

Abstract
Background
A fundamental precept of the carbohydrate-insulin model of obesity is that insulin secretion drives weight gain. However, fasting hyperinsulinemia can also be driven by obesity-induced insulin resistance. We used genetic variation to isolate and estimate the potentially causal effect of insulin secretion on body weight.
Methods
Genetic instruments of variation of insulin secretion (assessed as insulin concentration 30 minutes after oral glucose [insulin-30]) were used to estimate the causal relationship between increased insulin secretion and body mass index (BMI), using bidirectional Mendelian randomization analysis of genome-wide association studies. Data sources included summary results from the largest published meta-analyses of predominantly European ancestry for insulin secretion (n = 26,037) and BMI (n = 322,154), and individual-level data from the United Kingdom Biobank (n=138,541). Data from the Cardiology and Metabolic Patient Cohort study at Massachusetts General Hospital (n=1,675) were used to validate genetic associations with insulin secretion as well as test the observational association of insulin secretion and BMI.
Results
Higher genetically-determined insulin-30 was strongly associated with higher BMI (beta=0.098, P=2.2 × 10−21), consistent with a causal role in obesity. Similar positive associations were noted in sensitivity analyses using other genetic variants as instrumental variables. By contrast, higher genetically-determined BMI was not associated with insulin-30.
Conclusions
Mendelian randomization analyses provide evidence for a causal relationship of glucose-stimulated insulin secretion on body weight, consistent with the carbohydrate-insulin model of obesity.
Keywords: Obesity, insulin secretion, genetics
For most of the last 40 years, high dietary fat was considered a primary cause of obesity. Recently, attention has focused instead on high glycemic load foods (1), including fast-digesting carbohydrates like refined grains, potato products and added sugars.
According to the carbohydrate-insulin model (1–5), a high glycemic load diet – by increasing insulin secretion – alters substrate partitioning toward fat deposition and promotes weight gain. This hypothesis has received support from mechanistic studies of metabolic fuels (6), translational research (7), observational studies (8,9), and clinical trials (10). Consistent with prediction, several systematic reviews and meta-analyses have shown that lower-carbohydrate diets are superior to low-fat diets for weight loss (11–14). In particular, individuals with high insulin secretion appear particularly responsive to dietary carbohydrate content (8–10,15).
Despite these findings, the carbohydrate-insulin model remains controversial in the absence of definitive metabolic studies. Results of recent feeding studies examining the effect of dietary composition on energy expenditure are variable, showing advantages (16,17), disadvantages (18), or no effect of a lower-carbohydrate compared to a lower-fat diet (19,20). However, these very short protocols (some with duration of 1 week or less) may reflect short-term phenomena – specifically metabolic adaptations to higher fat intake (21–23)–rather than long-term effects of macronutrient composition on body composition.
Mendelian randomization offers a complementary approach to prior research on this topic, with the opportunity to examine relationships for longer than can be achieved in interventional trials, and is theoretically less susceptible to certain biases such as confounding and reverse causation present in conventional observational analyses. In Mendelian randomization, genetic predictors of an exposure of interest are used to assess the causal association between that exposure and an outcome (24). To clarify the causal direction of relationships between two observed traits (e.g., insulin secretion and obesity), a bidirectional strategy using a set of genetic instruments for each trait can be employed (25).
A recent Mendelian randomization analysis (26) cast doubt on the carbohydrate-insulin model, finding no evidence for a causal relationship between fasting insulin and BMI. However, in the carbohydrate-insulin model it is insulin secretion in response to carbohydrate, not fasting insulin, that influences weight gain, in part because the latter is strongly confounded by insulin resistance (5). Indeed, insulin resistance in adipose tissue protects against weight gain, as demonstrated by the fat-specific insulin receptor knockout mouse model (27). Therefore, we selected genetic variants identified by large genome-wide association studies associated with glucose-stimulated insulin secretion, not fasting insulin, to more appropriately test the carbohydrate-insulin model.
METHODS
Study design
Mendelian randomization is an analytic method that leverages the random assignment of genetic variants at conception to infer causal effects. Because genetic variants are randomly assorted during meiosis, this approach can be understood as analogous to an instrumental variable analysis of a randomized trial with non-adherence, in which exposure groups are defined by genotype rather than an assigned intervention. One limitation of Mendelian randomization is difficulty in distinguishing the directionality of causation, because genetic variants could have their primary influence on either trait. A bidirectional strategy (Figure 1) can be used in this situation, using separate sets of instruments for each trait to perform analyses in both directions (25). This strategy has been used by others to demonstrate that obesity is causal for traits such as high C-reactive protein (28), and high uric acid (29). We used bidirectional Mendelian randomization to assess the causal relationship between glucose-stimulated insulin secretion (as quantified by insulin levels 30 minutes after oral glucose, insulin-30) and obesity (as quantified by body mass index [BMI]).
Figure 1. Overview of bidirectional Mendelian randomization.
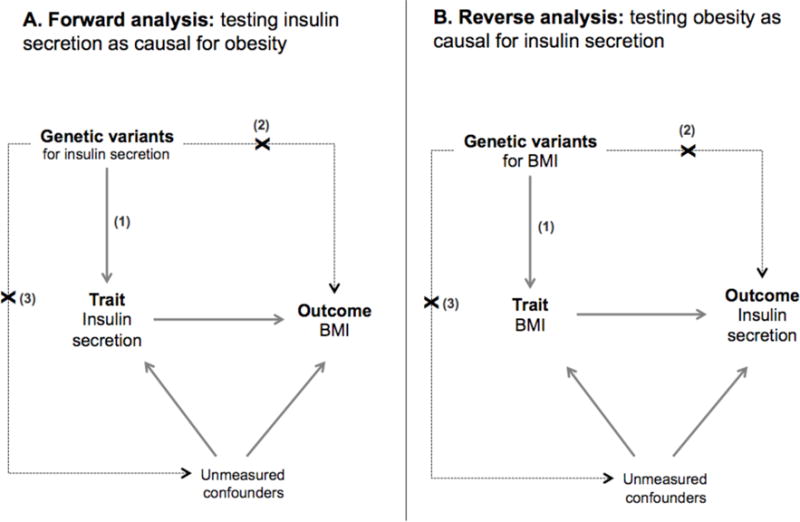
(A) Forward analysis estimates the causal effect of insulin secretion on obesity; (B) reverse analysis estimates the causal effect of obesity on insulin secretion. Key assumptions: (1) variants are reliably associated with the intermediate trait, (2) variants affect the outcome only through the intermediate trait, and (3) variants are independent of confounders.
We used publicly available, summary-level data from large-scale genome-wide association studies to identify single nucleotide polymorphisms (SNPs) that would serve as genetic instruments for each trait (insulin-30 and BMI), and individual-level data to validate the association of the instruments with the intermediate phenotype (Figure 2). The analyses were performed on summary-level data; the use of summary-level data enables more powerful analyses with larger sample size, and is comparable to individual-level analysis for larger sample sizes (30,31).
Figure 2. Study overview.
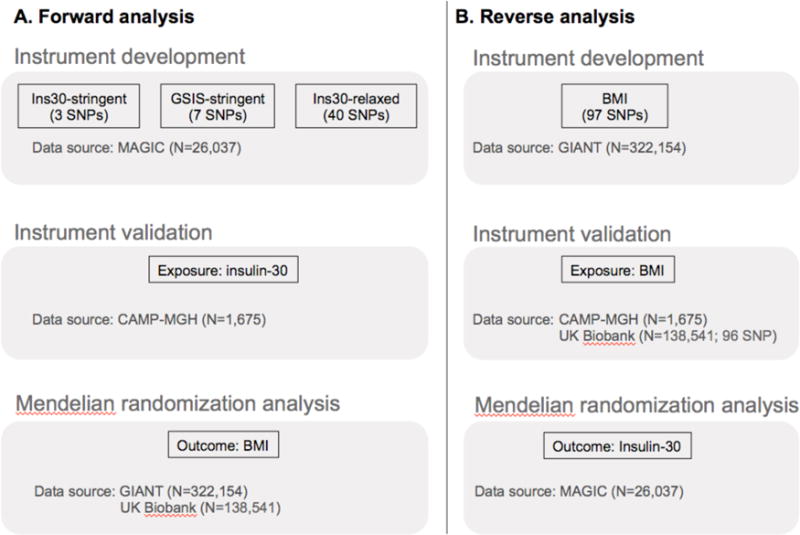
We developed and validated instrument sets for glucose-stimulated insulin secretion (GSIS) (A) and body mass index (BMI) (B) using published genome-wide association study data and independent data sets. Three GSIS sets were assembled using narrow and broad trait definitions (insulin-30 or any GSIS) with stringent and relaxed signficiance thresholds (P ≤ 5×10−8 or 5×10−5). Instrument sets were used to estimate causal association in the bidirectional Mendelian randomization analyses. SNPs, single nucleotide polymorphisms; Insulin-30, insulin concentration 30 min after oral glucose.
Data Sources and Study Subjects
We used summary-level data from the Meta-Analysis of Glucose- and Insulin-related traits Consortium (MAGIC) and the Genetic Investigation of ANthropometric Traits (GIANT) consortium for glucose-stimulated insulin secretion and BMI, respectively (32,33). MAGIC included up to 26,037 non-diabetic individuals of European ancestry who underwent oral glucose tolerance testing (OGTT) and either genome-wide, CardioMetabochip (Ilumina), or specific targeted genotyping of candidate loci (32). GIANT included up to 322,154 individuals of European ancestry from population-based and case-control studies with anthropometric data and either genome-wide or CardioMetabochip genotyping (33). Contributing studies to each consortium received approval from their respective institutional review boards.
To validate SNP-trait association measures for variants in genetic risk scores, as well as to test the observational association of insulin secretion and BMI, we used individual-level data from 1,675 non-Hispanic white participants in the Cardiology and Metabolic Patient Cohort study at Massachusetts General Hospital (CAMP-MGH). Participants all underwent a 75-g OGTT in the absence of anti-hyperglycemic therapies, in which plasma glucose was measured by hexokinase assay (Abbott), and insulin international units were determined using a radio-immunoassay (Roche). Informed consent was obtained from all study participants and the Partners Health Care institutional review board approved the protocol.
We used individual-level data from the United Kingdom Biobank (UK Biobank) to validate SNP-trait association measures for variants in the BMI genetic risk score and to perform the forward analysis (the test of insulin-30 as causal for BMI). Of more than 500,000 participants who were recruited from 2006 through 2010, N=138,541 participants had available genetic data, self-identified as British, Irish or other white background, passed quality control (omitting samples with poor heterozygosity, reported and genetic sex mismatch, first principal component equal to zero, principal component more than 4 standard deviations from the mean, or information score less than 0.3), and had the required covariates (height in cm by Seca 202 device, weight in kg, BMI, age, sex) measured at the baseline assessment. Individuals were not censored for current or future cardio-metabolic risk factors. UK Biobank obtained approval from its governing Research Ethics Committee. Analysis of the UK Biobank data was approved by the Broad Institute institutional review board.
Instrument Selection and Validation
To assess insulin secretion, we used as instrumental variables genetic variants associated with insulin-30 (34,35). Insulin-30 was chosen because it has been shown to be an effect modifier of the relationship between high glycemic-load carbohydrate and weight gain in translational research, observational studies and clinical trials (7–10,15).
Because analysis by the MAGIC consortium identified only three variants associated with insulin-30 at genome-wide significance (P ≤5 × 10−8; Supplemental Table 1) (32), we employed two strategies to expand the set of genetic instruments. First, we augmented our list to include variants with sub-genome-wide (P ≤5 × 10−5) association with insulin-30 (Supplemental Table 1). Second, we included variants associated at genome-wide significance for five additional glucose-stimulated insulin secretion (GSIS) traits: corrected insulin response, corrected insulin response adjusted for insulin sensitivity index, change in insulin at 30 minutes from baseline, disposition index, and insulin-30 adjusted for BMI (Supplemental Table 1). This selection scheme resulted in three insulin-30 genetic instrument sets: insulin-30-stringent (3 variants), insulin-30-relaxed (40 variants), and GSIS-stringent (7 variants).
For BMI, we selected the 97 variants reported by the GIANT consortium to be associated with BMI at genome-wide level of significance (33) (Supplemental Table 2). One BMI variant (rs2033529) was not present in the imputed UK Biobank data and did not have a suitable proxy SNP, and therefore was omitted from the analysis in this cohort.
Genetic risk scores (GRSs) for each trait were created using weights equal to the effect estimates from the MAGIC association study (for the insulin-30 GRSs) or GIANT association study (for the BMI GRS) (32,33). We validated our instrument sets by estimating the variance explained by the GRSs for each trait in cohorts that were independent from those used to estimate the weights. The GRSs for insulin-30 were validated in CAMP-MGH, while the GRS for BMI was validated in both CAMP-MGH and UK Biobank. Variance explained by the GRSs in the validation cohorts was compared to the estimated variance explained from the association study (calculated as the square of the effect estimate multiplied by the variance of a Binomial random variable with probability of success equal to the minor allele frequency). For the UK Biobank analysis, GRS was normalized by the mean effect size of all instruments, such that the linear regression coefficient reflected the mean per allele effect.
Statistical analysis
In the CAMP-MGH cohort, we carried out association analysis on insulin-30 after natural log transformation with adjustment for age and sex, as in the MAGIC analysis (32). We tested association for each individual SNP in the insulin-30 instrument set under an additive genetic model. We validated the insulin-30 and BMI GRSs as predictors of transformed insulin-30 and BMI, respectively, using linear regression.
In UK Biobank, we carried out association analysis on BMI after regression on covariates (age, age squared, and the first 10 principal components) and inverse normalized transformation of residuals, stratified by gender, as in the GIANT analysis (33). Genome-wide association was performed under an additive genetic model and effect estimates for males and females were combined by meta-analysis. We meta-analyzed UK Biobank effect estimates for males and females with GIANT summary statistics. We modeled covariate-adjusted BMI (age, age squared, gender) in the UK Biobank as a continuous outcome with linear regression using the weighted insulin-30 and BMI GRSs as predictors.
We estimated the causal effect of an exposure on an outcome by calculating the ratio of SNP-outcome to SNP-exposure effect estimates using the Genetics ToolboX package (https://CRAN.R-project.org/package=gtx). This approach is equivalent to an inverse variance weighted fixed-effects meta-analysis model of ratio estimates across all variants in the instrumental variable set (31). We used bidirectional Mendelian randomization to estimate the effect of insulin-30 on BMI (Figure 1A) and the effect of BMI on insulin-30 (Figure 1B). To create effect estimates that were more clinically interpretable, we scaled insulin-30 effect estimates from the MAGIC association study by the standard deviation of natural log insulin-30 estimated in CAMP-MGH (i.e. SNP-insulin-30 association divided by 0.66). BMI effect estimates have similar interpretation, as BMI was inverse normalized transformed in the GIANT association study.
To assess for violations of Mendelian randomization assumptions, we tested for heterogeneity and for directional pleiotropy, under the Instrument Strength Independent of Direct Effect or InSIDE assumption, according the method of Bowden et al (36). We also performed sensitivity analyses by omitting instrument outliers and comparing results to the simple median estimator (37).
RESULTS
Validation of Insulin-30 and BMI Instrument Sets
We assembled three different lists of genetic variants that have been associated at different levels of stringency with measures of glucose-stimulated insulin secretion (GSIS); we named these insulin-30-stringent (3 variants), insulin-30-relaxed (40 variants), and GSIS-stringent (7 variants; see Figure 2A and Methods for details). To validate these sets of genetic variants for use as instruments in Mendelian randomization, we examined their effects on insulin-30 in the independent CAMP-MGH cohort (Table 1). In this cohort, the insulin-30-stringent, insulin-30-relaxed, and GSIS-stringent instrument sets accounted for 0.9%, 0.05%, and 1.1% of the phenotypic variance in insulin-30 respectively, compared with 0.6%, 9.0%, and 0.7% in the MAGIC consortium. Individual SNPs were directionally consistent within the primary instrument set (insulin-30 stringent), and for the majority (6 of 7) variants in the GSIS-stringent set (Supplemental Table 3). In contrast, only 26 of 40 of the insulin-30-relaxed set were directionally consistent with the published genome-wide association study effects (Supplemental Table 3). Thus, relaxing the significance threshold appeared to increase the number of potential instruments at the expense of including false positives, as demonstrated by both the more modest directional consistency and the difference in variance explained in the discovery and validation data sets. We also validated the published set of 97 BMI-associated variants (Figure 2B); these explained 1.0% of the variation in BMI in the CAMP-MGH and 1.8% of variation in the UK Biobank cohort (described in Table 1), compared with 2.7% in the GIANT consortium. Nearly all BMI instruments (95 of 96) were directionally consistent in UK Biobank; one BMI instrument (rs11126666) was not statistically associated with BMI (P = 0.89).
Table 1.
Characteristics of CAMP-MGH and UK Biobank Participants
| CAMP-MGH | UK Biobank | |
|---|---|---|
| No. of individuals | 1,675 | 138,341 |
| Age, mean (SD), years | 57.9 (11.4) | 56.8 (8.0) |
| Men, no. (%) | 970 (57.9) | 65,119 (47) |
| BMI, mean (SD), kg/m2 | 28.4 (5.6) | 27.5 (4.8) |
Abbreviations: BMI, body mass index; SD, standard deviation; CAMP-MGH, Cardiology and Metabolic Patient Cohort study at Massachusetts General Hospital; UK Biobank, United Kingdom Biobank.
Observational association of insulin-30 with BMI
As in other correlational epidemiological studies, we found a positive correlation of BMI with insulin-30 in the CAMP-MGH cohort (r= 0.35, P< 0.0001). BMI was also correlated with baseline insulin, age, and gender (Table 2). Higher BMI remained associated with higher insulin-30 levels (P< 0.0001) even adjusting for age and gender in a multivariate model.
Table 2.
Relationships of insulin measures, body mass index, age, and sex in CAMP-MGH.
| BMI | Baseline insulin | Insulin-30 | Age | Male Sex | |
|---|---|---|---|---|---|
| BMI | 1 | 0.4544*** | 0.3322*** | 0.0605 | 0.1333*** |
| Baseline insulin | 1 | 0.5194*** | 0.0635** | 0.1372*** | |
| Insulin-30 | 1 | 0.046 | 0.0949** | ||
| Age | 1 | 0.056 | |||
| Sex | 1 |
All correlations, except insulin-30 with age, are nominally significant (P<0.05).
P<0.001
P<0.0001.
Abbreviations: BMI, body mass index; Insulin-30, insulin concentration 30 min after oral glucose.
GRS insulin-30 associated with higher BMI
Using linear regression, the insulin-30-stringent GRS (beta=0.095 kg/m2 per insulin-30 increasing allele, P=1.1 × 10−12) was associated with higher covariate-adjusted BMI in the UK Biobank. Neither the alternative insulin secretion GRS including GSIS-stringent GRS (P=0.26) or insulin-30-relaxed GRS (P=0.18) were found statistically associated with BMI. The association of higher BMI being found with higher genetic risk for early insulin secretion suggests that factors downstream of the insulin-30 genetic instruments may promote obesity.
Genetically determined insulin-30 associated with higher BMI
To further test for a causal role for insulin secretion in obesity, we used the insulin-30 instrumental sets in a Mendelian randomization analysis (Figure 1A, Figure 2A). Higher genetically determined insulin-30 was associated with higher genetically determined BMI using all three of the insulin secretion instrument sets in the GIANT dataset, (insulin-30-stringent beta=0.095, P=1.8 × 10−12; GSIS-stringent beta 0.055, P = 8.5 × 10−7; insulin-30-relaxed beta=0.01, P=0.02, Figure 3). The insulin-30-stringent instrument set, with instruments selected based on the narrowest GSIS trait definition and most stringent statistical significance threshold, yielded the highest effect estimates for insulin-30 on BMI. The broader (GSIS-stringent) and less stringent (insulin-30-relaxed) instrument sets produced progressively smaller effect estimates for insulin-30 on BMI.
Figure 3. Mendelian randomization effect estimates.
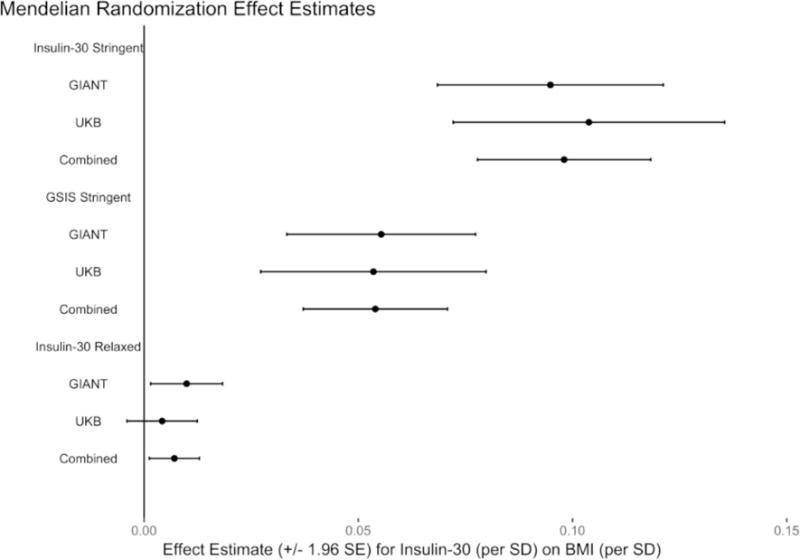
Estimates of higher insulin concentration 30 min after oral glucose on body mass index (BMI), using three different instrument sets (insulin-30-stringent, insulin-30-relaxed, and glucose-stimulated insulin secretion [GSIS]-stringent). GIANT, Genetic Investigation of Anthropometric Traits consortium; UKB, United Kingdom Biobank; SE, standard error; SD, standard deviation.
“This is an un-copyedited authored manuscript copyrighted by the American Association for Clinical Chemistry (AACC). This may not be duplicated or reproduced, other than for personal use or within the rule of ‘Fair Use of Copyrighted Materials’ (section 107, Title 17, U.S. Code) without permission of the copyright owner, AACC. The AACC disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The final publisher-authenticated version of the article is available at http://www.clinchem.org.”
Mendelian randomization effect estimates using the UK Biobank (insulin-30-stringent beta=0.104, P=1.3 × 10−10), a population-based cohort independent from the data used to identify insulin secretion instruments, or UK Biobank meta-analyzed with GIANT (insulin-30-stringent beta=0.098, P=2.2 × 10−21) were consistent with those obtained using GIANT alone. We obtained similar effect estimates and trends for the other two insulin-30 instrument sets using the UK Biobank and meta-analyzed data (Supplemental Table 4). These positive effect sizes are consistent with a causal role for insulin-30 on body weight, with increased early insulin secretion leading to higher BMI.
We performed two analyses to test the robustness of these results. We noted substantial heterogeneity in effect estimates for the variants in each of the three instruments related to insulin secretion (heterogeneity P values ranging from to 7.3 × 10−23 to 2.2 × 10−5). We therefore considered the possibility that the results were driven by a subset of SNPs within each instrument. We omitted the variant that individually produced the strongest effect estimate (QPCTL/GIPR, rs2287019) from the insulin-30-stringent instrument, and found that heterogeneity was eliminated while the two-instrument set was still statistically significantly associated with BMI, albeit with an attenuated effect size (beta=0.045, P=4.2 × 10−4, heterogeneity P=0.34, Supplemental Figure 1). Thus, the results are robust to removal of individual variants with outlier effect sizes. The simple median estimator – the median estimate of the individual SNP-outcome to SNP-exposure ratios – was positive for the insulin-30-stringent (beta=0.059) and GSIS-stringent (beta=0.016) instrument sets. The insulin-30-relaxed instrument simple median estimator was also positive, but close to zero (beta=0.00017).
We tested for pleiotropy, a known confounder of Mendelian randomization analyses, in which a subset of the variants in the instrument can be independently associated with both the exposure (insulin-30) and the outcome (BMI). The Egger test, which tests for directional pleiotropy, was not statistically significant for any of the instrument sets (Egger intercept insulin-30-stringent P=0.53, GSIS-stringent p=0.27, insulin-30-relaxed P=0.92).
Genetically determined BMI not associated with higher insulin-30
Finally, we used bidirectional Mendelian randomization to further examine the directionality of the relationship between insulin-30 and BMI (Figure 1B). Instead of using the combined insulin-30 associated variants as an instrument, we performed Mendelian randomization in the reverse direction, using 97 loci known to be associated with BMI in GIANT (33) as an instrument set (Figure 2B). We tested this set for association with early insulin secretion in the MAGIC data. We found no evidence of a reverse causal effect for BMI on insulin-30 (beta=0.115, P=0.43, heterogeneity P=0.13).
DISCUSSION
Our genetic-based analyses support a causal relationship of genetically determined glucose-stimulated insulin secretion with obesity. Genetic predisposition to higher levels of glucose-stimulated insulin secretion predicted higher adult BMI, while the reverse hypothesis—that genetic predisposition to obesity would predict higher levels of glucose-stimulated insulin secretion—did not hold true.
These results lead to two conclusions. First, it appears that a lifetime of high glucose-stimulated insulin secretion, likely in conjunction with typical diets consumed by the individuals in these cohorts, is obesogenic. An increase in log insulin-30 by one standard deviation (SD) was associated with 0.1 SD increase in covariate-adjusted BMI. This effect size roughly translates to a 160–180 cm person with below average insulin secretion (−1 SD) weighing 2.5–3.1 kg less than the same person with above average insulin secretion (+1 SD). These findings lend additional support to the carbohydrate-insulin model of weight regulation, which postulates that diets high in glycemic load promote weight gain through the anabolic effects of increased insulin secretion.
Second, our findings suggest a role for “precision medicine” in dietary interventions. Two clinical trials indicate that individuals with high baseline insulin-30 have greater weight loss on low-glycemic load diets than those with low insulin-30 (10,15). Consistent with recent conjecture, consideration of obesity subtypes might lead to more effective long-term treatment. While measurement of insulin-30 with OGT may not always be feasible, genetic testing for insulin secretion associated polymorphisms may still be of value as the GRS for insulin-30 was associated with insulin-30 and BMI.
Our study has potential limitations, as the ability to leverage genetic instruments to assess causal relationships with Mendelian randomization relies on two principles (Figure 1): the strength of the instrument relative to other sources of variation in the exposure (assumption 1), and the absence of instrument-outcome associations not mediated by the exposure-outcome causal effect of interest (assumptions 2 and 3). The insulin-30 and BMI genetic instruments explained a relatively small fraction (approximately 1–3%) of variation in the respective exposures. Large sample sizes will enhance power when using an instrument that only explains a small fraction of total variation in traits. An F-statistic greater than 10 has been typically used as a threshold for a strong instrument relative to sample size in the Mendelian randomization literature (38). Estimated F-statistics based on the variance explained and the sample size in UK Biobank are over 1000, indicating that these instruments are of sufficient strength to be used in to estimate the exposure-outcome association.
As the biological mechanisms are unknown for many of the genetic variants used in our analyses, we cannot know for certain that the second principle of Mendelian randomization, the absence of off-target instrument-outcome associations (assumptions 2 & 3 in Figure 1) such as pleiotropy, holds true. In the absence of knowledge of gene-trait biology, we used two statistical techniques, the Egger test and the removal of heterogeneous instruments, to test these assumptions. The Egger test has been developed to assess for violations of a special case termed directional pleiotropy (under the InSIDE assumption).
The Egger test was not statistically significant for any of the analyses, although this test is not well-powered and may not be valid in the context of substantial heterogeneity and/or violations of the InSIDE assumption (36). Tests for heterogeneity were statistically significant for all the instrument sets, which may be a marker of the true underlying biology, sample size for the testing data, or violation of the assumption for Mendelian randomization. Our results were robust to removal of the variants that contributed to the heterogeneity.
By studying genetic associations with more than one trait, genetic epidemiology studies can elucidate underlying biology of genetic variants. The SNP rs2287019, which is most proximal to the QPCTL and GIPR genes, stands out in our analysis as both a strong instrument for insulin-30 and a strong predictor of BMI. It was significant at a genome-wide level for both traits (32,33), as would be expected if there were a true causal effect of insulin secretion on BMI, or pleiotropy. There are good reasons that this instrument likely acts, at least in part, through insulin-30. GIPR encodes the receptor for the glucose-dependent insulinotropic peptide (GIP), an incretin hormone produced by the K cells in the intestines (39). GIP mediates early insulin secretion after enteral nutrition, particularly carbohydrate intake (40). Thus, the known biology suggests that this variant is a particularly relevant instrument for testing the effects of glucose-mediated insulin secretion. Effect estimates for this instrument strongly support the hypothesis that genetically determined insulin-30 has a large effect on BMI, and Mendelian randomization effect estimates are higher for this instrument than that for any other instrument. Therefore, this instrument is particularly plausible and lends strong support to the carbohydrate-insulin model. Another implication of this finding is that individuals with variation in the GIP receptor that increase both BMI and insulin-30 may be especially sensitive to the effects of a high glycemic load diet.
In summary, we found that genetic predisposition to higher glucose-stimulated insulin secretion was associated with higher BMI. This finding supports the carbohydrate-insulin model of obesity. These results underscore the need for adequately powered clinical trials and mechanistic studies of sufficient duration to explore how individuals with low vs high insulin secretion status respond to diets differing in glycemic load.
Supplementary Material
Acknowledgments
This research has been conducted using the UK Biobank Resource under Application Number 1189.
Glossary
LIST OF ABBREVIATIONS
- BMI
body mass index
- CAMP-MGH
Cardiology and Metabolic Patient Cohort study at Massachusetts General Hospital
- GIANT
Genetic Investigation of ANthropometric Traits consortium
- GRS
genetic risk score
- GSIS
glucose-stimulated insulin secretion
- Insulin-30
insulin concentration 30 minutes after oral glucose
- MAGIC
Meta-Analysis of Glucose- and Insulin-related traits Consortium
- OGTT
oral glucose tolerance testing
- SD
standard deviation
- SNP
single nucleotide polymorphism
- UK
Biobank, United Kingdom Biobank
Footnotes
Author contributions:
JNT and CMA designed the study under the guidance of JCF, JNH, and DSL; CMA, JNT, RS, PLH and SV acquired data and/or performed analyses; CMA, JNT, and DSL wrote the paper; JNH, DSL, and JCF had primary responsibility for final content. All authors read and approved the final manuscript.
References
- 1.Ludwig DS. The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA. 2002;287:2414–23. doi: 10.1001/jama.287.18.2414. [DOI] [PubMed] [Google Scholar]
- 2.Lucan SC, DiNicolantonio JJ. How calorie-focused thinking about obesity and related diseases may mislead and harm public health. An alternative. Public Health Nutr. 2015;18:571–81. doi: 10.1017/S1368980014002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taubes G. The science of obesity: what do we really know about what makes us fat? An essay by Gary Taubes. BMJ. 2013;346:f1050. doi: 10.1136/bmj.f1050. [DOI] [PubMed] [Google Scholar]
- 4.Templeman NM, Skovsø S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232:R173–83. doi: 10.1530/JOE-16-0449. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig DS, Friedman MI. Increasing Adiposity: consequence or cause of overeating? JAMA. 2014;311:2167. doi: 10.1001/jama.2014.4133. [DOI] [PubMed] [Google Scholar]
- 6.Walsh CO, Ebbeling CB, Swain JF, Markowitz RL, Feldman HA, Ludwig DS. Effects of diet composition on postprandial energy availability during weight loss maintenance. In: Votruba SB, editor. PLoS One. Vol. 8. 2013. p. e58172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawlak DB, Kushner JA, Ludwig DS. Effects of dietary glycaemic index on adiposity, glucose homoeostasis, and plasma lipids in animals. Lancet. 2004;364:778–85. doi: 10.1016/S0140-6736(04)16937-7. [DOI] [PubMed] [Google Scholar]
- 8.Chaput J-P, Tremblay A, Rimm EB, Bouchard C, Ludwig DS. A novel interaction between dietary composition and insulin secretion: effects on weight gain in the Quebec Family Study. Am J Clin Nutr. 2008;87:303–9. doi: 10.1093/ajcn/87.2.303. [DOI] [PubMed] [Google Scholar]
- 9.Hron BM, Ebbeling CB, Feldman HA, Ludwig DS. Relationship of insulin dynamics to body composition and resting energy expenditure following weight loss. Obesity. 2015;23:2216–22. doi: 10.1002/oby.21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebbeling CB, Leidig MM, Feldman HA, Lovesky MM, Ludwig DS. Effects of a low-glycemic load vs low-fat diet in obese young adults: a randomized trial. JAMA. 2007;297:2092–102. doi: 10.1001/jama.297.19.2092. [DOI] [PubMed] [Google Scholar]
- 11.Bueno NB, de Melo ISV, de Oliveira SL, da Rocha Ataide T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: a meta-analysis of randomised controlled trials Br J Nutr. 2013;110:1178–87. doi: 10.1017/S0007114513000548. [DOI] [PubMed] [Google Scholar]
- 12.Mancini JG, Filion KB, Atallah R, Eisenberg MJ. Systematic Review of the Mediterranean Diet for Long-Term Weight Loss. Am J Med. 2016;129:407–415.e4. doi: 10.1016/j.amjmed.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 13.Mansoor N, Vinknes KJ, Veierød MB, Retterstøl K. Effects of low-carbohydrate diets v. low-fat diets on body weight and cardiovascular risk factors: a meta-analysis of randomised controlled trials. Br J Nutr. 2016;115:466–79. doi: 10.1017/S0007114515004699. [DOI] [PubMed] [Google Scholar]
- 14.Tobias DK, Chen M, Manson JE, Ludwig DS, Willett W, Hu FB. Effect of low-fat diet interventions versus other diet interventions on long-term weight change in adults: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015;3:968–79. doi: 10.1016/S2213-8587(15)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pittas AG, Das SK, Hajduk CL, Golden J, Saltzman E, Stark PC, et al. A low-glycemic load diet facilitates greater weight loss in overweight adults with high insulin secretion but not in overweight adults with low insulin secretion in the CALERIE Trial. Diabetes Care. 2005;28:2939–41. doi: 10.2337/diacare.28.12.2939. [DOI] [PubMed] [Google Scholar]
- 16.Ebbeling CB, Swain JF, Feldman HA, Wong WW, Hachey DL, Garcia-Lago E, et al. Effects of dietary composition on energy expenditure during weight-loss maintenance. JAMA. 2012;307:2627–34. doi: 10.1001/jama.2012.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall KD, Chen KY, Guo J, Lam YY, Leibel RL, Mayer LE, et al. Energy expenditure and body composition changes after an isocaloric ketogenic diet in overweight and obese men. Am J Clin Nutr. 2016;104:324–33. doi: 10.3945/ajcn.116.133561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall KD, Bemis T, Brychta R, Chen KY, Courville A, Crayner EJ, et al. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. Cell Metab. 2015;22:427–36. doi: 10.1016/j.cmet.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergouignan A, Gozansky WS, Barry DW, Leitner W, MacLean PS, Hill JO, et al. Increasing Dietary Fat Elicits Similar Changes in Fat Oxidation and Markers of Muscle Oxidative Capacity in Lean and Obese Humans. In: Blanc S, editor. PLoS One. Vol. 7. 2012. p. e30164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thearle MS, Pannacciulli N, Bonfiglio S, Pacak K, Krakoff J. Extent and determinants of thermogenic responses to 24 hours of fasting, energy balance, and five different overfeeding diets in humans. J Clin Endocrinol Metab. 2013;98:2791–9. doi: 10.1210/jc.2013-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen OE, Caprio S, Reichard GA, Mozzoli MA, Boden G, Owen RS. Ketosis of starvation: a revisit and new perspectives. Clin Endocrinol Metab. 1983;12:359–79. doi: 10.1016/s0300-595x(83)80046-2. [DOI] [PubMed] [Google Scholar]
- 22.Yang MU, Van Itallie TB. Composition of weight lost during short-term weight reduction. Metabolic responses of obese subjects to starvation and low-calorie ketogenic and nonketogenic diets. J Clin Invest. 1976;58:722–30. doi: 10.1172/JCI108519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazquez JA, Adibi SA. Protein sparing during treatment of obesity: ketogenic versus nonketogenic very low calorie diet. Metabolism. 1992;41:406–14. doi: 10.1016/0026-0495(92)90076-m. [DOI] [PubMed] [Google Scholar]
- 24.Davey Smith G, Ebrahim S. Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 25.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:89–98. doi: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richmond R, Wade K, Corbin L, Bowden J, Hemani G, Timpson N, et al. Investigating the role of insulin in increased adiposity: Bi-directional Mendelian randomization study. bioRxiv. 2017:1–18. [Google Scholar]
- 27.Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–4. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 28.Welsh P, Polisecki E, Robertson M, Jahn S, Buckley BM, de Craen AJM, et al. Unraveling the directional link between adiposity and inflammation: a bidirectional Mendelian randomization approach. J Clin Endocrinol Metab. 2010;95:93–9. doi: 10.1210/jc.2009-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyngdoh T, Vuistiner P, Marques-Vidal P, Rousson V, Waeber G, Vollenweider P, et al. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. In: Kronenberg F, editor. PLoS One. Vol. 7. 2012. p. e39321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, EPIC-InterAct Consortium Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30:543–52. doi: 10.1007/s10654-015-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prokopenko I, Poon W, Mägi R, Prasad BR, Salehi SA, Almgren P, et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 2014;10:e1004235. doi: 10.1371/journal.pgen.1004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiu KC, Martinez DS, Yoon C, Chuang L-M. Relative contribution of insulin sensitivity and beta-cell function to plasma glucose and insulin concentrations during the oral glucose tolerance test. Metabolism. 2002;51:115–20. doi: 10.1053/meta.2002.29027. [DOI] [PubMed] [Google Scholar]
- 35.Phillips DI, Clark PM, Hales CN, Osmond C. Understanding oral glucose tolerance: comparison of glucose or insulin measurements during the oral glucose tolerance test with specific measurements of insulin resistance and insulin secretion. Diabet Med. 1994;11:286–92. doi: 10.1111/j.1464-5491.1994.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 36.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han C. Detecting invalid instruments using L1-GMM. Econ Lett. 2008;101:285–7. [Google Scholar]
- 38.Pierce BL, Ahsan H, VanderWeele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40:740–52. doi: 10.1093/ije/dyq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–37. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 40.Seino Y, Maekawa R, Ogata H, Hayashi Y. Carbohydrate-induced secretion of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1. J Diabetes Investig. 2016;7(Suppl 1):27–32. doi: 10.1111/jdi.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.