), Tubulin (Sigma, T9026 at 1:10,000), Brd4 (Bethyl Laboratories, A301-985A100 at 1:2,000), Nucleolin (Abcam, ab22758, 1:5000), Nanog (Ebioscience, 14-5761-80) and Actin (Sigma, A3854 at 1:20,000).
Growth curves
ESCs were seeded at a concentration of 30,000-40,000 cells per well of a 12-well plate. The next day, three wells of each line were counted to determine the starting cell number. The remaining cells were washed with PBS and put in fresh medium with indicated inhibitors or relevant vehicle control. Cells were counted on the indicated days thereafter using a Beckman Multisizer 3 with a cell volume gate of 400 – 10,000 fL. Medium was changed every day. For growth curves using cells expressing shRNA, cells were pre-treated with or without doxycycline for 48 h prior to seeding for curve and subsequent cell counts were normalized to seeding density. All curves were performed at least two independent times.
Quantification of gene expression
RNA was isolated from six-well plates using Trizol (Invitrogen) according to manufacturer instructions and 200 ng RNA was used for cDNA synthesis using iScript (BioRad). Quantitative real-time PCR analysis was performed in technical triplicate using QuantStudio 7 Flex (Applied Biosystems) with Power SYBR Green (Life Technologies). All data were generated using cDNA from triplicate wells for each condition. Actin was used as an endogenous control for all experiments except Supplementary Fig. 7i, for which 18S was used. Primers used for qRT-PCR are shown in Supplementary Table 3. For RNA-sequencing, RNA was isolated from ESCs using Trizol and then samples were prepared as instructed using the TruSeq RNA Sample Preparation Kit v2 (Illumina) in accordance with the manufacturer’s instructions. RNA-seq samples were sequenced using Illumina NextSeq 500 generating 75-bp single-end reads. Fastq files were aligned to genome build mm9 using TopHat with default parameters. Aligned features were counted with htseq-count and differential expression was determined using the edgeR package in Bioconductor as previously described63.
Colony formation assays
Cells were seeded at the indicated density in six-well plates. For experiments with continuous JQ1, 200 cells were seeded per well. The next day, cells were washed with PBS and put into fresh medium with DMSO or 500 nM JQ1 and medium was changed daily thereafter. For experiments with acute JQ1 treatment, 200 cells were seeded per well and medium was refreshed every 2-3 days. 48 h prior to fixation, cells were given fresh medium containing DMSO or 500 nM JQ1. For experiments with FACS sorted cells transduced with GFP-Cas9 and the indicated gRNAs, 10,000 GFP-positive cells were seeded in duplicate. For experiments with clonal CRISPR/Cas9-edited cell lines, ESCs from each individual clone were cultured in S/L or S/L+2i for 48 hours and then seeded at a density of 200 cells/well. Medium was refreshed every 2-3 days. For all experiments, seven days after initial seeding, wells were fixed with citrate/acetone/3% formaldehyde for 30 seconds and stained using the Leukocyte Alkaline Phosphatase Kit (Sigma) according to manufacturer instructions. For colony formation assays in S/L medium, colonies were scored manually in a blinded fashion. For colony formation assays in S/L+2i medium, alkaline phosphatase-positive colonies were scored automatically using ImageJ (NIH). Briefly, images of each well were binarized using the MaxEntropy function and particles greater than 12 pixels with a circularity of 0.5-1.0 were counted.
Chimera assays
Mice and embryos
Animal experiments comply with all relevant ethical regulations. All animal experiments were carried out according to the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medical College (protocol number: 2014-0061). ICR female mice were purchased from Taconic Farms (Germantown, NY). Females were superovulated at 6-8 weeks with 10 IU PMSG (pregnant mare serum gonadotrophin, Sigma-Aldrich) and 10 IU hCG (human chorionic gonadotrophin, Sigma-Aldrich) at intervals of 48 h. The females were mated individually to males and checked for the presence of a vaginal plug the following morning. Plugged females were sacrificed at 1.5 days after hCG injection for collection of 2-cell embryos. These embryos were flushed from the oviducts with KSOM+AA (Specialty Media) and cultured in KSOM for 2.5 days in vitro at 37 °C under 5% CO2 in air to the blastocyst stage.
Blastocyst injection and embryo transfer
ESCs were treated with DMSO or JQ1 as described above and kept on ice until injection. A flat tip microinjection pipette was used for ESC injection using piezo micro manipulator (Prime Tech) to break the embryos. ESCs were picked up in the end of the injection pipette and 15-20 ESCs were injected into each blastocyst. The injection pipette was also used to collect ESCs as a clump and to place them close to the inner cell mass of the blastocyst. The injected blastocysts were kept in KSOM + AA until embryo transfer. Ten injected blastocysts were transferred into each uterine horn of 2.5 dpc pseudopregnant ICR females.
Fluorescence activated cell sorting of Nanog reporter lines
Nanog-GFP ESCs64 were seeded at a concentration of 40,000 cells per well of a 12-well plate. The next day, cells were washed with PBS and put in fresh medium with indicated inhibitors or relevant vehicle control. 24 h later, cells were trypsinized and resuspended in PBS + 0.5% BSA and evaluated for GFP expression on a LSRFortessa machine (Beckman Dickinson). Analysis of GFP mean fluorescence intensity was performed using FlowJo v9.0. All experiments were performed at least two independent times.
Statistics and Reproducibility
GraphPad PRISM 7 software was used for statistical analyses. Error bars, p-values and statistical tests are reported in figure legends. Statistical tests include paired or unpaired two-tailed Student’s t-test, one-way ANOVA, two-way ANOVA and Fisher’s exact test. All experiments (except sequencing experiments, which were done once) were performed independently at least two times.
Data availability
RNA-seq, ChIP-seq and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the accession codes GSE88760 and GSE88769. Previously published sequencing data that were re-analysed here are available under accession codes GSE49847, GSE26833, GSE90895 and GSE56312. Source data for Fig. 4h, Fig. 6a and Supplementary Fig. 4a,h,i have been provided as Supplementary Table 5. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We thank the Thompson and Allis labs for discussion. We thank Rudolf Jaenisch for the iPSCs and Austin Smith for the cDNA encoding the chimeric LIF receptor. L.F. is the Jack Sorrell Fellow of the Damon Runyon Cancer Research Foundation (DRG- 2144-13). B.C. received support from HHMI and the Jane Coffin Childs Memorial Research Fund. D.A.-C. is recipient of a postdoctoral fellowship from the Ramón Areces Foundation. This work was supported by a grant from the Tri-Institutional Stem Cell Initiative (2014-034 to C.B.T., C.D.A and L.F.) and the Memorial Sloan Kettering Cancer Center Support Grant P30 CA008748. C.B.T. is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. He also serves on the board of directors of Merck and Charles River Laboratories.
Footnotes
Author Contributions
L.F., S.V. and B.C. designed, performed and analyzed all experiments under the guidance of C.D.A. and C.B.T. R.K. analyzed ChIP-Seq and ATAC-Seq data. B.K. assisted with RNA-Seq analyses. D.A.C. and S.L. assisted with generation of Brd4 shRNA and CRISPR-edited cell lines. D.W. and S.R. performed chimera experiments. Y.C. and M.R. provided technical assistance. L.F., S.V., B.C., C.D.A. and C.B.T. wrote the manuscript.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Nichols J, Smith A. Pluripotency in the embryo and in culture. Cold Spring Harbor perspectives in biology. 2012;4:a008128. doi: 10.1101/cshperspect.a008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev. 2016;37:76–81. doi: 10.1016/j.gde.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tee WW, Reinberg D. Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development. 2014;141:2376–2390. doi: 10.1242/dev.096982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nature genetics. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 7.Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Micco R, et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014;9:234–247. doi: 10.1016/j.celrep.2014.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan Y, Xue Y, Song C, Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11493–11498. doi: 10.1073/pnas.1309914110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martello G, Smith A. The nature of embryonic stem cells. Annual review of cell and developmental biology. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- 11.Boroviak T, Loos R, Bertone P, Smith A, Nichols J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol. 2014;16:516–528. doi: 10.1038/ncb2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hackett JA, et al. Synergistic Mechanisms of DNA Demethylation during Transition to Ground-State Pluripotency. Stem cell reports. 2013;1:518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ficz G, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell stem cell. 2013;13:351–359. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habibi E, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell stem cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Leitch HG, et al. Naive pluripotency is associated with global DNA hypomethylation. Nature structural & molecular biology. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen MT, et al. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016;35:1550–1564. doi: 10.15252/embj.201593317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singer ZS, et al. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Molecular cell. 2014;55:319–331. doi: 10.1016/j.molcel.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagwat AS, et al. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016;15:519–530. doi: 10.1016/j.celrep.2016.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell. 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzales-Cope M, Sidoli S, Bhanu NV, Won KJ, Garcia BA. Histone H4 acetylation and the epigenetic reader Brd4 are critical regulators of pluripotency in embryonic stem cells. BMC Genomics. 2016;17:95. doi: 10.1186/s12864-016-2414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horne GA, et al. Nanog requires BRD4 to maintain murine embryonic stem cell pluripotency and is suppressed by bromodomain inhibitor JQ1 together with Lefty1. Stem Cells Dev. 2015;24:879–891. doi: 10.1089/scd.2014.0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houzelstein D, et al. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Molecular and cellular biology. 2002;22:3794–3802. doi: 10.1128/MCB.22.11.3794-3802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu T, Pinto HB, Kamikawa YF, Donohoe ME. The BET family member BRD4 interacts with OCT4 and regulates pluripotency gene expression. Stem cell reports. 2015;4:390–403. doi: 10.1016/j.stemcr.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Q, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue F, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi J, et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naive pluripotency. Science. 2014;344:1156–1160. doi: 10.1126/science.1248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharov AA, et al. Identification of Pou5f1, Sox2, and Nanog downstream target genes with statistical confidence by applying a novel algorithm to time course microarray and genome-wide chromatin immunoprecipitation data. BMC Genomics. 2008;9:269. doi: 10.1186/1471-2164-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davie K, et al. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet. 2015;11:e1004994. doi: 10.1371/journal.pgen.1004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Urbanucci A, et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017;19:2045–2059. doi: 10.1016/j.celrep.2017.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 36.Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nature reviews. Molecular cell biology. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwiatkowski N, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parry D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Molecular cancer therapeutics. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 39.Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. The Journal of biological chemistry. 1998;273:13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- 40.Serizawa H, et al. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature. 1995;374:280–282. doi: 10.1038/374280a0. [DOI] [PubMed] [Google Scholar]
- 41.van Oosten AL, Costa Y, Smith A, Silva JC. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nature communications. 2012;3:817. doi: 10.1038/ncomms1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chronis C, et al. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017;168:442–459 e420. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galonska C, Ziller MJ, Karnik R, Meissner A. Ground State Conditions Induce Rapid Reorganization of Core Pluripotency Factor Binding before Global Epigenetic Reprogramming. Cell stem cell. 2015;17:462–470. doi: 10.1016/j.stem.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Meyenn F, et al. Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Molecular cell. 2016;62:983. doi: 10.1016/j.molcel.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa Y, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu H, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Developmental cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chapuy B, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rathert P, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525:543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes & development. 2013;27:1974–1985. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muller FJ, et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455:401–405. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong DJ, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell stem cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dow LE, et al. Conditional reverse tet-transactivator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PloS one. 2014;9:e95236. doi: 10.1371/journal.pone.0095236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, et al. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell stem cell. 2010;7:319–328. doi: 10.1016/j.stem.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dow LE, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dow LE, et al. A pipeline for the generation of shRNA transgenic mice. Nature protocols. 2012;7:374–393. doi: 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 62.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nature protocols. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 64.Faddah DA, et al. Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell stem cell. 2013;13:23–29. doi: 10.1016/j.stem.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ChIP-seq and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under the accession codes GSE88760 and GSE88769. Previously published sequencing data that were re-analysed here are available under accession codes GSE49847, GSE26833, GSE90895 and GSE56312. Source data for Fig. 4h, Fig. 6a and Supplementary Fig. 4a,h,i have been provided as Supplementary Table 5. All other data supporting the findings of this study are available from the corresponding author on reasonable request.