

Study of gene expression has been arguably the most active research field in functional genomics. Over the last two decades, various high-throughput technologies, from gene expression microarray to RNA-seq, have been widely applied to the whole-genome profiling of gene expression. The commonality of these experiments is that they measure the gene expression levels of “bulk” sample, which pools a large number (often in the scale of millions) of cells, and thus the measurements reflect the average expression of a population of cells.
The recently developed single-cell RNA sequencing technology (scRNA-seq) allows the transcriptomic profiling at the single-cell level (Tang et al. 2009). Compared with the bulk experiments, scRNA-seq provides important information for inter-cellular transcriptomic heterogeneity, adding another dimension to understand gene expression regulation and dynamics. The technology has gained considerable interests recently, and a number of experiments have been performed to study highly heterogeneous samples such as cancer and brain (Patel et al. 2014, Zeisel et al. 2015).
One major goal of scRNA-seq experiment is to characterize the heterogeneity of gene expression among cells, and then relate that to phenotypic variation. So far, the scRNA-seq data analyses have been mainly focused on cell clustering (Bendall et al. 2014, Trapnell et al. 2014) and differential expression (Kharchenko et al. 2014). However, since genes function through a complex biological system, another important aspect of gene expression analysis is to reconstruct and detect changes in the gene networks (Hase et al. 2013, Siegenthaler and Gunawan 2014). Differential network analysis can reveal biological responses to stimuli through the re-wiring of biological network (Ideker and Krogan 2012). So far, the network analysis of scRNA-seq data has not been fully explored.
Traditionally, from bulk expression data, the network construction and comparison are based on repeated measure of expression such as time courses or population studies. Studying differential network using population data is based on between-person correlation patterns, which may not agree with the within-person dynamics of genes, especially when the population is heterogeneous, such as cancer. This is similar to the situation in genetic association study, where the association could be masked by sample heterogeneity (Wills et al. 2013). Using scRNA-seq data, existing methods can be applied to study gene networks. Since the cellular heterogeneity is no longer an issue, other confounding factors (such as age and demographics) from the population studies are mostly removed, the gene network constructed from scRNA-seq data can potentially provide cleaner signals and more biologically plausible results compared with that from the bulk expression data.
In this work, we conducted proof-of-concept analyses of scRNA-seq data to construct and compare gene networks from distinct biological conditions. We performed analyses on two datasets: one for human brain cancer and the other for mouse embryonic stem cell differentiation. We found that results from both studies are biologically meaningful: genes showing different levels of connectivity on the network are related to the phenotypes of interest. Overall, these results show that constructing and comparing gene network are very promising directions for scRNA-seq data analysis, and add another dimension to the understanding of inter-cellular gene expression dynamics.
For gene network construction, we assumed the input data is an N by p matrix for expression values from N cells and p genes denoted by X. The gene expression values could be sequence read counts or normalized counts such as RPKM (Reads Per Kilobase of transcript per Million mapped reads). Due to technical limitation, scRNA-seq data often contains unusually high number of genes with zero expression values, which could be from undetectable low expression or are missing due to technical error. In our analysis, the first step is to discard these genes because they undermine the signal to noise ratio. In the results presented, we set a “sparsity threshold” at 40%, meaning that genes with over 40% cells having expression level 0 will be discarded.
We followed the approach for single cell gene differential connectivity test. The approach was originally developed for gene expression microarray data (Gill et al. 2010). Assume X1 and X2 are scRNA-seq data from two groups. For each group, we first constructed a pair-wise gene connectivity matrix using Spearman's rank correlation for robustness. After this step, we had two p by p connectivity matrices and , where Ski is the connectivity score between gene k and i. Based on connectivity matrices, we applied the method developed in (Gill et al. 2010) to perform the differential connectivity test. First, mean absolute distance (MDA) statistics is defined to measure the mean difference in connectivity of the kth gene between G1 and G2:
P-value is calculated using the following permutation approach. X1 and X2 are pooled together to have the pooled data X(n+m)×p. The rows of X are then permuted, and the first n rows are deemed from condition 1, and the remaining m rows are deemed from condition 2. The permutated MDA statistics are then calculated from these data. Repeat this procedure for P times and the permutation p-value is calculated as:
where I() is identity function and P is set to be 1000. At last, we control the false discovery rate (FDR) (Benjamini 1995). Given a significance threshold, differentially connected genes (DCGs) can be selected.
Here, we present the results from the scRNA-seq data from a human brain cancer study (Patel et al. 2014). It focuses on the intratumoral diversity in primary glioblastoma (GBM), where the heterogeneity and the associated redundant signals in GBM often make the traditional therapies ineffective. The major goal of the study is to characterize the cellular-level gene expression variation in GBM. The study includes expression data from 430 single cells isolated from five GBM patients (MGH26, MGH28-31) and 102 single cells from two gliomasphere cell lines (GBM6, GBM8). The data are obtained from GEO (GSE57872).
After preprocessing to remove genes with excessive zero expression values, 5939 genes are retained for differential connectivity test. We first explored the distribution of the Spearman rank correlation among gene expression from all samples (five patients and two cell lines). Fig. 1A shows the marginal distribution of the correlation. It is clear that the correlation among five GBM patients are stochastically weaker than those from two cell lines (density curves are more concentrated at zero), which is reasonable since one would expect greater heterogeneity from primary cancer samples.
Fig. 1.
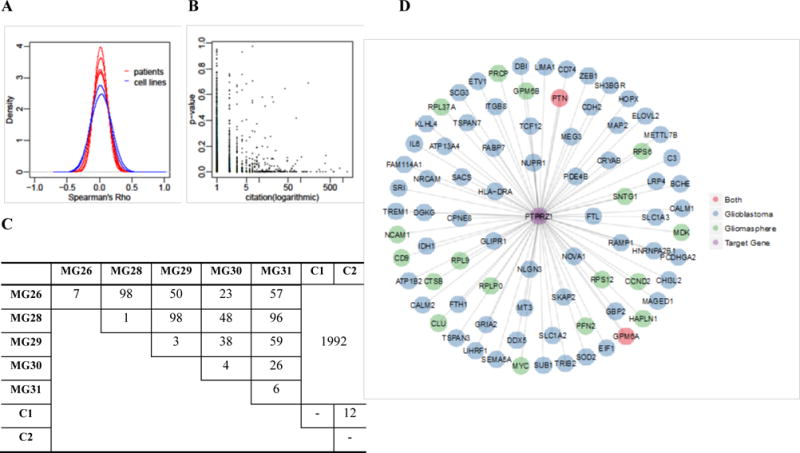
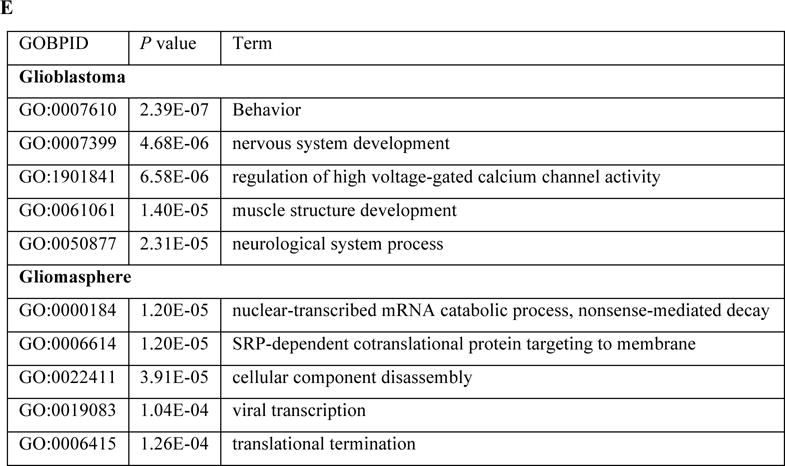
Differential single cell gene connectivity analysis of human glioblastomas data A: Histogram of the Spearman' Rho on five patients and two cell lines; B: Consistency analysis between the testing significance with the number of citation from PubMed; C: The number of significant differentially connected genes based on local false discovery rate (lfdr) < 0.001; D: Different connectivity pattern of PTPRZ1 under two conditions (glioblastoma and gliomasphere), where the edge is thresholded by fdr < 0.05; E: Top five GO biological processes of the genes connected to PTPRZ1 under two conditions, after manually removing redundancies.
Next, we conducted differential connectivity analyses under several settings: (1) the overall connectivity difference between primary GBM (five patients) and gliomasphere (two cell lines); (2) the pair-wise difference among all five GBM patients; (3) the difference between two gliomasphere cell lines. As a control, we tested each of the five patients by randomly spliting the cells into two sub-samples with equal sizes and test between them.
We used a stringent criterion to select DCGs (FDR < 0.001). The numbers of DCGs from all tests are summarized in Fig. 1C. First, there are substantial number of genes showing differential connectivity between primary GBM and two cell lines. We discovered 1992 DCGs which is 33.5% of the total number of genes tested. This demonstrates that the gene networks are significantly different between the primary tumors and the cell lines. The tests among GBM patients yield much less DCGs: on average there are about 50 DCGs which is about 1% of all genes. This is consistent with the results reported in the original publication (Patel et al. 2014), that the intratumoral heterogeneity was found to be greater than normal oligodendrocytes. In addition, many cells from one tumor crossed into the transcriptional space of another tumor. On the other hand, even though the GBM shows greater heterogeneity than the cell lines, the differences in gene connectivity patterns among GBM patients are not as pronounced as between GBM and cell lines. These results indicate that the cell lines may not be sufficiently representative of the primary tumor, at least in the sense of gene co-expression patterns. The comparison between the two cell lines results in 12 DCGs, which is reasonable since we expect less diversity from the cell lines. Finally, the within-patient comparison (shown in the diagonal elements of the table) gives a very small number of DCGs, which are false positives. This demonstrates that the false positive rates are very low from the differential connectivity test procedure. Overall, the numbers of DCGs discovered from the tests make biological sense.
To check whether the detected DCGs are biologically relevant or not, we searched each gene plus a specific keyword, the biological condition of the study, on PubMed and utilize the number of returned citations as an indicator of the previous exposure. For consistency analysis, we conducted a Poisson regression:
where i is gene index and α1 can be viewed as an indicator of the reliability of the results.
Here, we use “glioblastoma” as keyword in the PubMed search. Fig. 1B shows the p-values of the DCGs versus the numbers of citations returned from the search. A clear negative association can be observed. Based on Poisson regression, the association between p-value and Citation is significantly negative, where α1 = −2.06 with p-value <2×10−16. This indicates that genes with smaller p-values (showing greater differences in connectivity between GBM and gliomasphere cell lines) tend to be reported more previously. These results further validate the biological relevance of the differential connectivity results from this dataset. We took the DCGs between primary GBM (five patients) and gliomasphere (two cell lines) and performed the GO biological process enrichment analysis using GOstats (Falcon and Gentleman 2007). The top GO biological processes are focused around energy production, ion transport, RNA/protein synthesis, and small molecule metabolism.
To further investigate the different connectivity patterns, we select the significantly associated genes with any DCG at a local false discovery rate (lfdr) < 0.05 in either of the two groups, glioblastomas and gliomasphere. The lfdr values were obtained by fitting a mixture model based on Spearman’s Rho values using fdrtool package (Strimmer 2008). We took PTPRZ1 as an example. PTPRZ1 is a receptor protein tyrosine phosphatase that mainly functions in the central nervous system (CNS). Its normal function is regulating developmental processes in the CNS (Wang et al. 2010). As shown in Fig. 1D, the connectivity pattern of PTPRZ1 differs substantially between two conditions. Fig. 1E shows the top five GO biological process terms associated with the PTPRZ1-connected genes under two conditions. In glioblastoma cells, PTPRZ1 is correlated with genes that function in nervous system development, which is close to its normal biological function. On the other hand, in gliomasphere cells, PTRPZ1 is correlated with large molecule metabolism and localization. The results indicate that in gliomasphere cells, genes associated with tissue-specific functionalities may be dysregulated. The full results of gene-level connectivity pattern change and related biological functions can be found at http://web1.sph.emory.edu/users/tyu8/SCDGN/GBM.html. We note that having a large number of changed connections does not necessarily imply that the DCG is a functional hub, but only represents that the detected DCG has different significantly connected genes under different conditions, which may reflect the change of an underlying regulatory mechanism as demonstrated in the PTPRZ1 example.
We further conducted cell clustering using monocle (Trapnell et al. 2014) based on the expression values of the DCGs (Fig. S1). The DCGs can separate the samples reasonably well. The cell line data points form very close clusters, and the two cell line clusters are very close to each other. The primary tumor data points roughly separate by patients, and the data points are more spread than the cell line data, supporting the conclusion that the cell line data are more homogeneous than the primary tumor data. There are some cross-over of data points between primary tumors, which was also observed in Patel et al.’s original publication (Patel et al. 2014) using all genes with a different dimension reduction method.
Another study of mouse ES cells and mouse embryonic fibroblasts (MFE) can be found in Supplementary data. In this work, we performed gene network analysis from scRNA-seq data. The main purpose of this work is to perform proof-of-concept analyses to demonstrate and validate the possibility of differential network analysis from single cell data. Gene network construction and comparison have been widely applied in bulk expression data to discover gene regulation mechanism. The network analysis from bulk expression suffers several drawbacks. First, gene networks constructed from the bulk expression is based on averaged gene expressions of a large number of cells. In highly heterogeneous samples such as cancer, the heterogeneity could mask the true biological signals and provide biased results. Secondly, the bulk data were often performed in several batches, and the measurements could potentially be contaminated by technical artifacts such as batch effects. Furthermore, other confounding factors in the bulk data such as age, gender, disease status, could further weaken the biological signals. In contrast, scRNA-seq experiments are well-controlled for those artifacts, thus providing cleaner results.
As the analyses of scRNA-seq data are mostly focused on clustering and differential expression so far, our discoveries and results provide new perspective to the analysis of scRNA-seq data. Similar principals can potentially be applied to other single cell genomics data (Buenrostro et al. 2015) to further mine the rich information provided by this new and exciting technology. Our analyses are performed for all cells from an experiment, which could be heterogeneous and each subtype of cells has its own gene network. It will be better to perform cell clustering, and then perform network analysis within each subtype. This will be our research plan in the near future.
Supplementary Material
Acknowledgments
This work was partially supported by NIH grants (Nos. 2U19AI090023, 5P30AI50409, and R01GM122083).
References
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GPPe’er, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Gill R, Datta S, Datta S. A statistical framework for differential network analysis from microarray data. BMC Bioinformatics. 2010;11:95. doi: 10.1186/1471-2105-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase T, Ghosh S, Yamanaka R, Kitano H. Harnessing diversity towards the reconstructing of large scale gene regulatory networks. PLoS Comput Biol. 2013;9:e1003361. doi: 10.1371/journal.pcbi.1003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler C, Gunawan R. Assessment of network inference methods: how to cope with an underdetermined problem. PLoS One. 2014;9:e90481. doi: 10.1371/journal.pone.0090481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. Characterization of the activation of protein tyrosine phosphatase, receptor-type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor-2 alpha. PLoS One. 2010;5:e9641. doi: 10.1371/journal.pone.0009641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, Holmes C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol. 2013;31:748–752. doi: 10.1038/nbt.2642. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.