

Abstract
Staphylococcus aureus is an important human pathogen that is responsible for the vast majority of bacterial skin and soft tissue infections in humans. S. aureus can also become more invasive and cause life-threatening infections such as bacteremia, pneumonia, abscesses of various organs, meningitis, osteomyelitis, endocarditis, and sepsis. These infections represent a major public health threat due to the enormous numbers of these infections and the widespread emergence of methicillin-resistant S. aureus (MRSA) strains. MSRA is endemic in hospitals worldwide and is rapidly spreading throughout the normal human population in the community. The increasing frequency of MRSA infections has complicated treatment as these strains are more virulent and are increasingly becoming resistant to multiple different classes of antibiotics. The important role of the immune response against S. aureus infections cannot be overemphasized as humans with certain genetic and acquired immunodeficiency disorders are at an increased risk for infection. Understanding the cutaneous immune responses against S. aureus is essential as most of these infections occur or originate from a site of infection or colonization of the skin and mucosa. This review will summarize the innate immune responses against S. aureus skin infections, including antimicrobial peptides that have direct antimicrobial activity against S. aureus as well as pattern recognition receptors and proinflammatory cytokines that promote neutrophil abscess formation in the skin, which is required for bacterial clearance. Finally, we will discuss the recent discoveries involving IL-17-mediated responses, which provide a key link between cutaneous innate and adaptive immune responses against S. aureus skin infections.
Keywords: Staphylococcus aureus, Skin, Cutaneous, Infection, Immunology
Introduction
Staphylococcus aureus is a gram-positive extracellular bacterium that is the leading cause of skin and soft tissue infections, which include superficial skin infections such as impetigo and infected abrasions as well as more complicated skin infections such as cellulitis, folliculitis/furunculosis, subcutaneous abscesses, and infected ulcers and wounds (Fig. 1) [1, 2]. In addition, S. aureus can cause invasive and often life-threatening infections such as bacteremia, pneumonia, abscesses of various organs, meningitis, osteomyelitis, endocarditis, and sepsis [3]. A large epidemiologic study conducted in the USA found that S. aureus skin and soft tissue infections account for 11.6 million outpatient and emergency room visits and nearly 500,000 hospital admissions per year [1]. These S. aureus skin infections represent a major threat to public health given the massive numbers of infections as well as the widespread emergence of antibiotic resistant strains such as methicillin-resistant S. aureus (MRSA), including hospital- and community-acquired MRSA (CA-MRSA) infections [4–6]. A recent study demonstrated that 76% of all bacterial skin and soft tissue infections presenting to emergency rooms in 11 major USA cities were due to S. aureus and 78% of these infections were due to MRSA [2]. This indicates that MRSA is now responsible for more than 50% of all skin and soft-tissue infections presenting to emergency rooms in the USA [2]. Furthermore, the USA300 MRSA isolate, which is the most common community-acquired MRSA strain in the USA, is highly virulent and frequently associated with skin and soft tissue infections [4, 5]. S. aureus can also colonize the skin and mucosa of humans. In the USA, it is estimated that up to 30% of healthy individuals in the normal population are colonized with S. aureus and this is important because colonization is a risk factor for subsequent infection [7, 8].
Fig. 1.
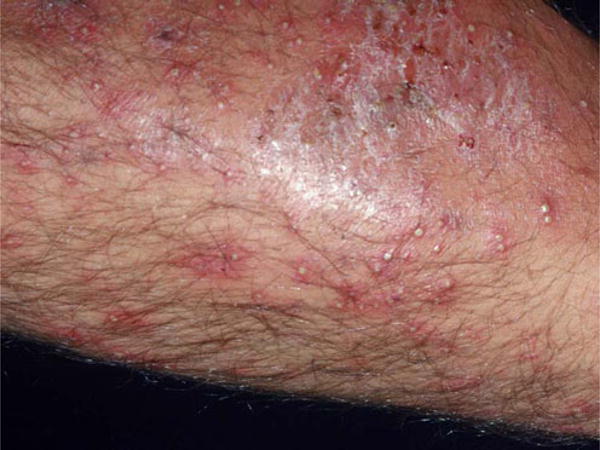
S. aureus folliculitis. Numerous infected hair follicles present as follicularly based erythematous, warm, edematous papules and pustules on this extremity (Courtesy of the Victor D. Newcomer collection at UCLA and Logical Images, Inc.)
It is essential to understand the protective cutaneous immune responses against S. aureus because most of these infections occur or originate from a site of infection or colonization in the skin and mucosa. Furthermore, the information gained from this area of investigation may provide the groundwork for future immunomodulatory therapies to help combat complicated S. aureus skin and soft tissue infections (and at other sites of infection) or vaccination strategies to help prevent S. aureus infections and colonization [9]. In this review, we will focus on the specific elements of cutaneous host immune responses that contribute to host defense against S. aureus skin infection and colonization. This will include a discussion of key innate immune responses, including the initial sensing of a S. aureus infection in the skin, neutrophil recruitment from the circulation to the skin, and the important role of antimicrobial peptides and proinflammatory cytokines. In addition, the adaptive immune responses mediated by B and T cells that play a role in the cutaneous immune responses against S. aureus will also be reviewed.
The physical and immune barrier of the skin
The skin is the primary barrier that protects the body from pathogenic microorganisms encountered in the environment (Fig. 2) [10, 11]. The corneal layer is the outermost layer of the epidermis and is a unique layer that is not found in other epithelia that are exposed to the environment such as the lung and gut epithelia [10, 11]. The corneal layer is comprised of essentially dead terminally differentiated keratinocytes that are devoid of organelles and contain highly crosslinked keratin fibrils [10, 11]. Beneath the corneal layer are the other layers of the epidermis, including the granular, spinous, and basal layers, which consist of keratinocytes at various stages of differentiation [10, 11]. The skin is continuously being turned over as keratinocytes migrate from the basal layer to the corneal layer, after which they are eventually shed [10, 11]. Beneath the epidermis is the dermis, which contains a fibrous stroma composed of collagen and elastin fibers [10, 11]. There are many different types of immune cells that reside in normal human epidermis and dermis [10, 11]. These include Langerhans cells in the epidermidis and macrophages, myeloid and plasmacytoid dendritic cells, mast cells, T and B lymphocytes, plasma cells, and NK cells in the dermis [10, 11]. Skin appendages such as sweat glands (eccrine and apocrine), sebaceous glands, and hair follicles extend through the dermis and epidermis and open onto the skin surface [10, 11].
Fig. 2.
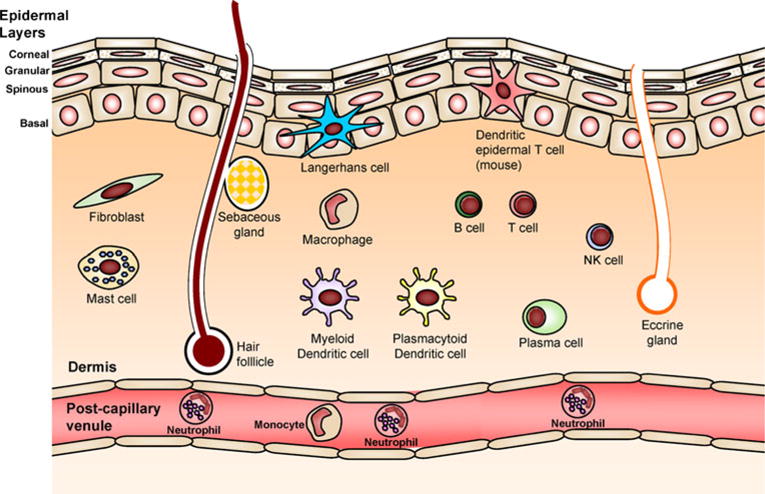
The physical and immune barrier of the skin. The epidermis is composed of layers of keratinocytes, including the corneal, granular, spinous, and basal layers. There are sweat glands, sebaceous glands, and hair follicles that span these layers. In addition, there are many resident immune cells in skin that participate in immune responses. In the epidermidis, there are Langerhans cells and in mouse epidermis there are DETCs. In the dermis, there are mast cells, macrophages, myeloid and plasmacytoid dendritic cells, B and T cells, NK cells, and plasma cells. Neutrophils, monocytes, and other immune cells are recruited from the circulation also participate in cutaneous immune responses
In addition to a physical barrier, the skin is in an opportune position to act as first responder and elicit early immune responses against invading pathogenic microorganisms from the environment such as S. aureus [10, 11]. Keratinocytes in the epidermis are equipped with mechanisms to detect conserved components of microorganisms and initiate early cutaneous innate immune responses such as the production of antimicrobial peptides and proinflammatory mediators [10, 11]. In addition, the resident immune system cells in the epidermis and dermis also participate in cutaneous immune responses [10, 11]. Thus, both the epithelial keratinocytes and immune cells that reside in the skin both contribute to cutaneous host defense mechanisms against S. aureus and other microbial pathogens.
Innate immune response against S. aureus skin infection
The cutaneous immune response to S. aureus involves both the innate and adaptive immune system. The innate immune system was once considered a nonspecific proinflammatory response, such as neutrophil and macrophage phagocytosis or complement activation [12], whereas the adaptive immune response was considered to be a highly specific response driven by recognition of antigens by B and T lymphocytes, which mediate antibody- and cell-mediated immune responses, respectively [13]. It is now known that the innate immune response has considerable specificity that is directed against conserved molecular patterns found in components of microorganisms called pathogen-associated molecular patterns (PAMPs) [12]. The receptors on host cells that recognize PAMPs are called pattern recognition receptors (PRRs) [12]. After recognition of PAMPs, PRRs trigger production of proinflammatory mediators such as cytokines, chemokines, and antimicrobial peptides to initiate early cutaneous immune responses, such as the recruitment of neutrophils from the circulation to the site of infection in the skin [12]. In this section, each of these components of the innate immune response against S. aureus skin infections will be discussed. The role of epidermal keratinocytes as sensors and initiators of immune mechanisms will be reviewed and the importance of neutrophils as the key effector cell against S. aureus infections will be discussed. This will be followed by an overview of antimicrobial peptides, PRRs that recognize S. aureus components, and IL-1R/MyD88 signaling, which is an essential cytokine response to cutaneous S. aureus infections that promotes neutrophil recruitment and bacterial clearance.
Keratinocytes
Epidermal keratinocytes have multiple mechanisms to promote early cutaneous innate immune responses against S. aureus. First, keratinocytes produce antimicrobial peptides that have direct bacteriostatic or bactericidal activity against S. aureus, including human β-defensin 2 (hBD2), hBD3, cathelicidin (LL-37), and RNase 7 [14–18]. Second, keratinocytes express PRRs such as Toll-like receptors (TLRs), including TLRs 1, 2, and 6, which recognize S. aureus lipopeptides, lipoteichoic acid, and peptidoglycan, and NOD2, which recognizes muramyl dipeptide (a breakdown product of S. aureus peptidoglycan) [19]. These PRRs promote innate immune responses such as the production of antimicrobial peptides, cytokines, and chemokines that promote neutrophil recruitment, which contribute to the initial defense against S. aureus [9]. Finally, keratinocytes can be activated by commensal skin bacteria such as S. epidermidis, which activates TLR2 on keratinocytes, leading to increased production of antimicrobial peptides (e.g., hBD2, hBD3, and RNase 7), which in turn promotes killing of S. aureus [20, 21]. Taken together, keratinocytes not only provide a physical barrier to prevent S. aureus infection but also promote early immune responses through the production of antimicrobial peptides, activation of PRRs, and production of proinflammatory cytokines and chemokines that promote recruitment of neutrophils and other immune cells to the skin.
Neutrophils
S. aureus cutaneous infections result in pyogenic lesions characterized by erythema, warmth, and induration with frequent ulceration and drainage of purulent material [4, 5, 22]. Microscopically, these lesions are composed primarily of collections of neutrophils, a hallmark of S. aureus infections, which are required for control and clearance of S. aureus infections [23–25]. Neutrophils represent the first-responder phagocytic cells that are recruited from the circulation to a site of infection or inflammation [26]. Once recruited, they have multiple mechanisms to help kill pathogens [26]. In this section, the important role of neutrophils in S. aureus infection will be discussed, including insights obtained from humans with genetic and acquired neutrophil disorders, mouse models of S. aureus skin infections, and mechanisms that S. aureus utilizes to evade the neutrophilic response.
Genetic and acquired conditions with defects in neutrophil number or function
The important role of neutrophils in host defense against S. aureus is demonstrated by the increased susceptibility to infection in patients with genetic or acquired conditions that result in decreased neutrophil numbers or function. In particular, patients with genetic conditions that result in decreased neutrophil numbers (e.g., severe congenital neutropenia) or defects in neutrophil function (e.g., chronic granulomatous disease, myeloperoxidase deficiency, leukocyte adhesion deficiency I, and neutrophil specific granule deficiency) are predisposed to S. aureus infections [27–29]. Acquired neutropenia seen in cancer patients on chemotherapy are also highly susceptible to S. aureus infections [30]. In addition to these disorders, patients with renal insufficiency and diabetes mellitus, which have a multitude of neutrophil functional impairments, including defects in neutrophil oxidative burst, chemotaxis, and phagocytosis, also have an increased risk for S. aureus infections [31, 32]. It should be noted that the impairments in neutrophil number and function in these diseases and disorders result in an increased susceptibility to S. aureus infections in a wide variety of tissues and organs, including the skin. However, there are a number of conditions that result in a preferential predisposition to S. aureus infections in the skin, including patients with defects in IL-1/TLR signaling (e.g., IRAK4- and MyD88-deficiency) [33–36] and patients with altered T cell responses (e.g., hyper-IgE syndrome, atopic dermatitis, and HIV disease) [37–42]. These specific human disorders have provided important new insights into the protective immune response against S. aureus infections in the skin and will be discussed in detail in this review.
The critical role of neutrophils in S. aureus infections in humans has been supported by studies of S. aureus infections in mice. For example, in a mouse model of S. aureus skin infection, neutrophil-depleted mice had a severe defect in bacterial clearance, resulting in non-healing skin lesions [24]. Furthermore, mice with defective neutrophil recruitment to the skin develop large skin lesions with increased bacterial burden and severe defects in bacterial clearance [23]. A recent study demonstrated that neutrophil abscess formation in mouse skin during cutaneous S. aureus-infected wounds was mediated by three mechanisms: (1) robust neutrophil recruitment to the skin from the circulation, (2) prolonged neutrophil survival within the abscess in the skin, and (3) homing of c-kit+ progenitor cells to the abscess where they locally produce mature neutrophils [43]. Thus, the evidence in humans and mice indicate that neutrophil recruitment to the skin is an essential immune mechanism that is required for immunity against S. aureus skin infections.
Neutrophil function and recruitment
Recruitment of neutrophils from the circulation to a site of S. aureus infection in the skin involves recognition of the pathogen by PRRs [44] and production/secretion of proinflammatory cytokines such as IL-1α, IL-1β, TNFα, and IL-6. These PRRs and cytokines induce upregulation of adhesion molecules such as P-selectin, E-selectin, and intercellular adhesion molecule 1 (ICAM1) on endothelium and L-selectin and lymphocyte function-associated antigen 1 (LFA1) on neutrophils to promote neutrophil rolling, adhesion and diapedesis so that the neutrophils can enter the infected tissue [45]. Furthermore, the PRRs and proinflammatory cytokines induce secretion of neutrophil-attracting chemokines such as CXC-chemokine ligand 1 (CXCL1; also known as Gro-α in humans and KC in mice), CXCL2 (also known as MIP2), CXCL5 (also known as ENA-78), and CXCL8 (also known as IL-8), which promote chemoattraction of neutrophils by activating CXC chemokine receptor 2 (CXCR2) expressed on neutrophils [45].
Once neutrophils encounter S. aureus, they use multiple mechanisms to facilitate bacterial killing (Fig. 3) [26]. Neutrophils express Fc and complement receptors that induce antibody- and complement-mediated phagocytosis to engulf opsonized bacteria into phagosomes [26]. Inside phagosomes, neutrophils have multiple mechanisms that contribute to bacterial killing [26]. These include oxidative burst to generate reactive oxygen species (O2−, H2O2, and hypochlorous acid [HOCl]), which induce direct bacterial killing but also produce a charge in the phagocytic membrane that activates enzymatic killing of the bacteria [26]. In addition, the phagosome contains several different types of antimicrobial peptides such as cathelicidin (LL-37), lysozyme, azurocidin, and α-defensins, which have direct bacteriostatic or bactericidal activity against S. aureus [26]. There are also numerous proteinases such as cathepsin G, elastase, gelatinase, collagenase, and proteinase 3 as well as acid hydrolases, which help degrade S. aureus bacterial proteins and components [26]. Furthermore, neutrophils contain factors that sequester essential nutrients to inhibit bacterial growth and survival, such as lactoferrin, which sequesters iron and copper, transcobalamin II, which binds to and sequesters vitamin B12, and neutrophil gelatinase associated lipocalin (NGAL), which binds to bacterial siderophores and prevents the bacteria from extracting iron [26]. If S. aureus bacteria enter the cytoplasm of neutrophils they encounter a protein complex called calprotectin (S100A8/S100A9), which inhibits S. aureus growth through chelation of Mn2+ and Zn2+ [46]. Finally, neutrophils release antimicrobial peptides, proteases and chromatin through neutrophil extracellular traps (NETs) that induce trapping and killing of S. aureus [47, 48].
Fig. 3.
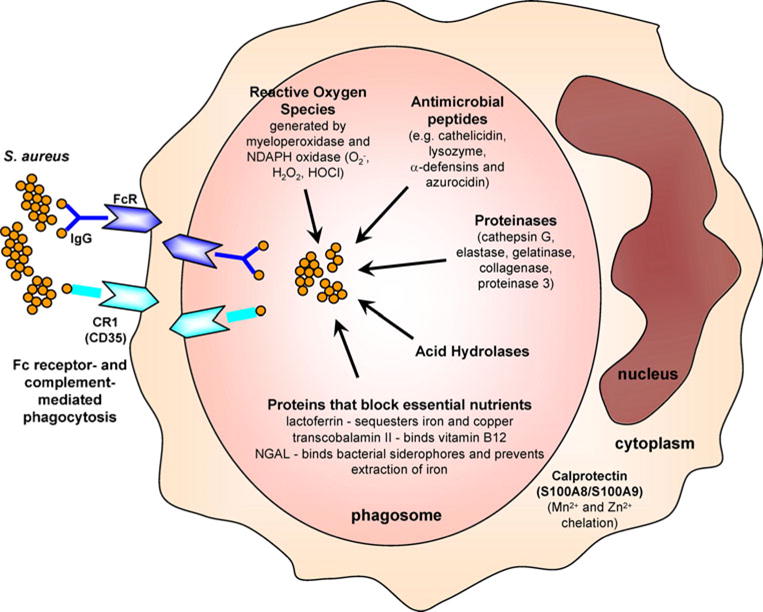
Neutrophil antimicrobial mechanisms against S. aureus. Neutrophils engulf opsonized bacteria using Fc and complement receptors into phagosomes. Inside phagosomes, neutrophils possess multiple distinct mechanisms to promote bacterial clearance. Oxidative burst generates reactive oxygen species (such as O2−, H2O2, and HOCl) through myeloperoxidase and NADPH oxidase, which can directly kill bacteria but also produce a charge in the phagosome membrane to induce enzymatic killing. Antimicrobial peptides such as cathelicidin (LL-37), lysozyme, α-defensins, and azurocidin also have direct antimicrobial activity. Proteinases such as cathepsin G, neutrophil elastase, gelatinase, collagenase and proteinase 3, and acid hydrolases degrade bacterial components. Neutrophils also express proteins that sequester essential nutrients from bacteria to limit their growth such as lactoferrin (which sequesters iron and copper), transcobalamin II (which binds vitamin B12), and neutrophil gelatinase associated lipocalin (NGAL; which binds bacterial siderophores, preventing the extraction of iron). If S. aureus enters the cytoplasm of neutrophils, there is an abundance of calprotectin, which sequesters Mn2+ and Zn2+ to inhibit bacterial growth
Mechanisms used by S. aureus to evade the neutrophilic response
The important role of neutrophils in host defense against S. aureus is further demonstrated by the existence of multiple mechanisms that S. aureus possesses that inhibit neutrophil recruitment and function [49]. First, S. aureus produces factors that block neutrophil chemotaxis. These factors include chemotaxis inhibitory protein of staphylococci (CHIPS) that interacts with the C5aR and the formylated peptide receptor to block neutrophil chemotaxis [50] and extracellular adherence protein (Eap), which binds to ICAM-1 and blocks neutrophil adhesion to endothelium and subsequent diapadesis and extravasation [51]. Second, S. aureus also produces factors that inhibit phagocytosis such as protein A, which binds the Fc portion of IgG in the incorrect orientation, preventing detection by Fc receptors [52, 53]. In addition, fibrinogen binding proteins and clumping factor A (ClfA) bind fibrinogen and impair phagocytosis [52, 53]. Third, S. aureus produces cytolytic toxins that damage membranes of host immune cells leading to osmotic lysis and the prevention of phagocytosis [52, 53]. The prototypical cytolytic toxin, α-toxin (or α-hemolysin), is secreted as a monomer but assembles into a 14-stranded β-barrel pore in the membrane of host cells resulting in cell lysis [52, 53]. In addition, there are biocomponent leukotoxins that are comprised of two subunits such as γ-toxin (or γ-hemolysin) and Panton–Valentine leukocidin (PVL) [52, 53]. Furthermore, CA-MRSA strains produce α-type phenol soluble modulins that lyse neutrophils and other host cells [54]. Although PVL has been epidemiologically linked with CA-MRSA cutaneous infections, α-toxin and α-type phenol soluble modulins may be more important determinants of virulence of CA-MRSA strains [55]. S. aureus evades reactive oxygen species via production of its carotenoid pigment, which is not only responsible for the golden color of S. aureus, but is also a potent antioxidant that neutralizes reactive oxygen species-mediated killing [56]. Moreover, S. aureus produces two superoxide dismutase enzymes that can degrade superoxide and impair reactive oxygen species-mediated killing [57]. Finally, S. aureus has been known to promote a survival program that allows it to survive inside of neutrophils to evade host defenses [58, 59]. Taken together, S. aureus possesses multiple mechanisms to inhibit or evade the neutrophilic response, providing further evidence of the important role of neutrophils in host defense against S. aureus.
Antimicrobial peptides
Antimicrobial peptides are a diverse group of polypeptides that are typically less than 50 amino acids in length and possess antimicrobial activity at physiologic conditions [60–62]. Although their precise mechanisms differ, most antimicrobial peptides are cationic peptides that interact with the anionic membrane surfaces of microbes leading to osmotic lysis [60–62]. Whereas some antimicrobial peptides are produced by keratinocytes and are present in normal skin, others are induced during infection and inflammation/wounding [63, 64]. In this section, we will focus on the specific antimicrobial peptides that are produced by keratinocytes and other immune cells that have been implicated in contributing to host defense against S. aureus skin infections (Table 1).
Table 1.
Antimicrobial peptides that contribute to host defense against S. aureus skin infection and colonization
| Cellular source in the skin | Mechanism of S. aureus evasion | References | |
|---|---|---|---|
| α-Defensins | Neutrophils | Staphylokinase, MprF, dltABCD operon | [65, 66, 68, 87, 90–92] |
| hBD2 | Keratinocytes, macrophages, and dendritic cells | IsdA | [18, 71, 73, 80, 81, 88] |
| hBD3 | Keratinocytes | [16, 17, 20, 21, 72–75, 81] | |
| hBD4 | Keratinocytes | [67] | |
| LL-37 | Keratinocytes, macrophages, and neutrophils | IsdA, Aureolysin, MprF, dltABCD operon | [15, 68, 73, 80, 81, 88–92] |
| Dermcidin | Sweat glands | Extracellular proteases | [69, 70, 93] |
| RNase 7 | Keratinocytes | [14, 21, 62] |
There are several antimicrobial peptides that are present in the skin during an infection that have bacteriostatic or bactericidal activity against S. aureus, including α-defensins (also called human neutrophil peptides [HNPs]), β-defensins, cathelicidin (LL-37), RNase7, and dermcidin (Table 1) [60–62]. Neutrophils express high levels of HNPs 1–3 and less amounts of HNP4, which together constitute nearly 50% of the peptides within neutrophil granules [65]. HNP2 has the highest degree of antimicrobial activity against S. aureus [66]. However, HNPs 1, 3, and 4 also exhibit antimicrobial activity against S. aureus [66]. There are four well-characterized human β-defensins (hBDs 1–4), which are expressed by epithelial cells, including keratinocytes, as well as by activated monocytes/macrophages and dendritic cells [60, 61]. hBD1 has no antimicrobial activity against S. aureus whereas hBD2 and hBD4 have a weak bacteriostatic effect against S. aureus in vitro [18, 67]. In contrast, hBD3 has strong bactericidal activity against S. aureus in vitro and in skin explants ex vivo [16, 17]. Human cathelicidin is also named LL-37 because it represents the 37 amino acid active antimicrobial peptide liberated from the C terminus of the parent protein, human cationic antimicrobial protein 18 kDa (hCAP-18) [60, 61]. LL-37, like hBD3, has potent bactericidal activity against S. aureus [15]. LL-37 is constitutively expressed in neutrophils and can be induced in epithelial cells, including keratinocytes [15, 68]. RNase 7 is another antimicrobial peptide that is produced by keratinocytes and has bactericidal activity against S. aureus [62]. A recent study found that high levels of RNase 7 in the stratum corneum could prevent colonization of skin explants by S. aureus [14]. Finally, dermcidin, which is produced by eccrine sweat glands and is found in human sweat, also has bactericidal activity against S. aureus [69, 70]. In keratinocytes, the production of hBD2, hBD3, and LL-37 can be induced by live or heat-killed S. aureus or by S. aureus components, including lipopeptides and lipoteichoic acid, which activate TLR2 [71–75]. Thus, human keratinocytes can upregulate the production of hBD2, hBD3, and LL-37 in response to S. aureus infections to enhance immune clearance of the pathogen. Furthermore, activation of the epidermal growth factor receptor by wounding of human skin also results in increased hBD3 production, providing another mechanism for enhancing antimicrobial activity against S. aureus [64, 76]. Although a link between vitamin D and host defense against S. aureus skin infection or colonization has yet to be demonstrated, vitamin D increases LL-37 production by keratinocytes, neutrophils, and monocytes/macrophages and thus vitamin D may also play a role in host defense against S. aureus skin infections [77–79].
Most of the antimicrobial activity of antimicrobial peptides against S. aureus has been demonstrated in vitro or ex vivo as described above. However, evidence that these antimicrobial peptides may be relevant to S. aureus skin infections in vivo in humans has come from the study of patients with two dermatologic diseases: (1) atopic dermatitis, which is an allergic Th2 inflammatory skin disease characterized by having an increased susceptibility to S. aureus skin infections, and (2) psoriasis, which is an autoimmune inflammatory skin disease with increased Th1 and Th17 responses that is resistant to S. aureus skin infections [80]. Although many different factors contribute to the difference in susceptibility to S. aureus skin infections between these two diseases, the expression of hBD2, hBD3, and LL-37 is significantly decreased in lesional skin from patients with atopic dermatitis compared with lesional skin from patients with psoriasis [80, 81]. These findings indicate that keratinocyte-derived antimicrobial peptides play a key role in controlling S. aureus infections of human skin in vivo. Finally, antimicrobial peptides not only have microbicidal activity against S. aureus, but they also promote the recruitment of immune cells to the site of infection. For example, HNPs promote recruitment of macrophages, T cells, and mast cells through a PKC-dependent mechanism [82]. hBD2 and hBD3 promote chemotaxis of immature dendritic cells and memory CD4+ T cells through CCR6 and promote chemotaxis of monocytes/macrophages through CCR2 [83, 84]. In addition, LL-37 promotes chemotaxis of neutrophils, monocytes and T cells by activating formyl peptide receptor-like 1 [85, 86]. Taken together, antimicrobial peptides may further enhance host defense mechanisms during S. aureus skin infections by inducing the recruitment of other immune cells.
The importance of antimicrobial peptides in host defense against S. aureus skin infections is further demonstrated by the presence of S. aureus evasion mechanisms that inhibit the activity of these antimicrobial peptides. For example, S. aureus produces staphylokinase, which binds to α-defensins and neutralizes their activity [87]. The S. aureus surface protein iron surface determinant A (IsdA) decreases bacterial cellular hydrophobicity rendering the bacteria resistant to hBD2 and LL-37 [88]. S. aureus also produces a metalloproteinase called aureolysin, which cleaves and inactivates LL-37 [89]. The S. aureus multiple peptide resistance factor protein (MprF) modifies the phospholipid phosphatidylglycerol in the membrane with L-lysine, which reduces the membrane anionic charge and inhibits killing of S. aureus by cationic antimicrobial peptides such as α-defensins and LL-37 [90, 91]. Similarly, products of the dltABCD operon attach positively charged D-alanine residues onto negatively charged phosphate groups in the backbone of teichoic acids, rendering the bacteria less susceptible to killing by α-defensins and LL-37 [68, 92]. S. aureus also secretes extracellular proteases that degrade dermcidin, thereby neutralizing its antimicrobial activity [93]. In summary, the existence of multiple mechanisms that S. aureus possesses to inhibit their activity provides evidence that antimicrobial peptides play an important role in host defense against S. aureus.
Recognition of S. aureus by pattern recognition receptors
PRRs are receptors on host cells that recognize conserved molecules of microorganisms called PAMPs [12]. After recognition, PRRs initiate downstream signaling events that promote the production of proinflammatory cytokines, chemokines, co-stimulatory, and adhesion molecules as well as antimicrobial peptides that contribute to innate and adaptive immune responses [12]. In this section, PRRs relevant to S. aureus skin infections will be discussed, including TLRs, nucleotide-binding oligomerization domain proteins (NODs), peptidoglycan recognition proteins (PGLYRPs) and tumor necrosis factor-α receptor 1 (TNFR1; Fig. 4).
Fig. 4.
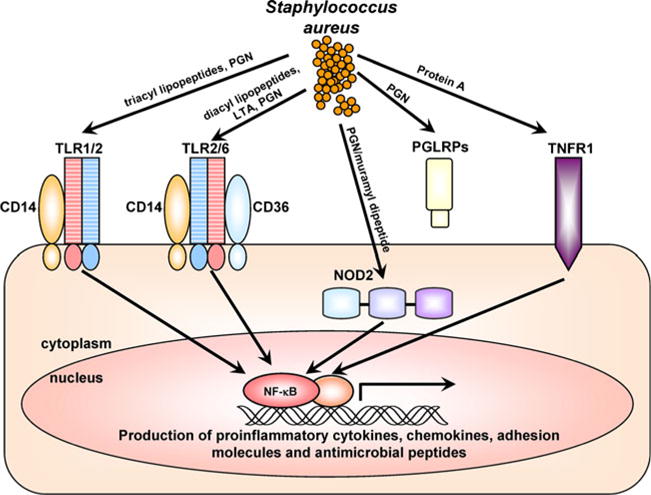
Pattern recognition receptors (PRRs) that recognize S. aureus. PRRs involved in recognizing components of S. aureus and cellular localization of these PRRs are shown. Toll-like receptor 2 (TLR2) heterodimerizes with TLR1 and TLR6 to recognize tri- and di-acyl lipopeptides, respectively (including S. aureus lipoteichoic acid [LTA], which is diacylated). TLR2 also recognizes S. aureus peptidoglycan (PGN) and CD14 and CD36 act as TLR2 co-receptors. Nucleotide-binding oligomerization domain containing 2 (NOD2) is an intracellular cytoplasmic receptor that recognizes the S. aureus peptidoglycan breakdown product muramyl dipeptide. Peptidoglycan receptor proteins (PGRPs or PGLYRPs) are secreted proteins that recognize S. aureus PGN. TNFR1 is a cell surface receptor that is activated by TNF-α but has also been shown to recognize S. aureus protein A. In general, signaling from these PRRs promotes activation of NF-κB and other transcription factors that induce transcription of proinflammatory cytokines, chemokines, adhesion molecules, and antimicrobial peptides that are involved in cutaneous host defense against S. aureus
Toll-like receptors
TLRs are important PRRs involved in host defense against S. aureus [94]. Activation of TLRs initiates several signaling cascades including nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) to initiate early immune responses [94]. Of all the known human TLRs (1–10), TLR2 has been the most implicated in host defense against S. aureus [9]. TLR2 is expressed on the cell surface of numerous cell types in the skin, including keratinocytes, Langerhans cells, monocytes/macrophages, dendritic cells, mast cells, endothelial cells, fibroblasts, and adipocytes [19]. TLR2 recognition of S. aureus components is complex and involves formation of heterodimers with TLR1 or TLR6 to recognize tri- or di-acyl lipopetides (including lipoteichoic acid, which is diacylated), respectively [95–97]. In addition, TLR2 interacts with co-receptors CD14 and CD36 to facilitate recognition and signaling [95, 98]. Although it is somewhat controversial due to potential lipopeptide contaminants in S. aureus peptidoglycan preparations, several studies have found that TLR2 also recognizes S. aureus peptidoglycan [99, 100]. In addition to recognizing and responding to S. aureus components, there is in vivo evidence in mice demonstrating that TLR2 plays an important host defense role against S. aureus skin infections. Our laboratory found that TLR2-deficient mice developed larger skin lesions with increased bacterial burden compared with wildtype mice during a S. aureus skin infection [23]. Although humans with complete TLR2 deficiency have not yet been identified, polymorphisms in TLR2 have been associated with an increased severity of atopic dermatitis, which may help explain the predisposition to S. aureus infections in these patients [101, 102].
Nucleotide-binding oligomerization domain proteins
In contrast to TLRs, which are found in either the cell membrane or membranes of endosomes, nucleotide-binding oligomerization domain proteins (NOD1 and NOD2) are PRRs that are found free in the cytosol [103]. NOD1 recognizes breakdown products of gram-negative peptidoglycan whereas NOD2 recognizes muramyl dipeptide, which is a breakdown product of peptidoglycan of both gram-positive and gram-negative bacteria [103]. Upon recognition, NOD1 and NOD2 (like TLRs) activate NF-κB and MAPK pathways to promote immune responses [103]. Although S. aureus is classically considered an extracellular pathogen, it is now clear that S. aureus can invade the cytoplasm of many different cell types where its muramyl dipeptide has the opportunity to interact with NOD2 [104]. For example, after a S. aureus cutaneous challenge, mice deficient in NOD2 developed larger skin ulcerations with impaired bacterial clearance compared with wildtype mice [105]. In this mouse model, activation of NOD2 resulted in increased IL-1β-induced IL-6 production, which enhanced bacterial killing by neutrophils [105]. Furthermore, the NOD2-mediated host immune response was dependent upon S. aureus α-toxin, which produced pores in cell membrane and facilitated entry of S. aureus into the cytoplasm [105]. Thus, NOD2 represents an important intracellular PRR that recognizes S. aureus muramyl dipeptide from bacteria that enters the cytoplasm of cells.
Peptidoglycan recognition proteins
In humans, there are four peptidoglycan recognition proteins (PGRPs or PGLYRPs)—PGLYRP1, PGRYRP2, PGLYRP3, and PGRYRP4 (formerly termed PGRP-S, -L, -Iα, and -Iβ) [106]. All of these PGRPs are secreted proteins and the relevance of these receptors in cutaneous defense against S. aureus is not fully known [44]. PGLYRP1, though not expressed in skin, is highly expressed in neutrophils, where it binds S. aureus peptidoglycan within tertiary granules and exerts bactericidal activity [107, 108]. PGLYRP2, which is expressed by skin keratinocytes, is distinct in that it has an active amidase that cleaves S. aureus peptidoglycan [109]. However, PGLYRP2 may not be involved in the immune response against S. aureus since there was no difference in cytokine production (IL-6 and TNF-α) or in susceptibility to infection between PGLYRP2-deficient mice and wildtype mice after systemic challenge with S. aureus [110]. Lastly, PGLYRP3 and PGLYRP4 are expressed in the epidermis, hair follicles, sebaceous glands, and sweat glands, and although their expression can be induced by certain bacteria, it is unclear if this is relevant to S. aureus skin infections [106].
Tumor necrosis factor-α receptor 1
Protein A is a S. aureus surface protein that binds immunoglobulins in the incorrect orientation, which represents an immune evasion mechanism because it inhibits antibody-mediated phagocytosis [52, 53]. It has been known that TNFR1, which is a receptor for TNF-α, also binds S. aureus protein A and this interaction plays an important role in host defense against S. aureus pneumonia [111]. Recently, it was shown that TNFR1 on the surface of keratinocytes also binds protein A and this interaction initiates NF-κB activation, leading to keratinocyte production of proinflammatory cytokines and chemokines such as IL-8 [112]. Although this study suggests a role for TNFR1 in host defense against S. aureus skin infections, TNFR1-deficient mice had no defects in neutrophil recruitment or host defense compared with wildtype mice after an in vivo S. aureus skin infection [23]. Thus it is unclear whether the TNFR1 interaction with protein A plays a major role in host defense against S. aureus infections in the skin.
IL-1-mediated neutrophil recruitment response
In this section, we will focus on the role of IL-1α and IL-1β, which signal via IL-1R, as critical promoters of neutrophil recruitment and cutaneous host immune defense against S. aureus. Understanding of the role of IL-1α and IL-1β and IL-1R signaling was obtained from insights from humans with genetic deficiencies in TLR and IL-1R signaling molecules and corroborated by studies in mouse models of S. aureus cutaneous infections and human culture systems.
As described earlier, patients with genetic or acquired impairments in neutrophil number and function are predisposed to S. aureus infections in various organs and tissues and are not limited to skin infections [27–29]. However, pediatric patients with genetic deficiencies in signaling molecules downstream of TLRs and IL-1R, namely MyD88 and IRAK4, are predisposed to S. aureus infections at cutaneous sites of infection [33–36]. TLR and IL-1R family members signal through MyD88, which subsequently interacts with IRAK4, to activate TRAF6 and induce transcription of NF-κB- and MAPK-dependent proinflammatory genes [9]. The patients with MyD88 or IRAK4 deficiency were found to have the same phenotype. Approximately one third suffered from severe and recurrent S. aureus skin infections, 80% developed S. pneumoniae pneumonia and about half the patients died from pneumococcal sepsis before they were 8 years old [33–36]. Furthermore, the frequency of infections decreased with time with no infections seen after the age of 14 years [33–36]. The fact that these patients were not susceptible to other bacterial, fungal, and viral infections indicates that TLR and IL-1R signaling is redundant against most infections but is essential for immunity against pyogenic infections (especially S. aureus and Streptococcus pneumoniae) at epithelial sites [113]. Since S. aureus skin infections require neutrophil recruitment and abscess formation for host defense and bacterial clearance, these findings provide evidence that TLR and IL-1R family members are critical in promoting this protective neutrophilic response against S. aureus skin infections in humans.
It is not known which of the MyD88- and IRAK4-dependent TLR and IL-1R family members contribute to the neutrophilic response against S. aureus skin infections, especially since MyD88/IRAK4 signaling is utilized by all TLRs (except TLR3, which signals via TRIF instead of MyD88) and the IL-1R family members, including IL-1R, IL-18R, and IL-33R [113]. However, data from mouse models of S. aureus skin infection and human cell cultures suggest that IL-1R signaling may be an important determinant for this response. In our laboratory, we compared neutrophil recruitment to the skin and host defense against a cutaneous S. aureus infection in MyD88-, TLR2-, and IL-1R-deficient mice [23]. We focused our efforts on studying TLR2 because of its known role in the recognition of S. aureus lipopeptides, lipoteichoic acid, and peptidoglycan as discussed earlier. We found that IL-1R-deficient mice had a similar phenotype as MyD88-deficient mice, which included a profound impairment in bacterial clearance and neutrophil recruitment to the skin [23]. In contrast, TLR2-deficient mice had only modest impairment in bacterial clearance and no defect in neutrophil recruitment [23]. Thus, IL-1R/MyD88 signaling is a more important determinant than TLR2/MyD88 signaling for neutrophil recruitment and host defense against S. aureus skin infections. We further demonstrated using bone marrow chimeric mice that the activity of IL-1R and MyD88 was dependent upon expression of these molecules by cells that reside in the skin [23]. In contrast, expression of IL-1R or MyD88 by bone marrow recruited cells (such as recruited neutrophils and monocytes) was dispensible for neutrophil recruitment and host defense [23].
It should be mentioned that TLR2 likely contributes to IL-1R-dependent neutrophil recruitment because IL-1β production in TLR2-deficient mice was impaired at 6 h after infection (but not at 24 hours after infection), indicating that TLR2 is one of the PRRs that induces production of IL-1β during S. aureus skin infections [23]. However, since IL-1β was still produced during the infection in TLR2-deficient mice, mechanisms other than TLR2 exist to induce IL-1β during S. aureus skin infections [23]. One such mechanism involves NOD2 activation, since NOD2-deficient mice also had decreased IL-1β production in mouse skin in response to S. aureus infection [105].
In subsequent studies, our laboratory evaluated the contribution of IL-1α and IL-1β, two known IL-1R ligands, to IL-1R/MyD88-mediated neutrophil recruitment and host defense against S. aureus skin infection in mice. Although both IL-1α and IL-1β can activate IL-1R/MyD88 signaling, their cellular source and post-translational processing are different [10]. Premade stores of biologically active IL-1α are present in keratinocytes and are released upon nonspecific injury or infection [10]. In contrast, IL-1β is an inducible cytokine that is produced by many different cell types, including keratinocytes, macrophages, and dendritic cells [10]. Moreover, production of biologically active IL-1β requires proteolytic processing of pro-IL-1β by caspase-1, which is dependent upon formation of an intracellular complex of proteins known as the inflammasome [114]. During an intradermal S. aureus infection using mice deficient in IL-1α or IL-1β, we found that IL-1β-deficient mice had defective bacterial clearance and neutrophil recruitment to the skin whereas IL-1α-deficient mice had a similar phenotype as wildtype mice [115]. Furthermore, mice deficient in apoptosis-associated speck-like protein containing a CARD [caspase-recruitment domain] (ASC), which is a critical component and required for formation of the inflammasome, had the same defects in host defense against a S. aureus skin infection as IL-1β-deficient mice [115]. These data indicate that inflammasome-mediated IL-1β production plays a more important role than IL-1α in activating IL-1R-mediated neutrophil recruitment and host defense against an intradermal S. aureus skin infection [115]. Additional studies demonstrated that the mechanism of IL-1β production in the context of S. aureus infection involved the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, which has been shown to be triggered by pore-forming α-, β-, and γ-hemolysins of S. aureus [116, 117] and the digestion of S. aureus peptidoglycan in phagosomes by lysozyme [118].
In other experiments, we inoculated the S. aureus bacteria into superficial scalpel wounds (instead of intradermally) of the skin of mice deficient in IL-1α or IL-1β [119]. In this case, both IL-1α- and IL-1β-deficient mice had defects in host defense and bacterial clearance, suggesting that during a superficial infection, IL-1α plays a more prominent role in contributing to IL-1R-mediated immune responses [119]. Consistent with these findings, in human keratinocyte cultures, S. aureus (as well as S. aureus lipoteichoic acid and peptidoglycan) induced release of significantly higher amounts of IL-1α than IL-1β and activated an “IL-1α signaling loop,” which resulted in continuous secretion of neutrophil chemokines such as CXCL1, CXCL2, and IL-8 [120]. Taken together, during a deeper intradermal S. aureus skin infection (such as cellulitis or a skin abscess), IL-1β is the predominant IL-1R ligand that promotes neutrophil recruitment whereas during a superficial S. aureus infection (such as impetigo or infected cuts and abrasions), IL-1α, which is likely produced by keratinocytes, also contributes to neutrophil recruitment and host defense. The reason for the differential roles and compartmentalization of IL-1α and IL-1β are not entirely clear. However, it is likely that the pre-made stores of IL-1α in keratinocytes represent a rapid and early cutaneous immune response to combat the initial invasion of bacteria into the epidermis whereas the inducible IL-1β response is mobilized if the infection continues to spread and invades the dermis and subcutaneous tissue.
By combining all of the results from these studies, an important mechanism for promoting the neutrophilic response against S. aureus skin infections has been uncovered (Fig. 5a). This mechanism begins with IL-1α secretion from keratinocytes and PRR-dependent (e.g., TLR2 and NOD2) and inflammasome-mediated IL-1β production from resident skin cells, which activate the IL-1R and subsequent MyD88/IRAK4 signaling. The MyD88/IRAK4 signaling pathway results in production of cytokines, chemokines and adhesion molecules that promote neutrophil recruitment and abscess formation at the site of S. aureus infection in the skin.
Fig. 5.
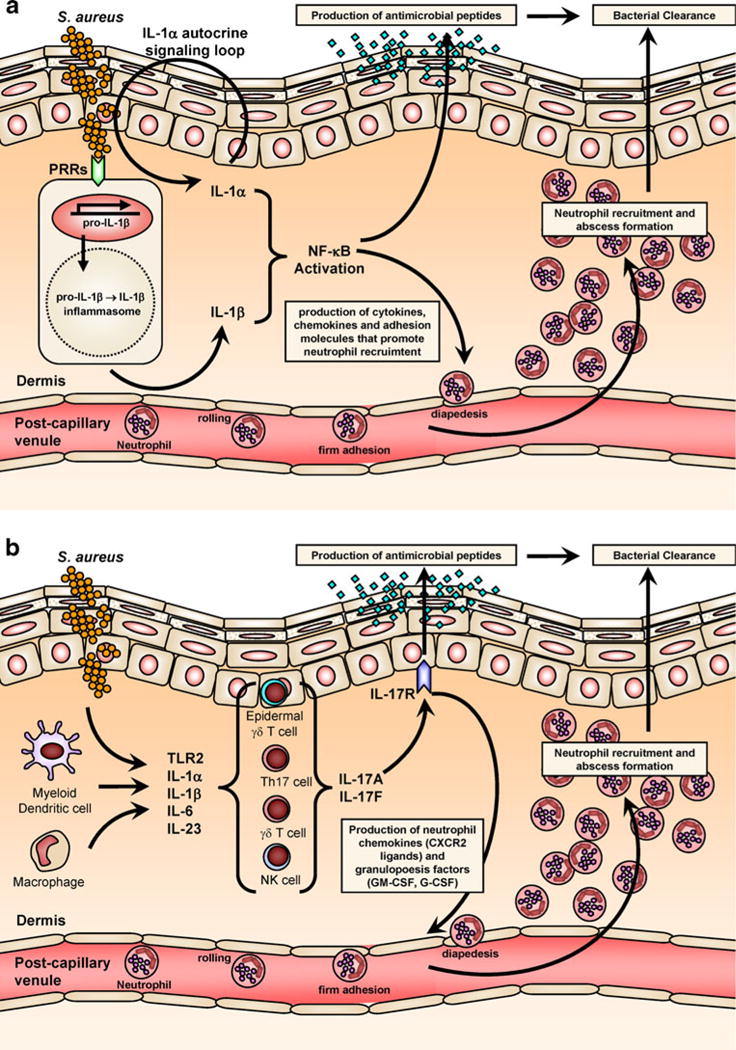
Cytokine responses that promote clearance of S. aureus skin infections. a IL-1 response against S. aureus. In response to S. aureus, IL-1α is produced and released by keratinocytes in an autocrine signaling loop. In addition, S. aureus skin infection results in activation of PRRs and the inflammasome by resident and recruited cells (such as macrophages and dendritic cells), which leads to production and secretion of active IL-1β. b IL-17 mediated response against S. aureus. IL-17A and IL-17F are produced by multiple different types of cells in the skin, including Th17 cells, γδ T cells, and NK cells (and perhaps mast cells and neutrophils) in response to different signals such as activation by TLR2, IL-1α/β, IL-6, and IL-23. IL-17A and IL-17F mediate immune responses by binding to the IL-17R, which is expressed primarily by keratinocytes. Both the IL-1 and IL-17 responses promote neutrophil recruitment and abscess formation and keratinocyte production of antimicrobial peptides (e.g., hBD2, hBD3, and LL-37) to help control S. aureus skin infections and mediate bacterial clearance
Adaptive immune responses to S. aureus
The innate immune system provides the first line of defense against microbial pathogens. In contrast, adaptive immune responses are recruited later on during an infection and are also responsible mediating immunologic memory responses [13]. The adaptive immune system can be divided into antibody- and cell-mediated immune responses, which are mediated by B cells and T cells, respectively [13]. In this section, B cell responses, especially antibody responses against S. aureus will be discussed. In addition, the emerging role of T cell responses will be examined in detail, with an emphasis on Th17 and IL-17 responses, which have recently been found to be essential in host defense against S. aureus cutaneous infections.
B cell responses
The B cell-mediated immune response against S. aureus involves the production of antibodies directed against specific antigens of components of S. aureus. These antibodies play an important role in opsonizing the bacteria and facilitating antibody-mediated (and complement-mediated) ingestion of the bacteria by phagocytes (e.g. neutrophils and macrophages) [121, 122]. After a S. aureus/MRSA infection in humans, many antibodies are generated, including antibodies against toxins (e.g., α-toxin, β and γ hemolysins, PVL and enterotoxins), virulence factors (e.g., aureolysin, IsdA, and superantigens), cell-wall proteins (e.g., clumping factors [ClfA/B] and fibronectin binding protein), and non-protein antigens (e.g., capsular polysaccharides, lipoteichoic acid, and peptidoglycan) [121]. The importance of B cell responses and antibodies in host defense against S. aureus infections is best illustrated by the existence of S. aureus protein A, which binds immunoglobulins in the incorrect orientation, thus enabling S. aureus to evade antibody detection and antibody-mediated phagocytosis [52, 53]. However, it is not clear if these antibody responses are indeed protective since antibody-based vaccines aimed at facilitating opsonization, such as vaccines against capsular polysaccharides (StaphVax®) or ClfA (Veronate®), have had limited efficacy in clinical trials [123, 124] and up to a third of patients with high antibody titers suffer from recurrent infections [125–127]. It is possible that newer antibody vaccines against multiple or different antigens may be more effective in the future [128, 129]. Some of these newer vaccines are not targeted against promoting antibody-mediated opsonization but instead target S. aureus virulence factors [122]. In particular, S. aureus antibody-based vaccines have been developed against the following antigens: α-toxin and PVL, which would inhibit the action of these cytolytic toxins [130–132]; protein A, which would block the ability of protein A to neutralize antibody opsonization and phagocytic responses [133]; IsdB, which would inhibit S. aureus uptake of the essential nutrient iron [134]; and clotting factors such as coagulase and von Willebrand factor binding protein, which would prevent abscess formation and facilitate access of host immune cells and antibiotics to kill the bacteria [135]. However, it is also unclear how efficacious these antibody-based vaccines will be in humans since many patients develop natural antibodies directed against the same components targeted by these newer vaccination strategies [121].
T cell responses
In this section, we will focus on the role of T cell responses in cutaneous host immune defense against S. aureus. In particular, we will review the important T helper (Th) cell subsets (CD4+ T cells), especially Th1, Th2, and Th17 cells, which have been implicated in the pathogenesis of S. aureus skin infections. Th1 cells produce IFN-γ and promote cell-mediated immune responses, Th2 produce IL-4 and IL-13 and promote antibody-mediated immune responses, and Th17 cells produce IL-17 (i.e., IL-17A and IL-17F) and IL-21 and IL-22 and IL-26 and promote neutrophil recruitment and abscess formation [136]. In general, a role for T helper cells in contributing to host defense against S. aureus has been suggested by studying human patient populations with certain T cell defects that are known to have an increased susceptibility to S. aureus skin infections. For example, patients with HIV/AIDS, who become deficient in CD4+ T cells during disease progression, are at an increased risk for colonization and skin infection with S. aureus [137–139].
Th1 and Th2 responses in the pathogenesis of S. aureus skin infections
With respect to Th1 responses, the Th1 cytokine IFNγ was found to promote neutrophil recruitment in a surgical thigh wound model in mice [58, 140] and protection against an intravenous S. aureus challenge in mice [141–143]. However, whether these responses relate to S. aureus skin infections in humans is unknown. In contrast, human skin lesions from the inflammatory skin disease atopic dermatitis, which are classically associated with a Th2 cytokine profile (i.e., IL-4, IL-13, and IL-10), have increased colonization and superinfection of their skin lesions with S. aureus [144]. The increased susceptibility of atopic dermatitis skin lesions to S. aureus colonization and superinfection is multifactorial and has been associated with a defective epidermal barrier and impaired innate immune responses such as decreased expression of antimicrobial peptides as discussed earlier [80, 81]. Indeed, the cytokine milieu (i.e., increased IL-4 and IL-13) in atopic dermatitis leads to decreased expression of hBD2 and hBD3 [81]. Furthermore, in mouse models of allergic skin inflammation, which mimics atopic dermatitis skin, IL-4 has been shown to increase expression of fibrinogen and fibronectin in the skin, which enhanced binding of S. aureus to the skin through S. aureus fibrinogen and fibronectin binding proteins [145]. Also, S. aureus produces superantigens, including staphylococcal enterotoxins A and B (SEA and SEB) and toxic shock syndrome toxin-1 (TSST-1), which nonspecifically activate T cells and exacerbate cutaneous inflammation [146]. In mouse skin sensitized to SEB, there was much more IL-4 produced than IFN-γ, suggesting that these superantigens may skew the cutaneous immune response towards a Th2 cytokine profile and contribute to the increased S. aureus colonization and superinfection in atopic dermatitis [147]. Taken together, based on findings in humans with atopic dermatitis and mouse models of allergic skin inflammation, Th2 cytokine responses are associated with increased susceptibility to S. aureus skin infections.
Th17 responses promote neutrophil recruitment against S. aureus cutaneous infections
Although these findings suggest that Th1 responses are protective and Th2 responses are deleterious against S. aureus skin infections, recent studies have also implicated Th17 cells as important mediators of neutrophil recruitment and host defense against S. aureus skin infections. Of the cytokines produced by Th17 cells, IL-17A, and IL-17F have been shown to promote neutrophil recruitment and abscess formation through the induction of CXCL1, CXCL2, CXCL5, and CXCL8, which induce chemotaxis of neutrophils, and granulopoesis factors (G-CSF and GM-CSF), which promote production and maturation of neutrophils [148, 149]. In addition, IL-17A, IL-17F, and IL-22 induce keratinocyte production of antimicrobial peptides such as LL-37 and hBD2 [150, 151]. Evidence for a role of Th17 cells in host defense against S. aureus cutaneous infections has been provided by studying patients with hyper-IgE syndrome, who suffer from recurrent and severe S. aureus and C. albicans cutaneous infections [152]. Recently, it was discovered that autosomal dominant and recessive forms of hyper-IgE syndrome have STAT3 [153, 154] and DOCK8 [155, 156] mutations that render these patients deficient in Th17 cells [39–42]. Since hyper-IgE patients have a profound defect in immunity against S. aureus skin infections, these findings provide key evidence that Th17 immune responses are critical for host defense against S. aureus skin infections in humans. In addition, humans with chronic mucocutaneous candidiasis who suffer from candidal infections at mucosal and cutaneous sites and to a lesser degree S. aureus skin infections were found to have autoantibodies specific for IL-17A, IL-17F, and IL-22, as well as mutations in the genes encoding IL-17F and IL-17RA [157–159], providing further evidence for a role of Th17 responses in host defense against S. aureus skin infections. Furthermore, this important role of Th17 cells may also be relevant to the increased susceptibility to S. aureus skin infections in patients with HIV/AIDS and atopic dermatitis, since there is a preferential loss of Th17 cells during the progression of HIV disease [38, 160, 161] and lesions of atopic dermatitis have decreased Th17 cells and responses [37, 162, 163]. Thus, although hyper-IgE syndrome, chronic mucocutaneous candidiasis, HIV/AIDS, and atopic dermatitis represent different diseases, they all share in common an increased susceptibility to S. aureus skin infections and a deficiency in Th17 cells, indicating the importance of Th17 cells in host defense against S. aureus skin infections in humans.
These findings in humans have been supported in mouse models of S. aureus skin infections. One study found that mice deficient in both IL-17A and IL-17F developed spontaneous S. aureus skin infections but did not have an increased susceptibility to a systemic S. aureus challenge [164]. These data in mice resemble the phenotype of humans with hyper-IgE syndrome, who suffer from S. aureus infections in the skin but not in other organs and tissues [152]. Although the reason for the increased susceptibility to S. aureus (and S. pneumoniae) in hyper-IgE syndrome at epithelial sites and not at other sites of infection is not entirely known, one study evaluated the dependence of different human cell types to activation by IL-17 and IL-22 [165]. In this study, human keratinocytes and bronchial epithelial cells were much more sensitive to activation by IL-17 and IL-22 in the induction of chemokines and antimicrobial peptides than other cell types such as fibroblasts and endothelial cells [165]. These findings suggest that Th17 responses are much more relevant for promoting host defense against pathogens in the skin (S. aureus) and the lung (S. pneumoniae) and not at other sites of infection, thus providing an explanation for the increased susceptibility to infection at epithelial sites in hyper-IgE syndrome.
Regarding the role of IL-17 against S. aureus cutaneous infections, in particular, our laboratory found that mice deficient in γδ T cells developed larger skin lesions, increased bacterial counts, decreased neutrophil chemokine production, and impaired neutrophil recruitment compared with wildtype mice [166]. In contrast, the phenotype of mice deficient in αβ T cells did not differ from wildtype mice [166]. In addition, the infected skin from γδ T cell deficient mice had a severe defect in induction of IL-17A and IL-17F early on after the infection (at 8 h) compared with wildtype mice, indicating that γδ T cell production of IL-17A and IL-17F was responsible for mediating neutrophil recruitment and host defense [166]. The early induction of IL-17A and IL-17F during S. aureus skin infection was also impaired in MyD88-, IL-1R-, TLR2-, and IL-23-deficient mice suggesting that IL-17A and IL-17F production was dependent upon MyD88, IL-1, TLR2, and IL-23 [166]. Furthermore, mice deficient in the IL-17R or wildtype mice treated with an anti-IL-17A neutralizing antibody were evaluated and both groups of mice had the same defects in host defense and bacterial clearance against the S. aureus skin infection as γδ T cell deficient mice [166]. Finally, local administration of a single dose of recombinant murine IL-17A to γδ T cell deficient mice at the time of S. aureus infection rescued these mice from any impairments in host defense [166]. These data corroborate the essential role of IL-17 in host defense against S. aureus skin infections described from studies in humans with hyper-IgE syndrome. However, it is not clear if γδ T cells play the same essential role against human S. aureus skin infections, especially since humans with hyper-IgE syndrome are deficient in Th17 cells and not γδ T cells [39–42]. Furthermore, mouse skin has a large resident γδ T cell population in the epidermis (called dendritic epidermal T cells [DETCs]), which is not present in human skin [167]. Therefore, it could be that Th17 cells (and perhaps other IL-17-producing cells such as dermal γδ T cells, NK cells, and NKT cells) may represent functional equivalents and serve as the source of IL-17 production during S. aureus skin infections in humans. It should be mentioned that recent reports have also demonstrated that neutrophils and mast cells also produce IL-17 in human skin and these cells could also contribute to the production of IL-17 during S. aureus skin infections [168, 169].
Taken together, these studies suggest a pathway in which IL-17-mediated neutrophil recruitment to a site of S. aureus infection in the skin begins with activation of TLR2, IL-1α/β, and IL-23 production, which stimulates IL-17A and IL-17F production by T cell and NK cell subsets in the skin (e.g., Th17 cells, γδ T cells, NK cells, NKT cells) and possibly neutrophils and mast cells (Fig. 5b). IL-17A and IL-17F then activate the IL-17R, which is present on keratinocytes, to produce proinflammatory cytokines, chemokines, and adhesion molecules that mediate neutrophil recruitment to a site of cutaneous S. aureus infection.
Conclusion
In summary, recent discoveries involving the innate and adaptive immune responses against cutaneous S. aureus infections have greatly increased our understanding of the immune mechanisms that help protect against these infections. These include innate immune responses, such as the recognition of S. aureus by PRRs, production of antimicrobial peptides, and IL-1α and IL-1β cytokine responses that promote neutrophil recruitment, which are critical in controlling S. aureus infection and colonization of the skin. In addition, adaptive immune responses such as antibody- and cell-mediated immune responses mediated by B and T cells, respectively, also contribute to host defense. In particular, the recent findings demonstrating that Th17 cells are essential in promoting neutrophil recruitment and abscess formation against S. aureus skin infections provided new insights into how the innate and adaptive immune responses interact to combat S. aureus cutaneous infections. As antibiotic resistance continues to increase, the mechanisms of protective immune responses against cutaneous S. aureus infections will be important to consider for the future development of immunomodulatory therapies and vaccination strategies to help prevent or treat S. aureus skin infections and colonization.
Acknowledgments
L.S.M. is supported by the United States National Institutes of Health (R01 AI078910).
Footnotes
This article is published as part of the Special Issue on Immunopathology of staphylococcal infections [34:3].
Contributor Information
Sheila Krishna, Division of Dermatology, Department of Medicine, University of California Los Angeles (UCLA), 52-121 Center for Health Sciences, 10833 Le Conte Avenue, Los Angeles, CA 90095, USA.
Lloyd S. Miller, Division of Dermatology, Department of Medicine, University of California Los Angeles (UCLA), 52-121 Center for Health Sciences, 10833 Le Conte Avenue, Los Angeles, CA 90095, USA Department of Orthopaedic Surgery, David Geffen School of Medicine, University of California Los Angeles (UCLA), Los Angeles, CA 90095, USA.
References
- 1.McCaig LF, McDonald LC, Mandal S, Jernigan DB. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg Infect Dis. 2006;12:1715–1723. doi: 10.3201/eid1211.060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2007;357:380–390. doi: 10.1056/NEJMcp070747. [DOI] [PubMed] [Google Scholar]
- 5.Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated methicillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak J, Talan DA, Chambers HF. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52:e18–e55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 7.Gorwitz RJ, Kruszon-Moran D, McAllister SK, McQuillan G, McDougal LK, Fosheim GE, Jensen BJ, Killgore G, Tenover FC, Kuehnert MJ. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. J Infect Dis. 2008;197:1226–1234. doi: 10.1086/533494. [DOI] [PubMed] [Google Scholar]
- 8.Hidron AI, Kourbatova EV, Halvosa JS, Terrell BJ, McDougal LK, Tenover FC, Blumberg HM, King MD. Risk factors for colonization with methicillin-resistant Staphylococcus aureus (MRSA) in patients admitted to an urban hospital: emergence of community-associated MRSA nasal carriage. Clin Infect Dis. 2005;41:159–166. doi: 10.1086/430910. [DOI] [PubMed] [Google Scholar]
- 9.Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004;4:211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nestle FO, Di MP, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Girardi M. Cutaneous perspectives on adaptive immunity. Clin Rev Allergy Immunol. 2007;33:4–14. doi: 10.1007/s12016-007-0040-9. [DOI] [PubMed] [Google Scholar]
- 14.Simanski M, Dressel S, Glaser R, Harder J. RNase 7 protects healthy skin from Staphylococcus aureus colonization. J Invest Dermatol. 2010;130:2836–2838. doi: 10.1038/jid.2010.217. [DOI] [PubMed] [Google Scholar]
- 15.Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect Immun. 2005;73:6771–6781. doi: 10.1128/IAI.73.10.6771-6781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kisich KO, Howell MD, Boguniewicz M, Heizer HR, Watson NU, Leung DY. The constitutive capacity of human keratinocytes to kill Staphylococcus aureus is dependent on beta-defensin 3. J Invest Dermatol. 2007;127:2368–2380. doi: 10.1038/sj.jid.5700861. [DOI] [PubMed] [Google Scholar]
- 17.Harder J, Bartels J, Christophers E, Schroder JM. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J Biol Chem. 2001;276:5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 18.Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 19.Miller LS. Toll-like receptors in skin. Adv Dermatol. 2008;24:71–87. doi: 10.1016/j.yadr.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di NA, Gallo RL. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. 2010;130:2211–2221. doi: 10.1038/jid.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, Schaller M, Schittek B. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol. 2011;131:382–390. doi: 10.1038/jid.2010.328. [DOI] [PubMed] [Google Scholar]
- 22.Elston DM. Community-acquired methicillin-resistant Staphylococcus aureus. J Am Acad Dermatol. 2007;56:1–16. doi: 10.1016/j.jaad.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Molne L, Verdrengh M, Tarkowski A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect Immun. 2000;68:6162–6167. doi: 10.1128/iai.68.11.6162-6167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdrengh M, Tarkowski A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect Immun. 1997;65:2517–2521. doi: 10.1128/iai.65.7.2517-2521.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouma G, Ancliff PJ, Thrasher AJ, Burns SO. Recent advances in the understanding of genetic defects of neutrophil number and function. Br J Haematol. 2010;151:312–326. doi: 10.1111/j.1365-2141.2010.08361.x. [DOI] [PubMed] [Google Scholar]
- 28.Lakshman R, Finn A. Neutrophil disorders and their management. J Clin Pathol. 2001;54:7–19. doi: 10.1136/jcp.54.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev. 2003;16:597–621. doi: 10.1128/CMR.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Barca E, Carratala J, Mykietiuk A, Fernandez-Sevilla A, Gudiol F. Predisposing factors and outcome of Staphylococcus aureus bacteremia in neutropenic patients with cancer. Eur J Clin Microbiol Infect Dis. 2001;20:117–119. doi: 10.1007/pl00011241. [DOI] [PubMed] [Google Scholar]
- 31.Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, Curi R, Sannomiya P. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res. 2007;40:1037–1044. doi: 10.1590/s0100-879x2006005000143. [DOI] [PubMed] [Google Scholar]
- 32.Chonchol M. Neutrophil dysfunction and infection risk in end-stage renal disease. Semin Dial. 2006;19:291–296. doi: 10.1111/j.1525-139X.2006.00175.x. [DOI] [PubMed] [Google Scholar]
- 33.Ku CL, von BH, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–2422. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 35.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, Vasconcelos J, Sirvent N, Guedes M, Vitor AB, Herrero-Mata MJ, Arostegui JI, Rodrigo C, Alsina L, Ruiz-Ortiz E, Juan M, Fortuny C, Yague J, Anton J, Pascal M, Chang HH, Janniere L, Rose Y, Garty BZ, Chapel H, Issekutz A, Marodi L, Rodriguez-Gallego C, Banchereau J, Abel L, Li X, Chaussabel D, Puel A, Casanova JL. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picard C, von BH, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, Creech CB, Ku CL, Ehl S, Marodi L, Al-Muhsen S, Al-Hajjar S, Al-Ghonaium A, Day-Good NK, Holland SM, Gallin JI, Chapel H, Speert DP, Rodriguez-Gallego C, Colino E, Garty BZ, Roifman C, Hara T, Yoshikawa H, Nonoyama S, Domachowske J, Issekutz AC, Tang M, Smart J, Zitnik SE, Hoarau C, Kumararatne DS, Thrasher AJ, Davies EG, Bethune C, Sirvent N, de RD, Camcioglu Y, Vasconcelos J, Guedes M, Vitor AB, Rodrigo C, Almazan F, Mendez M, Arostegui JI, Alsina L, Fortuny C, Reichenbach J, Verbsky JW, Bossuyt X, Doffinger R, Abel L, Puel A, Casanova JL. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Med (Baltimore) 2010;89:403–425. doi: 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, Khatcherian A, Novitskaya I, Carucci JA, Bergman R, Krueger JG. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. 2008;181:7420–7427. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prendergast A, Prado JG, Kang YH, Chen F, Riddell LA, Luzzi G, Goulder P, Klenerman P. HIV-1 infection is characterized by profound depletion of CD161+ Th17 cells and gradual decline in regulatory T cells. AIDS. 2010;24:491–502. doi: 10.1097/QAD.0b013e3283344895. [DOI] [PubMed] [Google Scholar]
- 39.Al KS, Keles S, Garcia-Lloret M, Karakoc-Aydiner E, Reisli I, Artac H, Camcioglu Y, Cokugras H, Somer A, Kutukculer N, Yilmaz M, Ikinciogullari A, Yegin O, Yuksek M, Genel F, Kucukosmanoglu E, Baki A, Bahceciler NN, Rambhatla A, Nickerson DW, McGhee S, Barlan IB, Chatila T. Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. J Allergy Clin Immunol. 2009;124(342–8):348. doi: 10.1016/j.jaci.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renner ED, Rylaarsdam S, nover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H) 17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122:181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim MH, Granick JL, Kwok C, Walker NJ, Borjesson DL, Curry FR, Miller LS, Simon SI. Neutrophil survival and c-kit+-progenitor proliferation in Staphylococcus aureus-infected skin wounds promote resolution. Blood. 2011;117:3343–3352. doi: 10.1182/blood-2010-07-296970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18:521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 46.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 47.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 48.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 49.Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell Microbiol. 2006;8:1687–1696. doi: 10.1111/j.1462-5822.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 50.Postma B, Poppelier MJ, van Galen JC, Prossnitz ER, van Strijp JA, de Haas CJ, van Kessel KP. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J Immunol. 2004;172:6994–7001. doi: 10.4049/jimmunol.172.11.6994. [DOI] [PubMed] [Google Scholar]
- 51.Athanasopoulos AN, Economopoulou M, Orlova VV, Sobke A, Schneider D, Weber H, Augustin HG, Eming SA, Schubert U, Linn T, Nawroth PP, Hussain M, Hammes HP, Herrmann M, Preissner KT, Chavakis T. The extracellular adherence protein (Eap) of Staphylococcus aureus inhibits wound healing by interfering with host defense and repair mechanisms. Blood. 2006;107:2720–2727. doi: 10.1182/blood-2005-08-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 53.Rooijakkers SH, van Kessel KP, van Strijp JA. Staphylococcal innate immune evasion. Trends Microbiol. 2005;13:596–601. doi: 10.1016/j.tim.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, Deleo FR, Otto M. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 55.Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- 56.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karavolos MH, Horsburgh MJ, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology. 2003;149:2749–2758. doi: 10.1099/mic.0.26353-0. [DOI] [PubMed] [Google Scholar]
- 58.McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J Immunol. 2008;181:1323–1332. doi: 10.4049/jimmunol.181.2.1323. [DOI] [PubMed] [Google Scholar]
- 59.Palazzolo-Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, Skaar EP, Deleo FR. Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J Immunol. 2008;180:500–509. doi: 10.4049/jimmunol.180.1.500. [DOI] [PubMed] [Google Scholar]
- 60.Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol. 2009;124:R13–R18. doi: 10.1016/j.jaci.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 61.Otto M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev Dermatol. 2010;5:183–195. doi: 10.1586/edm.10.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cho JS, Xuan C, Miller LS. Lucky number seven: RNase 7 can prevent Staphylococcus aureus skin colonization. J Invest Dermatol. 2010;130:2703–2706. doi: 10.1038/jid.2010.294. [DOI] [PubMed] [Google Scholar]
- 63.Miller LS, Sorensen OE, Liu PT, Jalian HR, Eshtiaghpour D, Behmanesh BE, Chung W, Starner TD, Kim J, Sieling PA, Ganz T, Modlin RL. TGF-a regulates TLR expression and function on epidermal keratinocytes. J Immunol. 2005;174:6137–6143. doi: 10.4049/jimmunol.174.10.6137. [DOI] [PubMed] [Google Scholar]
- 64.Sorensen OE, Thapa DR, Roupe KM, Valore EV, Sjobring U, Roberts AA, Schmidtchen A, Ganz T. Injury-induced innate immune response in human skin mediated by transactivation of the epidermal growth factor receptor. J Clin Invest. 2006;116:1878–1885. doi: 10.1172/JCI28422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lehrer RI. Multispecific myeloid defensins. Curr Opin Hematol. 2007;14:16–21. doi: 10.1097/00062752-200701000-00005. [DOI] [PubMed] [Google Scholar]
- 66.Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human a-defensins. Antimicrob Agents Chemother. 2005;49:269–275. doi: 10.1128/AAC.49.1.269-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia JR, Krause A, Schulz S, Rodriguez-Jimenez FJ, Kluver E, Adermann K, Forssmann U, Frimpong-Boateng A, Bals R, Forssmann WG. Human b-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J. 2001;15:1819–1821. [PubMed] [Google Scholar]
- 68.Jann NJ, Schmaler M, Kristian SA, Radek KA, Gallo RL, Nizet V, Peschel A, Landmann R. Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trap-associated cathelicidin. J Leukoc Biol. 2009;86:1159–1169. doi: 10.1189/jlb.0209053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rieg S, Steffen H, Seeber S, Humeny A, Kalbacher H, Dietz K, Garbe C, Schittek B. Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol. 2005;174:8003–8010. doi: 10.4049/jimmunol.174.12.8003. [DOI] [PubMed] [Google Scholar]
- 70.Steffen H, Rieg S, Wiedemann I, Kalbacher H, Deeg M, Sahl HG, Peschel A, Gotz F, Garbe C, Schittek B. Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob Agents Chemother. 2006;50:2608–2620. doi: 10.1128/AAC.00181-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dinulos JG, Mentele L, Fredericks LP, Dale BA, Darmstadt GL. Keratinocyte expression of human beta-defensin 2 following bacterial infection: role in cutaneous host defense. Clin Diagn Lab Immunol. 2003;10:161–166. doi: 10.1128/CDLI.10.1.161-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menzies BE, Kenoyer A. Signal transduction and nuclear responses in Staphylococcus aureus-induced expression of human beta-defensin 3 in skin keratinocytes. Infect Immun. 2006;74:6847–6854. doi: 10.1128/IAI.00389-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sayama K, Komatsuzawa H, Yamasaki K, Shirakata Y, Hanakawa Y, Ouhara K, Tokumaru S, Dai X, Tohyama M, Ten DP, Sugai M, Ichijo H, Hashimoto K. New mechanisms of skin innate immunity: ASK1-mediated keratinocyte differentiation regulates the expression of beta-defensins, LL37, and TLR2. Eur J Immunol. 2005;35:1886–1895. doi: 10.1002/eji.200425827. [DOI] [PubMed] [Google Scholar]
- 74.Sumikawa Y, Asada H, Hoshino K, Azukizawa H, Katayama I, Akira S, Itami S. Induction of beta-defensin 3 in keratinocytes stimulated by bacterial lipopeptides through Toll-like receptor 2. Microbes Infect. 2006;8:1513–1521. doi: 10.1016/j.micinf.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 75.Zanger P, Holzer J, Schleucher R, Scherbaum H, Schittek B, Gabrysch S. Severity of Staphylococcus aureus infection of the skin is associated with inducibility of human beta-defensin 3 but not human beta-defensin 2. Infect Immun. 2010;78:3112–3117. doi: 10.1128/IAI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miller LS, Modlin RL. Human keratinocyte Toll-like receptors promote distinct immune responses. J Invest Dermatol. 2007;127:262–263. doi: 10.1038/sj.jid.5700559. [DOI] [PubMed] [Google Scholar]
- 77.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 78.Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, Helfrich YR, Kang S, Elalieh HZ, Steinmeyer A, Zugel U, Bikle DD, Modlin RL, Gallo RL. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117:803–811. doi: 10.1172/JCI30142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173:2909–2912. doi: 10.4049/jimmunol.173.5.2909. [DOI] [PubMed] [Google Scholar]
- 80.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 81.Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, Darst MA, Gao B, Boguniewicz M, Travers JB, Leung DY. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. 2003;171:3262–3269. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 82.Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J. Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J Immunol. 2007;179:3958–3965. doi: 10.4049/jimmunol.179.6.3958. [DOI] [PubMed] [Google Scholar]
- 83.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 84.Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol. 2010;184:6688–6694. doi: 10.4049/jimmunol.0903984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol. 2006;140:103–112. doi: 10.1159/000092305. [DOI] [PubMed] [Google Scholar]
- 87.Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol. 2004;172:1169–1176. doi: 10.4049/jimmunol.172.2.1169. [DOI] [PubMed] [Google Scholar]
- 88.Clarke SR, Mohamed R, Bian L, Routh AF, Kokai-Kun JF, Mond JJ, Tarkowski A, Foster SJ. The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe. 2007;1:199–212. doi: 10.1016/j.chom.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 89.Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48:4673–4679. doi: 10.1128/AAC.48.12.4673-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ernst CM, Staubitz P, Mishra NN, Yang SJ, Hornig G, Kalbacher H, Bayer AS, Kraus D, Peschel A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009;5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 93.Lai Y, Villaruz AE, Li M, Cha DJ, Sturdevant DE, Otto M. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol Microbiol. 2007;63:497–506. doi: 10.1111/j.1365-2958.2006.05540.x. [DOI] [PubMed] [Google Scholar]
- 94.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 95.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 96.Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, Kirikae T, Gotz F. Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol. 2006;177:3162–3169. doi: 10.4049/jimmunol.177.5.3162. [DOI] [PubMed] [Google Scholar]
- 97.Takeda K, Takeuchi O, Akira S. Recognition of lipopeptides by Toll-like receptors. J Endotoxin Res. 2002;8:459–463. doi: 10.1179/096805102125001073. [DOI] [PubMed] [Google Scholar]
- 98.Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, von AS, Hartung T, Lien E, Bakke O, Espevik T. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol. 2008;84:280–291. doi: 10.1189/jlb.0907656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Muller-Anstett MA, Muller P, Albrecht T, Nega M, Wagener J, Gao Q, Kaesler S, Schaller M, Biedermann T, Gotz F. Staphylococcal peptidoglycan co-localizes with NOD2 and TLR2 and activates innate immune response via both receptors in primary murine keratinocytes. PLoS One. 2010;5:e13153. doi: 10.1371/journal.pone.0013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ahmad-Nejad P, Mrabet-Dahbi S, Breuer K, Klotz M, Werfel T, Herz U, Heeg K, Neumaier M, Renz H. The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J Allergy Clin Immunol. 2004;113:565–567. doi: 10.1016/j.jaci.2003.12.583. [DOI] [PubMed] [Google Scholar]
- 102.Oh DY, Schumann RR, Hamann L, Neumann K, Worm M, Heine G. Association of the toll-like receptor 2 A-16934T promoter polymorphism with severe atopic dermatitis. Allergy. 2009;64:1608–1615. doi: 10.1111/j.1398-9995.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- 103.Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity. 2011;34:665–679. doi: 10.1016/j.immuni.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 104.Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17:59–65. doi: 10.1016/j.tim.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 105.Hruz P, Zinkernagel AS, Jenikova G, Botwin GJ, Hugot JP, Karin M, Nizet V, Eckmann L. NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc Natl Acad Sci USA. 2009;106:12873–12878. doi: 10.1073/pnas.0904958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dziarski R, Gupta D. Review: mammalian peptidoglycan recognition proteins (PGRPs) in innate immunity. Innate Immun. 2010;16:168–174. doi: 10.1177/1753425910366059. [DOI] [PubMed] [Google Scholar]
- 107.Cho JH, Fraser IP, Fukase K, Kusumoto S, Fujimoto Y, Stahl GL, Ezekowitz RA. Human peptidoglycan recognition protein S is an effector of neutrophil-mediated innate immunity. Blood. 2005;106:2551–2558. doi: 10.1182/blood-2005-02-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dziarski R, Platt KA, Gelius E, Steiner H, Gupta D. Defect in neutrophil killing and increased susceptibility to infection with nonpathogenic gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood. 2003;102:689–697. doi: 10.1182/blood-2002-12-3853. [DOI] [PubMed] [Google Scholar]
- 109.Wang ZM, Li X, Cocklin RR, Wang M, Wang M, Fukase K, Inamura S, Kusumoto S, Gupta D, Dziarski R. Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-L-alanine amidase. J Biol Chem. 2003;278:49044–49052. doi: 10.1074/jbc.M307758200. [DOI] [PubMed] [Google Scholar]
- 110.Xu M, Wang Z, Locksley RM. Innate immune responses in peptidoglycan recognition protein L-deficient mice. Mol Cell Biol. 2004;24:7949–7957. doi: 10.1128/MCB.24.18.7949-7957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gomez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, Cheung A, Prince A. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10:842–848. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 112.Classen A, Kalali BN, Schnopp C, Andres C, Aguilar-Pimentel JA, Ring J, Ollert M, Mempel M. TNF receptor I on human keratinocytes is a binding partner for staphylococcal protein A resulting in the activation of NF kappa B, AP-1, and downstream gene transcription. Exp Dermatol. 2011;20:48–52. doi: 10.1111/j.1600-0625.2010.01174.x. [DOI] [PubMed] [Google Scholar]
- 113.Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol. 2011;29:447–491. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- 114.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 115.Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O’Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–6942. doi: 10.4049/jimmunol.179.10.6933. [DOI] [PubMed] [Google Scholar]
- 116.Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Craven RR, Gao X, Allen IC, Gris D, Bubeck WJ, McElvania-Tekippe E, Ting JP, Duncan JA. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cho JS, Zussman J, Donegan NP, Ramos RI, Garcia NC, Uslan DZ, Iwakura Y, Simon SI, Cheung AL, Modlin RL, Kim J, Miller LS. Noninvasive in vivo imaging to evaluate immune responses and antimicrobial therapy against Staphylococcus aureus and USA300 MRSA skin infections. J Invest Dermatol. 2011;131:907–915. doi: 10.1038/jid.2010.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Olaru F, Jensen LE. Staphylococcus aureus stimulates neutrophil targeting chemokine expression in keratinocytes through an autocrine IL-1alpha signaling loop. J Invest Dermatol. 2010;130:1866–1876. doi: 10.1038/jid.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Holtfreter S, Kolata J, Broker BM. Towards the immune proteome of Staphylococcus aureus—the anti-S. aureus antibody response. Int J Med Microbiol. 2010;300:176–192. doi: 10.1016/j.ijmm.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 122.Kobayashi SD, Deleo FR. A MRSA-terious enemy among us: boosting MRSA vaccines. Nat Med. 2011;17:168–169. doi: 10.1038/nm0211-168. [DOI] [PubMed] [Google Scholar]
- 123.Shinefield H, Black S, Fattom A, Horwith G, Rasgon S, Ordonez J, Yeoh H, Law D, Robbins JB, Schneerson R, Muenz L, Fuller S, Johnson J, Fireman B, Alcorn H, Naso R. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2002;346:491–496. doi: 10.1056/NEJMoa011297. [DOI] [PubMed] [Google Scholar]
- 124.DeJonge M, Burchfield D, Bloom B, Duenas M, Walker W, Polak M, Jung E, Millard D, Schelonka R, Eyal F, Morris A, Kapik B, Roberson D, Kesler K, Patti J, Hetherington S. Clinical trial of safety and efficacy of INH-A21 for the prevention of nosocomial staphylococcal bloodstream infection in premature infants. J Pediatr. 2007;151:260–265. doi: 10.1016/j.jpeds.2007.04.060. [DOI] [PubMed] [Google Scholar]
- 125.Miller LG, Quan C, Shay A, Mostafaie K, Bharadwa K, Tan N, Matayoshi K, Cronin J, Tan J, Tagudar G, Bayer AS. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant and -susceptible Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483–492. doi: 10.1086/511041. [DOI] [PubMed] [Google Scholar]
- 126.Huang SS, Platt R. Risk of methicillin-resistant Staphylococcus aureus infection after previous infection or colonization. Clin Infect Dis. 2003;36:281–285. doi: 10.1086/345955. [DOI] [PubMed] [Google Scholar]
- 127.Nguyen DM, Mascola L, Brancoft E. Recurring methicillin-resistant Staphylococcus aureus infections in a football team. Emerg Infect Dis. 2005;11:526–532. doi: 10.3201/eid1104.041094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Deresinski S, Herrera V. Immunotherapies for Staphylococcus aureus: current challenges and future prospects. Infect Control Hosp Epidemiol. 2010;31(Suppl 1):S45–S47. doi: 10.1086/655992. [DOI] [PubMed] [Google Scholar]
- 129.Schaffer AC, Lee JC. Vaccination and passive immunisation against Staphylococcus aureus. Int J Antimicrob Agents. 2008;32(Suppl 1):S71–S78. doi: 10.1016/j.ijantimicag.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 130.Kennedy AD, Bubeck WJ, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, Deleo FR. Targeting of a-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis. 2010;202:1050–1058. doi: 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bubeck WJ, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med. 2008;205:287–294. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brown EL, Dumitrescu O, Thomas D, Badiou C, Koers EM, Choudhury P, Vazquez V, Etienne J, Lina G, Vandenesch F, Bowden MG. The Panton–Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin Microbiol Infect. 2009;15:156–164. doi: 10.1111/j.1469-0691.2008.02648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim HK, Cheng AG, Kim HY, Missiakas DM, Schneewind O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J Exp Med. 2010;207:1863–1870. doi: 10.1084/jem.20092514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Harro C, Betts R, Orenstein W, Kwak EJ, Greenberg HE, Onorato MT, Hartzel J, Lipka J, DiNubile MJ, Kartsonis N. Safety and immunogenicity of a novel Staphylococcus aureus vaccine: results from the first study of the vaccine dose range in humans. Clin Vaccine Immunol. 2010;17:1868–1874. doi: 10.1128/CVI.00356-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 2010;6:e1001036. doi: 10.1371/journal.ppat.1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mathews WC, Caperna JC, Barber RE, Torriani FJ, Miller LG, May S, McCutchan JA. Incidence of and risk factors for clinically significant methicillin-resistant Staphylococcus aureus infection in a cohort of HIV-infected adults. J Acquir Immune Defic Syndr. 2005;40:155–160. doi: 10.1097/01.qai.0000179464.40948.b9. [DOI] [PubMed] [Google Scholar]
- 138.Skiest D, Brown K, Hester J, Moore T, Crosby C, Mussa HR, Hoffman-Roberts H, Cooper T. Community-onset methicillin-resistant Staphylococcus aureus in an urban HIV clinic. HIV Med. 2006;7:361–368. doi: 10.1111/j.1468-1293.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- 139.Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168–172. doi: 10.1034/j.1600-0560.2002.290307.x. [DOI] [PubMed] [Google Scholar]
- 140.McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, Tzianabos AO, Lee JC. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc Natl Acad Sci USA. 2006;103:10408–10413. doi: 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE, Jr, Spellberg B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009;5:e1000703. doi: 10.1371/journal.ppat.1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gaudreau MC, Lacasse P, Talbot BG. Protective immune responses to a multi-gene DNA vaccine against Staphylococcus aureus. Vaccine. 2007;25:814–824. doi: 10.1016/j.vaccine.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 143.Zhao YX, Tarkowski A. Impact of interferon-gamma receptor deficiency on experimental Staphylococcus aureus septicemia and arthritis. J Immunol. 1995;155:5736–5742. [PubMed] [Google Scholar]
- 144.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 145.Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol. 2001;116:658–663. doi: 10.1046/j.0022-202x.2001.01331.x. [DOI] [PubMed] [Google Scholar]
- 146.Leung DY, Hauk P, Strickland I, Travers JB, Norris DA. The role of superantigens in human diseases: therapeutic implications for the treatment of skin diseases. Br J Dermatol. 1998;139(Suppl 53):17–29. doi: 10.1046/j.1365-2133.1998.1390s3017.x. [DOI] [PubMed] [Google Scholar]
- 147.Laouini D, Kawamoto S, Yalcindag A, Bryce P, Mizoguchi E, Oettgen H, Geha RS. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J Allergy Clin Immunol. 2003;112:981–987. doi: 10.1016/j.jaci.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 148.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 149.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 150.Peric M, Koglin S, Kim SM, Morizane S, Besch R, Prinz JC, Ruzicka T, Gallo RL, Schauber J. IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J Immunol. 2008;181:8504–8512. doi: 10.4049/jimmunol.181.12.8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 153.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 154.Holland SM, Deleo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 155.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, Chen A, Kim HS, Lloret MG, Schulze I, Ehl S, Thiel J, Pfeifer D, Veelken H, Niehues T, Siepermann K, Weinspach S, Reisli I, Keles S, Genel F, Kutukculer N, Camcioglu Y, Somer A, Karakoc-Aydiner E, Barlan I, Gennery A, Metin A, Degerliyurt A, Pietrogrande MC, Yeganeh M, Baz Z, Al-Tamemi S, Klein C, Puck JM, Holland SM, McCabe ER, Grimbacher B, Chatila TA. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–1302. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, Matthews HF, Davis J, Turner ML, Uzel G, Holland SM, Su HC. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cecchinato V, Franchini G. Th17 cells in pathogenic simian immunodeficiency virus infection of macaques. Curr Opin HIV AIDS. 2010;5:141–145. doi: 10.1097/COH.0b013e32833653ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, Mold J, Swainson L, Barbour JD, Baskin CR, Palermo R, Pandrea I, Miller CJ, Katze MG, McCune JM. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009;5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hayashida S, Uchi H, Moroi Y, Furue M. Decrease in circulating Th17 cells correlates with increased levels of CCL17, IgE and eosinophils in atopic dermatitis. J Dermatol Sci. 2011;61:180–186. doi: 10.1016/j.jdermsci.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 163.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, Ramon M, Bergman R, Krueger JG, Guttman-Yassky E. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–1252. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 165.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, Tsuge I, Karasuyama H. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206:1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, Cheng G, Modlin RL, Miller LS. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Girardi M. Immunosurveillance and immunoregulation by gammadelta T cells. J Invest Dermatol. 2006;126:25–31. doi: 10.1038/sj.jid.5700003. [DOI] [PubMed] [Google Scholar]
- 168.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187:490–500. doi: 10.4049/jimmunol.1100123. [DOI] [PMC free article] [PubMed] [Google Scholar]