

Abstract
Insulin resistance is associated with sleep apnoea, leading us to hypothesise that it is also associated with elevations in pharyngeal collapsibility, even in the absence of sleep apnoea.
90 bariatric patients were characterised for sleep apnoea, pharyngeal collapsibility and insulin resistance. Patients with a respiratory disturbance index (RDI) >10 events·h−1, diabetes mellitus, tonsillar hypertrophy and pulmonary disease were excluded. The remaining 14 females underwent collapsibility measurements (passive critical pressure, Pcritp) during non-rapid eye movement sleep. The homeostasis model assessment (HOMA) index, a measure of insulin resistance, was derived from measurements of fasting glucose and insulin levels, and compared to Pcritp.
Groups with high Pcritp compared to low Pcritp did not differ in age, body mass index or RDI. HOMA and insulin were elevated in the high Pcritp group compared to the low Pcritp group (p<0.02). Pcritp correlated with HOMA (Spearman’s ρ=0.565, 95% CI 0.104–0.862; p=0.035) and insulin (Spearman’s ρ=0.609 95% CI 0.196–0.835; p=0.021).
Obese insulin-resistant subjects without frank diabetes or sleep apnoea demonstrate preclinical elevations in pharyngeal collapsibility, which may increase their susceptibility to sleep apnoea. Our findings suggest that insulin resistance could play a significant role in sleep apnoea pathogenesis by generating requisite elevations in pharyngeal collapsibility.
Introduction
Obstructive sleep apnoea (OSA) is characterised by recurrent episodes of upper airway obstruction that result from increases in upper airway collapsibility during sleep [1]. Recent evidence suggests that increases in airway collapsibility are related to disturbances in pharyngeal structural and neuromuscular control [2]. Structural loads have been linked to obesity and central adiposity [3, 4] which are potent risk factors for OSA [5–7]. Nevertheless, mechanisms linking obesity with increased airway collapsibility remain poorly understood.
In addition, sleep apnoea and obesity have been linked to insulin resistance [8–12]. Insulin resistance has been associated with central adiposity and ectopic fat deposition in a broad array of skeletal muscles [13–16]. Similarly, sleep apnoea has been associated with central adiposity [17, 18] and excess fat deposits in upper airway structures including the tongue [19, 20], which can increase pharyngeal collapsibility and sleep apnoea susceptibility [19, 21, 22]. It is therefore possible that insulin resistance is associated with elevations in upper airway collapsibility in obesity, which could in turn predispose to OSA. However, in OSA patients, intermittent oxyhaemoglobin desaturations and sleep fragmentation can aggravate insulin resistance [23, 24] and thereby confound the relationship between insulin resistance and sleep apnoea susceptibility. Thus, it is not clear whether insulin resistance predisposes to sleep apnoea (by increasing upper airway collapsibility), or vice versa.
The major goal of the present study was to examine the link between insulin resistance and upper airway collapsibility. We hypothesised that insulin resistance is associated with elevations in pharyngeal collapsibility in obesity that is unrelated to concomitant OSA. To minimise potential confounding by sleep apnoea [11, 25], we examined this relationship in a select group of obese patients who were free of significant sleep apnoea by quantifying upper airway dysfunction with measurements of passive critical pressure (Pcritp) during sleep.
Methods
Participants
A cross-sectional study was conducted in 90 patients presenting to the Johns Hopkins Bayview Medical Center bariatric surgery clinic (Baltimore, MD, USA) between 2005 and 2012. Inclusion criteria included age >21 years and a body mass index (BMI) >35 kg·m−2. All volunteers were screened for sleep apnoea and were included if they had an overall respiratory disturbance index (RDI) of <10 events·h−1 (n=29). The remaining participants were excluded if they had asthma (diagnosed using pulmonary function tests and/or current use of bronchodilators; n=3), diabetes mellitus (defined by prior clinical diagnosis, use of hypoglycaemic agents or documented fasting glucose >126 mg·dL−1; n=2) or palatine tonsillar hypertrophy (grade ⩾II tonsils; n=5) [26]. One patient met two exclusion criteria (palatine tonsillar hypertrophy and diabetes mellitus). None of the remaining subjects were pregnant or had unstable cardiovascular disease, documented prior use of nasal continuous positive airway pressure (CPAP) within the past 3 months, moderate or severe chronic obstructive pulmonary disease (defined by Global Initiative for Chronic Obstructive Lung Disease criteria [27–29]), current use of systemic steroids, liver disease, history of HIV disease or use of home supplemental oxygen. Of the remaining 20 participants, Pcritp was successfully determined in 14 subjects [30], who comprised the final study group (figure 1). Those with and without Pcritp measurements did not differ in age (mean±SD 39.2±8.7 years versus 39.8±8.8 years, p=0.743) or BMI (mean±SD 47.5±4.3 kg·m−2 versus 43.6±4 kg·m−2, p=0.094). Informed consent was obtained for a protocol that was approved by the Johns Hopkins Medical Institutions institutional review board.
FIGURE 1.
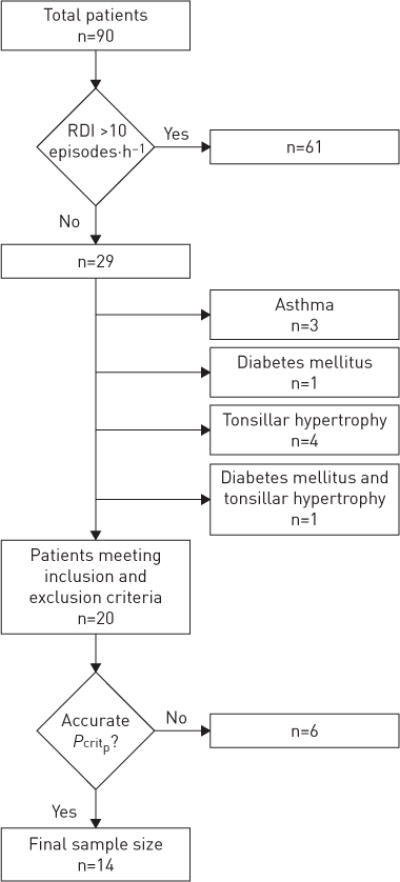
Flow chart illustrating how the final study sample was derived from bariatric patients, after excluding those with sleep apnoea, other medical conditions and loss of nocturnal data.
Study design
The relationship between measures of insulin resistance (fasting glucose and insulin, and the homeostasis model assessment (HOMA) index) was examined in the final patient sample. We also accounted for potential confounds of these relationships using measures of body anthropometry and sleep apnoea severity (RDI) in these patients.
Study procedures and protocols
Anthropometrics
Measurements of obesity and regional adiposity were performed, including weight, height and neck, waist and hip circumference, BMI and waist-to-hip ratio.
Baseline assessment of sleep and breathing patterns
A full-night sleep study was performed from 22:00 h to 06:00 h to characterise sleep and breathing patterns, as previously described [31]. In brief, digitised signals included left and right electro-oculogram, submental electromyogram, electroencephalogram, arterial oxygen saturation, nasal pressure, chest and abdominal plethysmography and video monitoring to determine body position throughout the night. Patients were instructed to sleep in the supine position whenever possible. Sleep staging, sleep disordered breathing episodes and arousals were scored according to standard criteria [31]. Hypopnoea was defined as a ⩾30% decrease in airflow for ⩾10 s that was accompanied by either an oxyhaemoglobin desaturation of 4% or arousal. The RDI was quantified for the entire night, and separately for rapid eye movement (REM) and non-REM (NREM) sleep. A fasting blood sample was drawn at 07:00 h after the baseline sleep study.
Assessing upper airway mechanical loads (Pcritp)
Within a month of the initial sleep study, subjects underwent another sleep study to assess mechanical loads from the upper airway pressure–flow relationship in NREM sleep using a validated protocol [30]. In brief, nasal pressure was elevated initially to abolish airflow obstruction completely during NREM sleep. The nasal pressure was then lowered abruptly for five breaths to characterise the level of maximal inspiratory airflow across a range of nasal pressures encompassing the pressure at which airflow fell to zero. Strict criteria were implemented to determine whether sufficient data were obtained to generate an accurate upper airway pressure–flow relationship with minimal extrapolation [30]. In 14 out of 20 eligible patients, these relationships were deemed adequate to derive the Pcritp as the nasal pressure at zero flow, a measure of pharyngeal mechanical loads [2, 30, 32].
Blood work
Fasting morning serum was analysed using ELISA to measure insulin (Linco Research Inc., St Charles, MO, USA) and glucose (in the Johns Hopkins Bayview Medical Center clinical laboratory). The HOMA index was calculated as fasting serum insulin (μU·mL−1) × fasting blood glucose (mmol·L−1)/22.5 [33]. The HOMA-IR provides a validated index of insulin resistance against euglycaemic hyperinsulinaemic clamp studies [34] in a broad range of research subjects.
Data analysis
All anthropometric, demographic and sleep study variables are presented as median (interquartile range) (table 1). Our analyses were designed to characterise associations between measures of insulin resistance and its major predictor, Pcritp, which was treated as both a dichotomous and continuous variable. Dichotomous comparisons were based on whether Pcritp fell above or below the median value of −1.98 cmH2O for the entire group. Nonparametric Mann–Whitney rank sum tests were used to compare parameters between groups with high and low Pcritp. Continuous comparisons of Pcritp with metabolic parameters were also examined using Spearman’s ρ (rank correlation coefficient). Significance was inferred for p<0.05. Analyses were conducted using SPSS (version 22; IBM, Armonk, NY, USA).
TABLE 1.
Demographic, anthropometries and steep disordered breathing parameters
| Entire group | Low Pcritp | High Pcritp | p-value | |
|---|---|---|---|---|
| Subjects | 14 | 7 | 7 | |
| Age years | 37.2 (34.0-43.2) | 42.3 (34.0-48.4) | 34.9 (34.0-43.2) | 0.710 |
| Anthropometries | ||||
| Weight kg | 127.8 (122.3-134.2) | 125.9 (122.3-128.3) | 132.7 (1 19.6-136.9) | 0.456 |
| BMI kg·m−2 | 48.6 (45.9-50.1) | 49.1 (44.0-51.3) | 48.1 (47.6-50.1) | 1 |
| Neck circumference cm | 39.7 (36.8-41.3) | 39.8 (37.0-42.2) | 39.5 (36.0-41.3) | 0.710 |
| Waist circumference# cm | 126.8 (120.8-134.8) | 129.7 (120.8-136.5) | 126.8 (1 12.0-132.7) | 0.534 |
| Hip circumference# cm | 143.8 (136.5-145.8) | 140.2 (135.0-145.5) | 144.3 (141.0-148.7) | 0.628 |
| Waist/hip ratio# | 0.91 (0.86-0.94) | 0.94 (0.91-0.96) | 0.87 (0.85-0.92) | 0.138 |
| Respiratory disturbance index events-h−1 | ||||
| NREM | 3.1 (1.6-4.5) | 2.3 (0.7-4.5) | 3.3 (3.0-5.2) | 0.165 |
| REM | 10.6 (7.5-22.9) | 22.9 (2.3-43.8) | 10.5 (7.5-10.6) | 0.259 |
| Total¶ | 4.4 (2.5-6.6) | 4.2 (2.3-9.5) | 4.5 (3.9-6.5) | 1 |
| Oxygen desaturation index (4% SpO2 drop)+ events·h−1 | ||||
| NREM | 1.7 (0.63-2.23) | 1.1 (0.44-2.51) | 1.9 (1.84-1.95) | 0.291 |
| REM | 1.8 (0.63-3.46) | 2.6 (0.54-5.94) | 1.2 (0.72-1.98) | 0.291 |
| Total¶ | 3.8 (2.03-5.50) | 3.8 (0.73-7.56) | 3.8 (2.58-3.95) | 0.808 |
| Peripheral capillary oxyhaemoglobin saturation % | ||||
| Baseline SpO2 | ||||
| NREM | 96.0 (94.8-96.8) | 95.6 (94.5-96.2) | 96.6 (94.8-97.7) | 0.209 |
| REM | 95.8 (95.3-96.6) | 95.6 (95.1-95.8) | 96.5 (95.8-96.7) | 0.128 |
| Total¶ | 95.8 (95.2-96.9) | 95.7 (95.2-95.8) | 96.6 (95.1-97.1) | 0.259 |
| Average low SpO2 | ||||
| NREM | 91.6 (90.2-93.0) | 91.0 (90.2-91.6) | 92.9 (90.1-93.6) | 0.366 |
| REM | 91.3 (90.5-91.8) | 90.7 (88.8-91.3) | 91.5 (90.5-92.7) | 0.128 |
| Total¶ | 91.4 (90.1-92.7) | 90.8 (89.2-91.4) | 92.5 (90.3-93.0) | 0.209 |
| Pcritp cmH2O | −1.98 (−5.22-−1.00) | −5.22 (−8.00-−3.01) | −1.00 (−1.55-1.30) | 0.001 |
Data are presented as n or median (interquartile range], unless otherwise stated. Pcritp: passive critical pressure; BMI: body mass index; NREM: non-rapid eye movement (REM); SpO2: peripheral capillary oxyhaemoglobin saturation.
: one patient had no waist and hip measurements;
: NREM and REM values combined;
: oxygen desaturation indices could not be calculated in two patients.
Results
Patient characteristics
Demographic and anthropometric characteristics are shown for the 14 patients in table 1 for the entire group and for those with high and low Pcritp. All subjects were severely obese females with a median BMI of 48.1 kg·m−2. Four were African-American, nine were Caucasian, and one was Latino. Anthropometric and demographic parameters did not differ significantly between groups with high compared to low passive Pcritp. Specifically, these groups did not differ significantly in the degree of adiposity (BMI) or in measures of regional fat distribution (waist to hip ratio, and neck and waist circumferences), although contrary to expectations [21], those with low passive Pcritp were marginally, but not significantly, older (p=0.710), and had somewhat greater degrees of central adiposity (higher waist-to-hip ratio, p=0.138).
By design, patients with significant sleep apnoea (a total RDI ⩾10 events·h−1) were excluded from the study. When comparing RDI between groups with low and high Pcritp (table 1), we found no significant differences in NREM RDI, REM RDI or total RDI. Furthermore, four patients in the low Pcritp group and five patients in the high Pcritp group had a total RDI of <5 events·h−1. The baseline peripheral capillary oxyhaemoglobin saturation (SpO2) and the average low SpO2 during NREM, REM and total sleep were not significantly different between groups.
Upper airway function during sleep and metabolic disturbances
Comparing low and high Pcritp groups (figure 2), we found significantly increased insulin levels in those with higher compared to lower Pcritp (p=0.011), whereas no significant difference in fasting glucose was observed (p=0.902). Similarly, HOMA was elevated in the group with a high Pcritp compared to low Pcritp (p=0.017). Of note, no significant differences in anthropometric, demographic or sleep apnoea characteristics were found between groups with high and low HOMA values (table 2).
FIGURE 2.
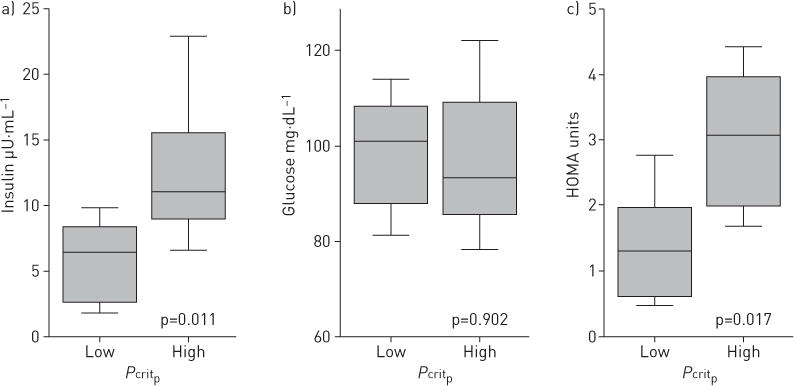
Fasting a) insulin and b) glucose and c) homeostasis model assessment (HOMA) in groups with high and low passive critical pressure (Pcritp) during non-rapid eye movement sleep. Significant elevations in insulin and HOMA were observed in the high Pcritp group versus the low Pcritp group.
TABLE 2.
Passive critical pressure (Pcritp) correlations with indices of glucose homeostasis
| Low HOMA | High HOMA | p-value | |
|---|---|---|---|
| Subjects | 7 | 7 | |
| Age years | 37.9 (34.6-42.3) | 34.9 (29.6-50.2) | 0.85 |
| Anthropometrics | |||
| Weight kg | 277 (263.0-281.7) | 292 (271.5-301.2) | 0.23 |
| BMI kg·m−2 | 48.1 (44.0-49.1) | 49.7 (47.6-51.6) | 0.34 |
| Neck circumference cm | 40 (36.5-42.2) | 39.2 (36.8-41.3) | 0.57 |
| Waist circumference# cm | 129.5 (120.8-136.5) | 126.8 (118.2-132.7) | 0.67 |
| Hip circumference# cm | 141.2 (135.0-143.8) | 145.8 (136.5-150) | 0.15 |
| Waist-to-hip ratio# | 0.94 (0.88-0.96) | 0.87 (0.85-0.92) | 0.12 |
| Respiratory disturbance index events·h−1 | |||
| NREM | 2.3 (0.7-3.3) | 4.5 (3.0-5.2) | 0.05 |
| REM | 7.7 (2.3-22.9) | 10.6 (10.5-23.4) | 0.28 |
| Total¶ | 3.9 (2.3-4.5) | 6.5 (4.0-9.5) | 0.06 |
| Peripheral capillary oxyhaemoglobin saturation % | |||
| Baseline SpO2 | |||
| NREM | 95.6 (94.5-97.7) | 96.2 (94.8-96.6) | 0.85 |
| REM | 95.8 (95.1-97.3) | 95.8 (95.6-96.6) | 0.95 |
| Total¶ | 95.8 (95.2-97.1) | 95.8 (95.1-96.6) | 0.61 |
| Average low SpO2 | |||
| NREM | 92.3 (90.2-94.6) | 91.6 (90.1-92.9) | 0.48 |
| REM | 91.3 (90.0-92.7) | 91.3 (90.5-91.8) | 0.85 |
| Total¶ | 91.3 (90.1-94.2) | 91.4 (89.8-92.5) | 0.66 |
Data are presented as n or median (interquartile range), unless otherwise stated. HOMA: homeostasis model assessment; BMI: body mass index; NREM: non-rapid eye movement (REM); SpO2: peripheral capillary oxyhaemoglobin saturation.
: one patient had no waist and hip measurements;
: NREM and REM values combined.
In addition, we observed that Pcritp was positively correlated with HOMA score (Spearman’s ρ=0.565, 95% CI 0.104–0.862; p=0.035) and with insulin serum level (Spearman’s ρ=0.609, 95% CI 0.196–0.835; p=0.021) (table 3 and figure 3). In contrast, Pcritp did not correlate with fasting glucose level. Pcritp did not correlate significantly with age, neck circumference, weight or BMI.
TABLE 3.
Passive critical pressure (Pcritp) and homeostasis model assessment (HOMA) correlations with body anthropometrics and demographics
| Parameters | Spearman’s ρ (95% CI) | p-value |
|---|---|---|
| Pcritp | ||
| Glucose homeostasis | ||
| Glucose | −0.145 (−0.720-0.584) | 0.620 |
| Insulin | 0.609 (0.196-0.835) | 0.021 |
| HOMA | 0.565 (0.104-0.862) | 0.035 |
| Anthropometrics/demographics | ||
| Age | 0.068 (−0.665-0.673) | 0.817 |
| Weight | 0.235 (−0.510-0.773) | 0.418 |
| BMI | −0.037 (−0.676-0.609) | 0.899 |
| Neck circumference | −0.134 (−0.668-0.451) | 0.648 |
| HOMA | ||
| Anthropometrics | ||
| Weight | 0.385 (−0.192-0.862) | 0.175 |
| BMI | 0.297 (−0.340-0.806) | 0.303 |
| Neck circumference | −0.103 (−0.805-0.646) | 0.725 |
| Waist circumference | −0.022 (−0.594-0.695) | 0.943 |
| Hip circumference | 0.489 (0.021-0.731) | 0.090 |
| Waist-to-hip ratio | −0.440 (−0.835-0.362) | 0.133 |
BMI: body mass index.
FIGURE 3.
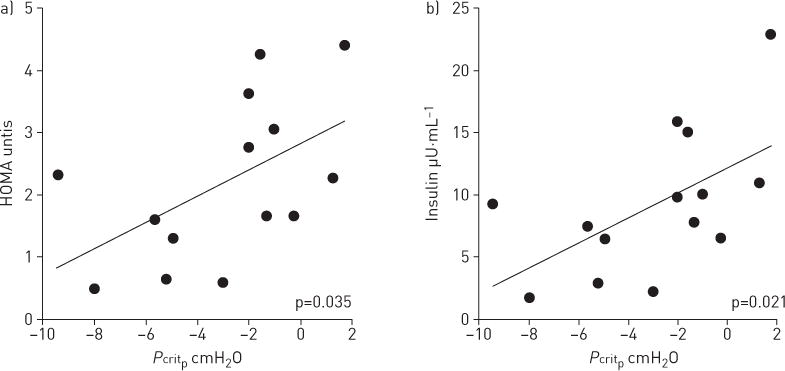
Scatter plots of insulin resistance metrics versus passive critical pressure (Pcritp). a) Homeostasis model assessment (HOMA); b) insulin.
HOMA did not correlate significantly with BMI (Spearman’s ρ=0.297, 95% CI −0.340–0.806; p=0.303), weight (Spearman’s ρ=0.385, 95% CI −0.192–0.862; p=0.175), neck circumference (Spearman’s ρ=−0.103, 95% CI −0.805–0.646; p=0.725), waist (Spearman’s ρ=−0.022, 95% CI −0.594–0.695; p=0.943), hip (Spearman’s ρ=0.489, 95% CI 0.021–0.731; p=0.090) or waist-to-hip ratio (Spearman’s ρ=−0.440, 95% CI −0.835–0.362; p=0.133).
Discussion
The aim of our study was to analyse the association between insulin resistance and upper airway collapsibility in a well-defined sample of obese individuals without overt diabetes or sleep apnoea. Our major finding was that insulin resistance was greater in the group with high compared to low Pcritp, and correlated directly with Pcritp, independently of demographic and anthropometric characteristics as well as RDI. These findings suggest that insulin resistance could confer underlying defects in pharyngeal collapsibility during sleep and increase sleep apnoea susceptibility in obese individuals.
Obesity imposes mechanical loads on the airway that increase its collapsibility (Pcritp) during sleep [2, 35]. As airflow obstruction ensues, active neuromuscular responses are recruited that can mitigate the obstruction and prevent the development of OSA [2, 36]. Current evidence suggests that elevations in mechanical loads and reductions in neuromuscular responses are both required for sleep apnoea pathogenesis [2, 37]. Other factors, including ventilatory demand, controller gain and arousal threshold can also modulate sleep apnoea severity [38]. These findings suggest that despite severe obesity, our subjects were protected from significant sleep apnoea for one of two reasons. In those with a relatively low Pcritp, structural stability of the pharynx prevented airway collapse during sleep. In those with a high Pcritp, compensatory neuromuscular responses would be required to offset mechanical loads and maintain airway patency during sleep. Thus, elevations in Pcritp place subjects at risk of OSA, which will ensue when neuromuscular responses are also lacking.
Two mechanisms can account for the observed associations between insulin resistance and upper airway collapsibility (figure 4). First, OSA and accompanying oxyhaemoglobin desaturations and arousals can exacerbate insulin resistance [23, 24, 39] (figure 4, arrow 1), which decreases with CPAP therapy.
FIGURE 4.
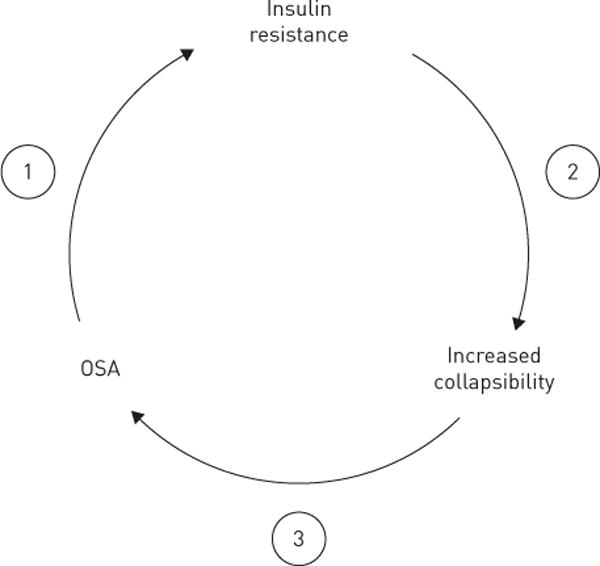
Putative pathogenic mechanisms leading to increased upper airway collapsibility and obstructive sleep apnoea (OSA) in obese insulin-resistant patients. OSA can worsen insulin resistance (arrow 1) and underlying defects in pharyngeal collapsibility (arrow 2), leading to further increases in severity of OSA (arrow 3).
Acute metabolic consequences of apnoeic activity can explain the high prevalence of sleep apnoea in type 2 diabetics [40]. However, even in the absence of sleep apnoea we found that insulin resistance was associated with underlying defects in upper airway mechanical function. This finding in a select group of obese nondiabetic, nonapnoeic subjects suggests that insulin resistance can predispose to upper airway obstruction during sleep (figure 4, arrow 2), thereby contributing to OSA pathogenesis. Its effect on upper airway collapsibility may be related to ectopic fat deposition, which has been demonstrated in heart [41], abdominal viscera [42] and skeletal muscle [13]. Ectopic fat accumulation in the tongue and peripharyngeal tissues in sleep apnoea patients [14, 19] can also account for observed elevations in upper airway collapsibility, and increase sleep apnoea susceptibility (figure 4, arrow 3).
In addition to ectopic fat deposits in upper airway structures, insulin resistance has been linked with central adiposity. Obesity and central adiposity have been associated with decreases in end-expiratory lung volume [43, 44], known to increase pharyngeal collapsibility during sleep [22]. Taken together, these findings suggest that in addition to obesity, insulin resistance may play a significant role in the pathogenesis of OSA by generating requisite elevations in pharyngeal collapsibility. Once sleep apnoea develops, insulin resistance can further increase upper airway collapsibility and perpetuate a vicious cycle of sleep apnoea progression.
Several limitations should be considered when reviewing our results. First, our inferences are based on a small sample of obese females, potentially limiting the generalizability of our findings. The exclusion of males from our sample was to be expected, given that patients with sleep apnoea were strictly excluded and that sleep apnoea is so highly prevalent in obese males. As noted, we sought to minimise comorbidities, including sleep apnoea, which could have confounded our assessment of metabolic determinants of upper airway function. Second, we acknowledge that we studied a convenience sample of patients presenting to a bariatric clinic, which may not accurately reflect characteristics of a community-based obese population. Nonetheless, our sample reflects the spectrum of obese patients, in whom differences in metabolic function may confer either protection or susceptibility to sleep apnoea. Third, although no anthropometric differences in obesity and regional adiposity were observed between groups, it is still possible that residual confounding by differences in body anthropometry exists, and/or that anthropometric differences would emerge with a greater sample size. It is also possible that imaging modalities, which offer greater precision in delineating regional fat, could have revealed subtle differences in fat content and distribution to account for variability in Pcritp. Fourth, we recognise that our study design was cross-sectional in nature, which precludes us from implicating causal mechanisms linking metabolic and upper airway dysfunction. Specifically, it is possible that visceral adiposity could lead to elevations in both HOMA-insulin resistance and Pcritp. If so, we would expect that these parameters would correlate with measures of regional adiposity, which was not the case. Fifth, it is possible that those with elevations in airway collapsibility (high Pcritp) were protected from developing sleep apnoea by active neuromuscular responses that preserved airway patency during sleep. Nevertheless, challenges in assessing these active responses experimentally precluded us from addressing this possibility. Sixth, we acknowledge that low levels of REM apnoea were present, which could account for differences in HOMA-insulin resistance between low and high Pcritp groups. Paradoxically, the amount of REM apnoea was greater in the low compared to the high Pcritp group, which would have only served to attenuate observed differences in HOMA-insulin resistance between these groups [11, 12, 39]. Finally, elevations in Pcritp could be related to disturbances in ventilatory control in insulin-resistant patients [45, 46], which could further reduce neuromuscular tone in the hypotonic (passive) pharyngeal airway. Nevertheless, Pcritp has been commonly used to integrate anatomical impediments to the maintenance of pharyngeal patency during sleep [38], and further work will be required to determine definitively whether insulin resistance confers further disturbances in pharyngeal neuromuscular control during sleep.
Our findings have important implications for clinical practice and research. First, the findings suggest that impaired glucose homeostasis, even in the absence of frank diabetes mellitus is a risk factor for OSA. They also imply that insulin resistance may play a causal role in sleep apnoea pathogenesis by generating requisite elevations in pharyngeal collapsibility. As sleep apnoea ensues, concomitant increases in insulin resistance could further increase upper airway collapsibility, perpetuating a vicious cycle of sleep apnoea progression (figure 4). Second, it is possible that measures which reduce insulin resistance could also lower pharyngeal collapsibility during sleep. Specifically, agents that target ectopic fat deposition in insulin-resistant patients may ultimately reverse defects in upper airway mechanical function and improve OSA.
Acknowledgments
National Institutes of Health: R01 HL50381, HL128970 and HL080105. Funding information for this article has been deposited with FundRef.
Footnotes
Conflict of interest: None declared.
References
- 1.Remmers JE, deGroot WJ, Sauerland EK, et al. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol. 1978;44:931–938. doi: 10.1152/jappl.1978.44.6.931. [DOI] [PubMed] [Google Scholar]
- 2.Patil SP, Schneider H, Marx JJ, et al. Neuromechanical control of upper airway patency during sleep. J Appl Physiol. 2007;102:547–556. doi: 10.1152/japplphysiol.00282.2006. [DOI] [PubMed] [Google Scholar]
- 3.Gleadhill IC, Schwartz AR, Schubert N, et al. Upper airway collapsibility in snorers and in patients with obstructive hypopnea and apnea. Am Rev Respir Dis. 1991;143:1300–1303. doi: 10.1164/ajrccm/143.6.1300. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz AR, Gold AR, Schubert N, et al. Effect of weight loss on upper airway collapsibility in obstructive sleep apnea. Am Rev Respir Dis. 1991;144:494–498. doi: 10.1164/ajrccm/144.3_Pt_1.494. [DOI] [PubMed] [Google Scholar]
- 5.Horner RL, Mohiaddin RH, Lowell DG, et al. Sites and sizes of fat deposits around the pharynx in obese patients with obstructive sleep apnoea and weight matched controls. Eur Respir J. 1989;2:613–622. [PubMed] [Google Scholar]
- 6.Millman RP, Carlisle CC, McGarvey ST, et al. Body fat distribution and sleep apnea severity in women. Chest. 1995;107:362–366. doi: 10.1378/chest.107.2.362. [DOI] [PubMed] [Google Scholar]
- 7.Whittle AT, Marshall I, Mortimore IL, et al. Neck soft tissue and fat distribution: comparison between normal men and women by magnetic resonance imaging. Thorax. 1999;54:323–328. doi: 10.1136/thx.54.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pallayova M, Steele KE, Magnuson TH, et al. Sleep apnea predicts distinct alterations in glucose homeostasis and biomarkers in obese adults with normal and impaired glucose metabolism. Cardiovasc Diabetol. 2010;9:83. doi: 10.1186/1475-2840-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polotsky VY, Patil SP, Savransky V, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med. 2009;179:228–234. doi: 10.1164/rccm.200804-608OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster GD, Borradaile KE, Sanders MH, et al. A randomized study on the effect of weight loss on obstructive sleep apnea among obese patients with type 2 diabetes: the Sleep AHEAD study. Arch Intern Med. 2009;169:1619–1626. doi: 10.1001/archinternmed.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harsch IA, Schahin SP, Brückner K, et al. The effect of continuous positive airway pressure treatment on insulin sensitivity in patients with obstructive sleep apnoea syndrome and type 2 diabetes. Respiration. 2004;71:252–259. doi: 10.1159/000077423. [DOI] [PubMed] [Google Scholar]
- 12.Punjabi NM, Ahmed MM, Polotsky VY, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance. Respir Physiol Neurobiol. 2003;136:167–178. doi: 10.1016/s1569-9048(03)00079-x. [DOI] [PubMed] [Google Scholar]
- 13.Miljkovic I, Cauley JA, Wang PY, et al. Abdominal myosteatosis is independently associated with hyperinsulinemia and insulin resistance among older men without diabetes. Obesity. 2013;21:2118–2125. doi: 10.1002/oby.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snel M, Jonker JT, Schoones J, et al. Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. Int J Endocrinol. 2012;2012:983814. doi: 10.1155/2012/983814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley DE, Slasky BS, Janosky J. Skeletal muscle density: effects of obesity and non-insulin-dependent diabetes mellitus. Am J Clin Nutr. 1991;54:509–515. doi: 10.1093/ajcn/54.3.509. [DOI] [PubMed] [Google Scholar]
- 16.Albu JB, Kovera AJ, Allen L, et al. Independent association of insulin resistance with larger amounts of intermuscular adipose tissue and a greater acute insulin response to glucose in African American than in white nondiabetic women. Am J Clin Nutr. 2005;82:1210–1217. doi: 10.1093/ajcn/82.6.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157:1746–1752. [PubMed] [Google Scholar]
- 18.Simpson L, Mukherjee S, Cooper MN, et al. Sex differences in the association of regional fat distribution with the severity of obstructive sleep apnea. Sleep. 2010;33:467–474. doi: 10.1093/sleep/33.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nashi N, Kang S, Barkdull GC, et al. Lingual fat at autopsy. Laryngoscope. 2007;117:1467–1473. doi: 10.1097/MLG.0b013e318068b566. [DOI] [PubMed] [Google Scholar]
- 20.Schwab RJ, Pasirstein M, Pierson R, et al. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Respir Crit Care Med. 2003;168:522–530. doi: 10.1164/rccm.200208-866OC. [DOI] [PubMed] [Google Scholar]
- 21.Kirkness JP, Schwartz AR, Schneider H, et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol. 2008;104:1618–1624. doi: 10.1152/japplphysiol.00045.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Squier SB, Patil SP, Schneider H, et al. Effect of end-expiratory lung volume on upper airway collapsibility in sleeping men and women. J Appl Physiol. 2010;109:977–985. doi: 10.1152/japplphysiol.00080.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis M, Punjabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol. 2009;106:1538–1544. doi: 10.1152/japplphysiol.91523.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamatakis KA, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest. 2010;137:95–101. doi: 10.1378/chest.09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schahin SP, Nechanitzky T, Dittel C, et al. Long-term improvement of insulin sensitivity during CPAP therapy in the obstructive sleep apnoea syndrome. Med Sci Monit. 2008;14:CR117–CR121. [PubMed] [Google Scholar]
- 26.Friedman M, Tanyeri H, La Rosa M, et al. Clinical predictors of obstructive sleep apnea. Laryngoscope. 1999;109:1901–1907. doi: 10.1097/00005537-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 28.Enright PL. GOLD stage 1 is crying wolf. Thorax. 2009;64:182–183. [PubMed] [Google Scholar]
- 29.Swanney MP, Ruppel G, Enright PL, et al. Using the lower limit of normal for the FEV1/FVC ratio reduces the misclassification of airway obstruction. Thorax. 2008;63:1046–1051. doi: 10.1136/thx.2008.098483. [DOI] [PubMed] [Google Scholar]
- 30.Patil SP, Punjabi NM, Schneider H, et al. A simplified method for measuring critical pressures during sleep in the clinical setting. Am J Respir Crit Care Med. 2004;170:86–93. doi: 10.1164/rccm.200309-1239OC. [DOI] [PubMed] [Google Scholar]
- 31.Iber C, Ancoli-Israel S, Chesson AL, et al. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. Westchester: American Academy of Sleep Medicine; 2007. [Google Scholar]
- 32.Schwartz AR, O’Donnell CP, Baron J, et al. The hypotonic upper airway in obstructive sleep apnea: role of structures and neuromuscular activity. Am J Respir Crit Care Med. 1998;157:1051–1057. doi: 10.1164/ajrccm.157.4.9706067. [DOI] [PubMed] [Google Scholar]
- 33.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 34.Bonora E, Targher G, Alberiche M, et al. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care. 2000;23:57–63. doi: 10.2337/diacare.23.1.57. [DOI] [PubMed] [Google Scholar]
- 35.Wellman A, Jordan AS, Malhotra A, et al. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170:1225–1232. doi: 10.1164/rccm.200404-510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin CH, Kirkness JP, Patil SP, et al. Compensatory responses to upper airway obstruction in obese apneic men and women. J Appl Physiol. 2012;112:403–410. doi: 10.1152/japplphysiol.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King ED, O’Donnell CP, Smith PL, et al. A model of obstructive sleep apnea in normal humans. Role of the upper airway. Am J Respir Crit Care Med. 2000;161:1979–1984. doi: 10.1164/ajrccm.161.6.9904096. [DOI] [PubMed] [Google Scholar]
- 38.Eckert DJ, White DP, Jordan AS, et al. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188:996–1004. doi: 10.1164/rccm.201303-0448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tasali E, Mokhlesi B, Van Cauter E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest. 2008;133:496–506. doi: 10.1378/chest.07-0828. [DOI] [PubMed] [Google Scholar]
- 40.Kuna ST, Reboussin DM, Borradaile KE, et al. Long-term effect of weight loss on obstructive sleep apnea severity in obese patients with type 2 diabetes. Sleep. 2013;36:641–649. doi: 10.5665/sleep.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sironi AM, Petz R, De Marchi D, et al. Impact of increased visceral and cardiac fat on cardiometabolic risk and disease. Diabet Med. 2012;29:622–627. doi: 10.1111/j.1464-5491.2011.03503.x. [DOI] [PubMed] [Google Scholar]
- 42.Jouët P, Sabaté JM, Maillard D, et al. Relationship between obstructive sleep apnea and liver abnormalities in morbidly obese patients: a prospective study. Obes Surg. 2007;17:478–485. doi: 10.1007/s11695-007-9085-3. [DOI] [PubMed] [Google Scholar]
- 43.Pelosi P, Croci M, Ravagnan I, et al. The effects of body mass on lung volumes, respiratory mechanics, and gas exchange during general anesthesia. Anesth Analg. 1998;87:654–660. doi: 10.1097/00000539-199809000-00031. [DOI] [PubMed] [Google Scholar]
- 44.Babb TG, Wyrick BL, DeLorey DS, et al. Fat distribution and end-expiratory lung volume in lean and obese men and women. Chest. 2008;134:704–711. doi: 10.1378/chest.07-1728. [DOI] [PubMed] [Google Scholar]
- 45.Polotsky VY, Wilson JA, Haines AS, et al. The impact of insulin-dependent diabetes on ventilatory control in the mouse. Am J Respir Crit Care Med. 2001;163:624–632. doi: 10.1164/ajrccm.163.3.2007120. [DOI] [PubMed] [Google Scholar]
- 46.Montserrat JM, Cochrane GM, Wolf C, et al. Ventilatory control in diabetes mellitus. Eur J Respir Dis. 1985;67:112–117. [PubMed] [Google Scholar]