

Abstract
A combined acid HCl/DMPU-acetic acid catalytic system was used in the hydrochlorination of a wide range of unactivated alkenes. This hydrochlorination strategy is remarkably greener than previous reported methods in terms of high atom efficiency, no toxic waste generated and metal-free process. The higher efficiency, compared with other commercially available HCl reagents, was augmented by the good regioselectivity and functionality tolerance found. A stepwise mechanism for this hydrochlorination process was proposed based on kinetic studies.
The hydrohalogenation of alkenes–one of the most fundamental organic reactions in elementary organic chemistry–has been extensively studied for over a century. However, in contrast to the facile hydrobromination and hydroiodination of alkenes,1 the hydrochlorination of alkenes, especially of unactivated monosubstituted alkenes, is still challenging. Although various indirect synthesis of alkyl chlorides, such as the reduction of alkenyl chlorides and the chlorination through C-H activation have been reported,2 the direct hydrochlorination of alkenes is still the most straightforward atom-economical method. HCl generated in situ from highly reactive chlorine-containing sources have been used for alkene hydrochlorinations, but the toxic waste and the limited scope restricted their practical uses.3 Notably, Carreira and co-workers described a direct hydrochlorination of challenging monosubstituted alkenes, catalyzed by cobalt, for a wide range of functional groups, including acid sensitive species (Scheme 1a). However, only the hydrochlorination of aliphatic alkenes were reported in their work.4 More recently, the Snyder group developed a novel chlorophosphonium pre-reagent for the addition of HCl to olefins. This reagent was easy to handle and smoothly delivered HCl to di- and tri-substituted aliphatic alkenes (Scheme 1b).5 However, the most convenient and widely available chlorine source, HCl, has been succinctly studied. Although HCl gas or liquid HCl have been utilized in this reaction,6 only a very limited substrate scope was reported. Meanwhile, storing and handling gaseous or liquid HCl is always a safety concern. A remarkable application of a supported HCl salt for the hydrochlorination of olefins was demonstrated by Nicewicz and co-workers, using photoredox catalysis.7 A series of substituted styrene substrates were converted to the corresponding chlorinated products in anti-Markovnikov manner under irradiation of blue LED using a HCl/2,6-lutidine salt as HCl source (Scheme 1c). In general, the success of the aforementioned hydrochlorinations depended on the structural and electronic nature of alkene substrates: highly substituted or strained alkenes and styrene-based substrates were usually necessary substrates. In this regard, an unbiased hydrochlorination of alkenes relying on easy-handling HCl reagents is still needed. But, to the best of our knowledge, no such general strategy for hydrochlorination of olefins has been reported.
Scheme 1.
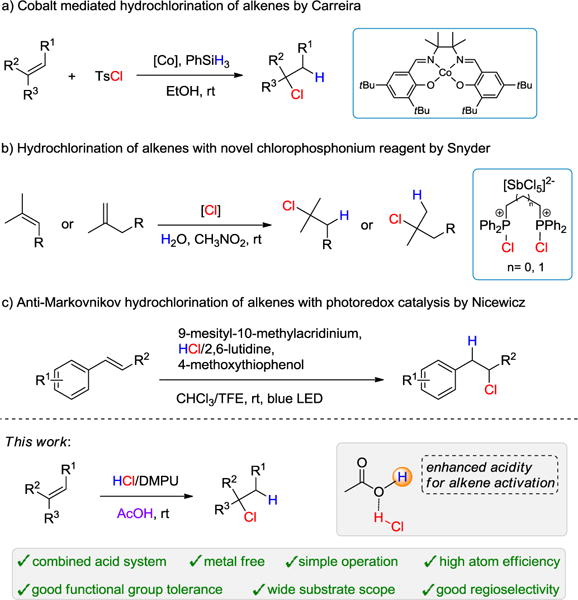
Notable examples of hydrochlorination of olefins.
In our continuing efforts to halogenate organic molecules with DMPU-supported hydrogen halides,1g, 8 we hypothesized that the highly active and concentrated HCl/DMPU8f, 8g might provide a more widely applicable protocol for addition of HCl to alkenes. However, in our initial attempts, various transition metal catalysts failed to give hydrochlorinated products, which underscored the inefficient activation of alkenes or the catalyst poisoning enabled by the high concentration of chlorine. These observations suggested that perhaps a metal-free, yet highly acidic system, could be appropriate for our designed hydrochlorination strategy. In our previous reports, we demonstrated an efficient alkyne hydration method through acid-assisted Brønsted acid catalysis, in which the activation of the alkyne was enabled by the enhanced acidity of the reaction system by the combination of two acids.9 Based on this concept, we now report a highly efficient strategy for the hydrochlorination of unactivated alkenes through a combined acid system (Scheme 1).
We commenced our studies using styrene 1a as model substrate and HCl/DMPU as HCl source (Table 1). As mentioned before, in our initial trials we examined the possibility of hydrochlorination using metal catalysis. A chlorine-tolerant gold catalytic system8g could not efficiently activate the C=C double bond (Table 1, entry 1). Other metal-based Lewis acid catalysts were also ineffective for the hydrochlorination of 1a (Table 1, entries 2-4). Brønsted acid catalysts were also tested (Table 1, entries 5-7) but TfOH, HNTf2 as well as a solid acid, Nafion NR50, failed to improve the yield of the desired product. We switched our strategy and used acidic solvents such as TFA and AcOH in the absence of strong acid catalysts (Table 1, entries 8-9). No chlorinated product was observed when TFA was used but a high yield (84%) of (1-chloroethyl)benzene 2a was obtained in AcOH. We later found that the reaction at room temperature also produced 2a in 57% yield (Table 1, entry 10). To increase the yield, more HCl/DMPU was utilized (Table 1, entries 11-12). Although there was no obvious difference in yield when 6 and 8 equivalents of HCl/DMPU were used, respectively, the latter was chosen to account for more challenging aliphatic alkenes that would be further examined.
Table 1.
Optimization for hydrochlorination of styrene with HCl/DMPUa

| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | x | catalyst | solvent | T (°C) | Yield (%) |
| 1 | 4 | AuPPh3Cl | HFIP | 70 | 5 |
| 2 | 4 | FeCl3 | DMF | 100 | 8 |
| 3 | 4 | CuI | DMF | 100 | 8 |
| 4 | 4 | Ga(OTf)3 | DMF | 100 | 3 |
| 5 | 4 | TfOH | DMF | 100 | 12 |
| 6 | 4 | HNTf2 | DMF | 100 | 10 |
| 7 | 4 | Nafion NR50 | DMF | 100 | n.r. |
| 8 | 4 | – | TFA | 70 | n.d. |
| 9 | 4 | – | AcOH | 100 | 84 |
| 10 | 4 | – | AcOH | rt | 57 |
| 11 | 6 | – | AcOH | rt | 95 |
| 12 | 8 | – | AcOH | rt | 98 |
Yield was determined by GCMS.
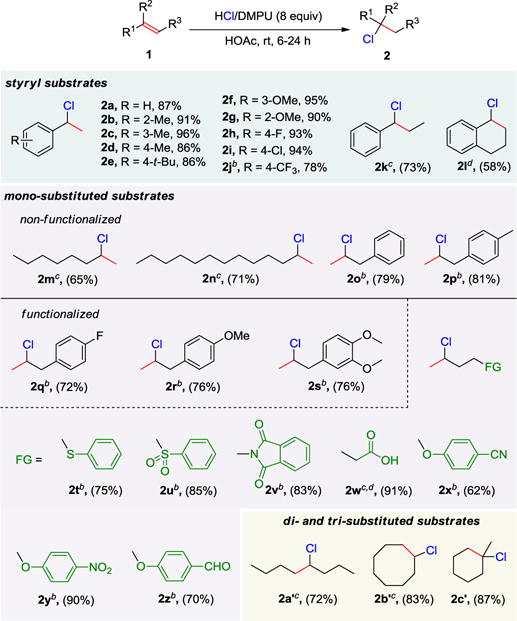
After establishing the optimal reaction conditions, the substrate scope was explored (Table 2). First of all, we investigated the styryl system (Table 2, 2a-2l). The efficacy of our protocol showed a dependence with the electronic properties of the aryl substituents. The substrates containing strong electron-withdrawing group, such as CF3, needed higher temperature to complete the conversion. But substrates with electron-donating groups underwent the reaction smoothly to give hydrochlorinated products in high yields. The scope of our strategy was further extended to the more challenging unactivated monosubstituted alkenes. Non-functionalized alkenes were efficiently converted to the corresponding chlorinated products (Table 2, 2m–2p). More importantly, a wide range of functional groups such as sulfide, sulfone, imide, carboxylic acid, cyano, nitro as well as carbonyl groups were well tolerated (Table 2, 2q–2z). In addition, this protocol could also be applied for the hydrochlorination of di- and tri-substituted alkenes with good efficiencies (Table 2, 2a′–2c′).
Table 2.
Substrate scope of hydrochlorination of alkenes with HCl/DMPUa
|
Isolated yields; Reaction condition: 1 (0.2 mmol) and HCl/DMPU (43% w/w, 1.6 mmol) in AcOH (0.5 ml) at room temperature.
100 °C
50 °C.
GCMS yield.
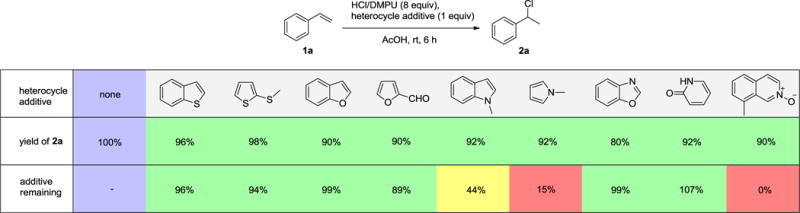
To gain insight on the versatility of our strategy towards heteroatomic ring systems, we conducted an additive-based reaction screening, a strategy regarded as a robust and high-throughput approach for the examination of functionality tolerance (Table 3).10 To our delight, the yields with all the tested additives ranged from 80% to 98%. Most of the heterocycles tested remained intact, including benzo[b]thiophene, 2-(methylthio)thiophene, benzofuran, furfural, benzo[d]oxazole, and 2-pyridone. 1-Methylindole and 1-methylpyrrole partially decomposed, with recoveries of 44% and 15%, respectively. On the other hand, 8-methylquinoline N-oxide was completely converted to its reduced form. Our reaction protocol was also easily utilized in large scale synthesis (eq 1).
 |
(eq 1) |
Table 3.
Examination of heteroarene compatibility through additive-based reaction screeninga
|
The assessment of results is represented with color code, the yield of the standard reaction in the absence of additive is showed in purple, the yield with additive is scaled relative to standard reaction. The product yield represents: > 70% in green, 40-70% in yellow and < 40% in red; the additive remaining represents: > 70% in green, 40-70% in yellow and < 40% in red.
Compared with other commercially available HCl sources, the observed superior performance of HCl/DMPU on the hydrochlorination of styrene (2a) was consistent with our previous results in the hydrochlorination of alkynes (Scheme 2A).8f,8g After demonstrating the generality of our hydrochlorination strategy, we turned our attention to its mechanism. First, we investigated the kinetic isotope effect (KIE) for the hydrochlorination of styrene (2a). A KIE of 2.2 was observed (Scheme S1), which indicated that the addition of hydrogen to alkene might be the rate-determining step. In addition, we found that the reaction rate was dependent on the concentration of chlorine (Scheme 2B). Based on these outcome and the observation that trace amounts of 1-phenylethyl acetate were found as byproduct, we hypothesized that a stepwise reaction took place: 1) the enhanced acidity, by a combined acid system, was able to activate the olefinic double bond, rendering it amenable for nucleophilic acetylation (Scheme 2C). In support of this step, we noticed that the formation of 1-phenylethyl acetate was very efficient in the presence of either a Brønsted acid such as TfOH, or a Lewis acid such as Ga(OTf)3; on the contrary, no desired product was generated without an acid promoter. Meanwhile, a KIE of 1.33 was obtained when Ga(OTf)3 was used as promoter (Scheme S2 in Supporting Information), a result that is consistent with the assumption that the addition of AcOH to alkene was the rate-determining step; 2) the nucleophilic substitution of 1-phenylethyl acetate with chloride afforded (1-chloroethyl)benzene (Scheme 2D). It was found that the presence of acetic acid significantly facilitated the substitution process due to the enhanced leaving group capacity of the acetoxy group. Moreover, an inverse kinetic isotope effect (KIE=0.59) was observed (Scheme S3 in Supporting Information), which indicated a rehybridization of the transition state involved in the substitution mechanism.11
Scheme 2.
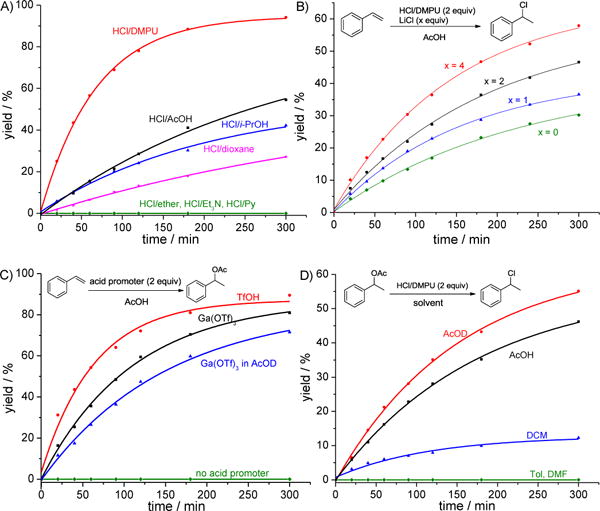
A) Comparison of efficiency among various HCl sources; B) The effect of chloride concentration; C) Step 1: acetylation of styrene with AcOH; D) Step 2: substitution of phenylethyl acetate with HCl.
In summary, we have developed a highly efficient and widely applicable metal-free protocol for the hydrochlorination of alkenes through a combined acid catalytic system. This strategy worked well for challenging monosubstituted alkenes, and exhibited good regioselectivity and functional group tolerance. A kinetic study suggested a sequential acetylation/chlorination pathway.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health for financial support (R01GM121660). Bo Xu is grateful to the National Science Foundation of China for financial support (NSFC-21472018).
Footnotes
Electronic supplementary information (ESI) available: Detailed experimental procedures, and characterization of compounds. See DOI: 10.1039/b000000x
Conflicts of interest
There are no conflicts interest to declare.
Notes and references
- 1.(a) Barluenga J, González JM, Campos PJ, Asensio G. Angew Chem Int Ed. 1985;24:319–320. [Google Scholar]; (b) Campos PJ, García B, Rodríguez MA. Tetrahedron Lett. 2002;43:6111–6112. [Google Scholar]; (c) Das B, Srinivas Y, Holla H, Narender R. Chem Lett. 2007;36:800–801. [Google Scholar]; (d) Reddy C Kishan, Periasamy M. Tetrahedron Lett. 1990;31:1919–1920. [Google Scholar]; (e) Pagni RM, Kabalka GW, Boothe R, Gaetano K, Stewart LJ, Conaway R, Dial C, Gray D, Larson S, Luidhardt T. J Org Chem. 1988;53:4477–4482. [Google Scholar]; (f) Irifune S, Kibayashi T, Ishii Y, Ogawa M. Synthesis. 1988;5:366–369. [Google Scholar]; (g) Li Z, Ebule R, Kostyo J, Hammond GB, Xu B. Chem Eur J. 2017;23:12739–12743. doi: 10.1002/chem.201703457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Weiss HM. J Chem Educ. 1995;72:848. [Google Scholar]; (i) Sanseverino AM, Mattos MCSd. J Braz Chem Soc. 2001;12:685–687. [Google Scholar]; (j) Galli M, Fletcher CJ, del Pozo M, Goldup SM. Org Biomol Chem. 2016;14:5622–5626. doi: 10.1039/c6ob00692b. [DOI] [PubMed] [Google Scholar]; (k) Nishio Y, Mifune R, Sato T, Ishikawa S-i, Matsubara H. Tetrahedron Lett. 2017;58:1190–1193. [Google Scholar]; (l) Isenberg N, Grdinic M. J Chem Educ. 1969;46:601. doi: 10.1021/ed049p392. [DOI] [PubMed] [Google Scholar]; (m) Mayo FR, Walling C. Chem Rev. 1940;27:351–412. [Google Scholar]
- 2.(a) King SM, Ma X, Herzon SB. J Am Chem Soc. 2014;136:6884–6887. doi: 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]; (b) Kundu R, Ball ZT. Org Lett. 2010;12:2460–2463. doi: 10.1021/ol100472t. [DOI] [PubMed] [Google Scholar]; (c) Liu W, Groves JT. J Am Chem Soc. 2010;132:12847–12849. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]; (d) Qin Q, Yu S. Org Lett. 2015;17:1894–1897. doi: 10.1021/acs.orglett.5b00582. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Li GX, Yang G, He G, Chen G. Chem Sci. 2016;7:2679–2683. doi: 10.1039/c5sc04169d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Reyes JR, Rawal VH. Angew Chem Int Ed. 2016;55:3077–3080. doi: 10.1002/anie.201510909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang Z, Sun Q, Xu D, Xia C, Sun W. Green Chem. 2016;18:5485–5492. [Google Scholar]; (h) Gazić Smilović I, Casas-Arcé E, Roseblade SJ, Nettekoven U, Zanotti-Gerosa A, Kovačevič M, Časar Z. Angew Chem Int Ed. 2012;51:1014–1018. doi: 10.1002/anie.201106262. [DOI] [PubMed] [Google Scholar]
- 3.(a) Boudjouk P, Kim BK, Han BH. Synth Commun. 1996;26:3479–3484. [Google Scholar]; (b) de Mattos MCS, Sanseverino AM. Synth Commun. 2000;30:1975–1983. [Google Scholar]; (c) Yadav Veejendra K, Babu KG. Eur J Org Chem. 2005;2005:452–456. [Google Scholar]; (d) Delaude L, Laszlo P. Tetrahedron Lett. 1991;32:3705–3708. [Google Scholar]; (e) Kropp PJ, Daus KA, Crawford SD, Tubergen MW, Kepler KD, Craig SL, Wilson VP. J Am Chem Soc. 1990;112:7433–7434. [Google Scholar]; (f) Kropp PJ, Daus KA, Tubergen MW, Kepler KD, Wilson VP, Craig SL, Baillargeon MM, Breton GW. J Am Chem Soc. 1993;115:3071–3079. [Google Scholar]
- 4.Gaspar B, Carreira EM. Angew Chem Int Ed. 2008;47:5758–5760. doi: 10.1002/anie.200801760. [DOI] [PubMed] [Google Scholar]
- 5.Schevenels FT, Shen M, Snyder SA. J Am Chem Soc. 2017;139:6329–6337. doi: 10.1021/jacs.6b12653. [DOI] [PubMed] [Google Scholar]
- 6.(a) Whitmore FC, Johnston F. J Am Chem Soc. 1933;55:5020–5022. [Google Scholar]; (b) Schmerling L. J Am Chem Soc. 1946;68:195–196. [Google Scholar]; (c) Dewar MJS, Fahey RC. J Am Chem Soc. 1963;85:2245–2248. [Google Scholar]; (d) Stille JK, Sonnenberg FM, Kinstle TH. J Am Chem Soc. 1966;88:4922–4925. [Google Scholar]; (e) Fahey RC, McPherson CA. J Am Chem Soc. 1971;93:2445–2453. [Google Scholar]; (f) Becker KB, Grob CA. Synthesis. 1973;1973:789–790. [Google Scholar]; (g) Fr⊘yen P, Skramstad J. Synth Commun. 1994;24:1871–1877. [Google Scholar]; (h) Alper H, Huang Y, Dell'Amico D Belli, Calderazzo F, Pasqualetti N, Veracini CA. Organometallics. 1991;10:1665–1671. [Google Scholar]; (i) Kennedy JP, Sivaram S. J Org Chem. 1973;38:2262–2264. [Google Scholar]; (j) Landini D, Rolla F. J Org Chem. 1980;45:3527–3529. [Google Scholar]
- 7.Wilger DJ, GrandjeanJean-Marc M, Lammert TR, Nicewicz DA. Nat Chem. 2014;6:720–726. doi: 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]
- 8.(a) Okoromoba OE, Han J, Hammond GB, Xu B. J Am Chem Soc. 2014;136:14381–14384. doi: 10.1021/ja508369z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Okoromoba OE, Li Z, Robertson N, Mashuta MS, Couto UR, Tormena CF, Xu B, Hammond GB. Chem Commun. 2016;52:13353–13356. doi: 10.1039/c6cc07855a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Okoromoba OE, Hammond GB, Xu B. Org Lett. 2015;17:3975–3977. doi: 10.1021/acs.orglett.5b01919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liang S, Hammond GB, Xu B. Chem Eur J. doi: 10.1002/chem.201702664,n/a-n/a. [DOI] [Google Scholar]; (e) Liang S, Barrios FJ, Okoromoba OE, Hetman Z, Xu B, Hammond GB. J Fluorine Chem. 2017;203:136–139. doi: 10.1016/j.jfluchem.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liang S, Ebule R, Hammond GB, Xu B. Org Lett. 2017;19:4524–4527. doi: 10.1021/acs.orglett.7b02101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ebule R, Liang S, Hammond GB, Xu B. ACS Catal. 2017;7:6798–6801. doi: 10.1021/acscatal.7b02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Yamamoto H, Futatsugi K. Angew Chem Int Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]; (b) Liang S, Hammond GB, Xu B. Chem Commun. 2015;51:903–906. doi: 10.1039/c4cc08938c. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gensch T, Teders M, Glorius F. J Org Chem. 2017;82:9154–9159. doi: 10.1021/acs.joc.7b01139. [DOI] [PubMed] [Google Scholar]; (b) Richardson J, Ruble JC, Love EA, Berritt S. J Org Chem. 2017;82:3741–3750. doi: 10.1021/acs.joc.7b00201. [DOI] [PubMed] [Google Scholar]; (c) Bayeh L, Le PQ, Tambar UK. Nature. 2017;547:196–200. doi: 10.1038/nature22805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Collins KD, Glorius F. Nat Chem. 2013;5:597–601. doi: 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]; (e) Collins KD, Rühling A, Glorius F. Nat Protocols. 2014;9:1348–1353. doi: 10.1038/nprot.2014.076. [DOI] [PubMed] [Google Scholar]; (f) Collins KD, Rühling A, Lied F, Glorius F. Chem Eur J. 2014;20:3800–3805. doi: 10.1002/chem.201304508. [DOI] [PubMed] [Google Scholar]; (g) Leger PR, Murphy RA, Pushkarskaya E, Sarpong R. Chem Eur J. 2015;21:4377–4383. doi: 10.1002/chem.201406242. [DOI] [PubMed] [Google Scholar]
- 11.(a) Marlier JF, Campbell E, Lai C, Weber M, Reinhardt LA, Cleland WW. J Org Chem. 2006;71:3829–3836. doi: 10.1021/jo060223t. [DOI] [PubMed] [Google Scholar]; (b) Marlier JF, Fogle EJ, Redman RL, Stillman AD, Denison MA, Robins LI. J Org Chem. 2015;80:1905–1908. doi: 10.1021/jo502472m. [DOI] [PubMed] [Google Scholar]; (c) Hogg JL, Rodgers J, Kovach I, Schowen RL. J Am Chem Soc. 1980;102:79–85. [Google Scholar]; (d) Lu Y, Qu F, Moore B, Endicott D, Kuester W. J Org Chem. 2008;73:4763–4770. doi: 10.1021/jo800820u. [DOI] [PubMed] [Google Scholar]; (e) Mitton CG, Gresser M, Schowen RL. J Am Chem Soc. 1969;91:2045–2047. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.