

Abstract
Background
Bacterial taxonomy aims to classify bacteria based on true evolutionary events and relies on a polyphasic approach that includes phenotypic, genotypic and chemotaxonomic analyses. Until now, complete genomes are largely ignored in taxonomy. The genus Lactobacillus consists of 173 species and many genomes are available to study taxonomy and evolutionary events.
Results
We analyzed and clustered 98 completely sequenced genomes of the genus Lactobacillus and 234 draft genomes of 5 different Lactobacillus species, i.e. L. reuteri, L. delbrueckii, L. plantarum, L. rhamnosus and L. helveticus. The core-genome of the genus Lactobacillus contains 266 genes and the pan-genome 20′800 genes. Clustering of the Lactobacillus pan- and core-genome resulted in two highly similar trees. This shows that evolutionary history is traceable in the core-genome and that clustering of the core-genome is sufficient to explore relationships. Clustering of core- and pan-genomes at species’ level resulted in similar trees as well. Detailed analyses of the core-genomes showed that the functional class “genetic information processing” is conserved in the core-genome but that “signaling and cellular processes” is not. The latter class encodes functions that are involved in environmental interactions. Evolution of lactobacilli seems therefore directed by the environment. The type species L. delbrueckii was analyzed in detail and its pan-genome based tree contained two major clades whose members contained different genes yet identical functions. In addition, evidence for horizontal gene transfer between strains of L. delbrueckii, L. plantarum, and L. rhamnosus, and between species of the genus Lactobacillus is presented. Our data provide evidence for evolution of some lactobacilli according to a parapatric-like model for species differentiation.
Conclusions
Core-genome trees are useful to detect evolutionary relationships in lactobacilli and might be useful in taxonomic analyses. Lactobacillus’ evolution is directed by the environment and HGT.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4601-5) contains supplementary material, which is available to authorized users.
Keywords: Comparative genomics, Lactobacillus, Core-genome, Pan-genome, Horizontal gene transfer
Background
In the last 10 years, sequencing of complete genomes developed from research that required a consortium to an effort that a single person can manage [1]. The decreasing costs and increasing speed of complete genomes sequencing have led to an enormous amount of data of various quality that is not completely analyzed yet [2]. Next generation sequencing (NGS) technology is highly useful for research on diseases or on phenotypic variations of specific genes. Such research; however, needs high quality data, i.e. a genome coverage that results in a reliable sequence [3]. In microbiology, high quality sequences are suitable for research such as sub-typing of Salmonella enterica strains to monitor outbreaks or for calculating bacterial pan-genomes [4, 5]. Complete genome sequences are only poorly applied in bacterial classification and phylogenetic studies [6]. Complete genomes are; however, most preferable for phylogenetic studies because evolutionary pressure works on the whole organism and not on a subset of genes.
A polyphasic approach is commonly used for bacterial classification and analysis of evolutionary relationships [7, 8]. Polyphasic approaches are not standardized and include phenotypic, genotypic and chemotaxonomic parameters to determine whether a bacterial isolate belongs to an existing species or if a new species has to be defined [6, 9]. Assignment of a strain to a species is based amongst others on two genotypic parameters: sequence similarity of more than 98.7% in the 16S rRNA gene and a DNA-DNA hybridization (DDH) degree of more than 70% [10, 11]. Other genetic parameters are also useful for bacterial classification. For example, EcoSNPs, SNPs that are specific for a dimorphic nucleotide position in a clade, can be used to build phylogenetic trees [12]. Comparison of complete genome sequences to 70% DNA-DNA hybridization levels is possible using defined parameters for conserved DNA regions and unique matches [13–15]. Additionally, an average nucleotide identity (ANI) value of 94% corresponds to 70% DNA-DNA hybridization and is thus also a usable parameter to define species.
The core-genome is the set of homologous genes that are present in all genomes of an analyzed dataset and the pan-genome is the set of all genes that are present in the analyzed dataset [16]. In addition, the softcore-genome is the set of genes, present in ≥95% of the genomes [17]. The softcore-genome is useful, because it circumvents the absolute impact of poor quality sequences on the core-genome. An open pan-genome is increasing with every new genome included whereas a closed pan-genome remains on a constant gene number after a certain number of genomes were included [18]. The status of the core- and pan-genomes depends on number of analyzed genomes and on the properties of the species analyzed, such as the ability of the species to integrate exogenous DNA and on the species’ lifestyle and environment [19–21]. Taxonomy of bacteria based on core- and pan-genome might be a powerful extension of the polyphasic approach. Pan-genome clustering of 29 Geobacillus genomes revealed horizontal gene transfer as a factor in evolution of Geobacillus and such transfer should be implemented in its taxonomy [22]. Horizontal gene transfer was also detected in a recently diverged Vibrio population, where ecological differentiation based on single nucleotide polymorphisms occurred [12].
The heterogeneous genus Lactobacillus (L.) contains 173 species not including 17 subspecies [23]. Lactobacilli have been isolated from a whole range of fermented food products such as yoghurt, cheese, vegetables, beverages, sausages and sourdough. Further, lactobacilli are also found in the human and animal gastro-intestinal tracts [24]. The Qualified Presumption of Safety (QPS) status from the European Food Safety Agency EFSA facilitates commercial use and acceptance of most Lactobacillus species. This makes them ideal candidates for the use as protective and starter cultures [25]. Aside from their preserving qualities, strains of some Lactobacillus species are also exploited for their health promoting potential as probiotics and vaccine carrier [26, 27]. In December 2016, a total of 121 completely sequenced and assembled genomes were available in public databases with sizes ranging from 1.37 Mpb for L. sanfranciscensis TMW1.1 to 3.74 Mbp for L. paracellinoides TMW1.1995 [28]. Lactobacillus and related genera were initially clustered into three subgroups based on 16S RNA gene comparison: the Lactobacillus delbrueckii group, the Lactobacillus casei-Pediococcus group and the Leuconostoc group [29, 30]. A recent 16S rRNA gene based clustering of the Lactobacillus type strains species resulted in a phylogenetic tree with 15 major groups [31]. There is; however, only moderate correlation between 16S rRNA gene sequence clustering and clustering based on fermentation type and metabolic properties.
The goal of this study was to analyze the phylogeny of the Lactobacillus genus and a dedicated set of species via core-, softcore- and pan-genome clustering. Such complete genome based clustering provides a detailed overview of gene contents of the core- and pan-genome and will provide insights on relationship of species and their gene exchange.
Methods
Genome sequences
A total of 98 complete sequenced Lactobacillus genomes and 202 draft genomes belonging to the species Lactobacillus plantarum, Lactobacillus helveticus, Lactobacillus delbrueckii, Lactobacillus reuteri and Lactobacillus rhamnosus were obtained from public databases (Additional file 1). To prevent too high impact of poorly assembled genomes for the Lactobacillus species calculation, draft genomes were only used if they fell within a range of ± 2σ around the average gene and protein number of the species.
Calculation of core- and pan-genome
Orthologous cluster were created using the Perl-script collection GET_HOMOLOGOUS [32] applying the following for identification and clustering CDS into orthologous groups: -E < 1e-05 for blastp searches and -C 75% minimum alignment coverage. The core-genome was determined using the Ortho Markov Cluster algorithm (OMCL) [33] and the pan-genome using the OMCL algorithm with –t 0; reporting all clusters in the pan-genome. A pan-genome matrix was created using the script compare_clusters with the settings: -d including only OMCL data, −m produce intersection in pan-genome matrix.
The core-genome was defined by genes present in all genomes, the softcore by genes present in 95–100% of the genomes, the shell by genes present in more than 2 genomes but less than 95% of the genomes and the cloud genes present in 2 or less of the genomes and calculated with the parse_pangenome_matrix script: -s report clusters.
The development-calculation of core- and pan-genome starts with comparing two genomes and including single genomes step-by-step until all genomes are integrated. The order of the included genome was randomized n-times (n = number of included genomes) and calculated with a home-made script in MATLAB R2014b based on the pangenome_matrix_t0. The home-made scripts used in this study are available in Additional file 2.
Clustering and analyses of core- and pan-genome
Protein-based clustering was performed with GET_HOMOLOGOUS [32] using the OMCL algorithm as follows: -t 0, −t all or -t n (n = 0.95 x number of included genomes) for clustering the pan-, core- and softcore-genome, −M; with the OMCL algorithm and – A; to create an average identity matrix. The created average identity matrix of clustered sequences was visualized using the script hcluster_matrix with the option –d gower; for selecting the gower distance calculation for clustering [34]. Core- and pan-genome (Additional file 1) were analyzed with the metagenome analysis tool GhostKOALA against “genus_prokaryotes + family_eukaryotes” database using the Brite, Pathway and Module reconstruction algorithm [35]. Brite reconstruction uses KEGG Brite hierarchies with combined sets of K numbers. Pathway reconstruction aligns gene to the KEGG pathway map and Module reconstruction uses sets of K numbers to evaluate if a block (pathway or structural complex) is complete. The relative increase of genes in a category compared to the complete increase of genes in core- and pan-genomes were calculated and analyzed with Fisher’s exact test in MATLAB R2014b (Additional file 2).
Identification of clade specific genes
Identification of clade specific genes in a set of bacterial isolates was performed using the parse_pangenome_matrix script of GET_HOMOLOGOUS [32] with option; −A a list of genomes in one clade; option –B a list genomes of another clade to compare against; −g finding genes present in genomes of clade A and absent in genomes of clade B; −e find gene family expansions in A with respect to B. To determine if a gene encodes a unique function in a clade that is not compensated by isoenzymes in the other clade, the core-genome of the clade was compared with the pan-genome of all other clades using GhostKOALA [35]. The presence of isoenzymes was analyzed for each gene manually.
Identification of representative genes for clustering the type species Lactobacillus delbrueckii
To identify which gene or set of genes represents most closely the pan-genome-phylogenetic tree of L. delbrueckii, the tree of each core gene was compared to the tree of the pan genome of L. delbrueckii using TOPD/FMTS [36] and CLC Workbench 8 (CLC Genomic, Aarhus, Denmark). Each homologous gene set from the core genome was imported as multi-entry FASTA into CLC Genomic Workbench 8. The genes were aligned using the “Create alignment” tool using standard parameters. Trees were created with the toolbox “Create Tree” using “Neighbor Joining” as tree construction method and “Jukes-Cantor” as nucleotide distance measure with a Bootstrap value of 100. Trees were exported as nexus files and compared to the pan-genome tree using TOPD/FMTS using the following parameters: -m nodal method of calculation; −n 10 number of random sequences; −c reference comparing all versus pan-genome tree. Identical trees have a nodal distance = 0. The higher the nodal distance is the less identical are the trees. The 5% and 95% percentile was calculated for all core gene nodal distances and the genes outside this range analyzed manually.
Identification of ecoSNPs in Lactobacillus delbrueckii
To analyze ecoSNP distribution, core gene alignments were exported from the CLC workbench as ClustalW files and imported into MATLAB R2014b to determine the consensus sequences. Each gene was compared with the consensus sequence and SNPs were determined and analyzed for its specificity to a clade in the pan-genome of L. delbrueckii.
Potential horizontal gene transfer within clades
For identification of potential horizontal gene transfer (HGT) events, genes with a 30–70% presence in all clades were selected. An absence-presence matrix for all genes and strains was constructed for each clade in MS Excel and genes within the 30–70% criterion selected.
Results
Calculation of core- and pan-genome of complete Lactobacillus genomes
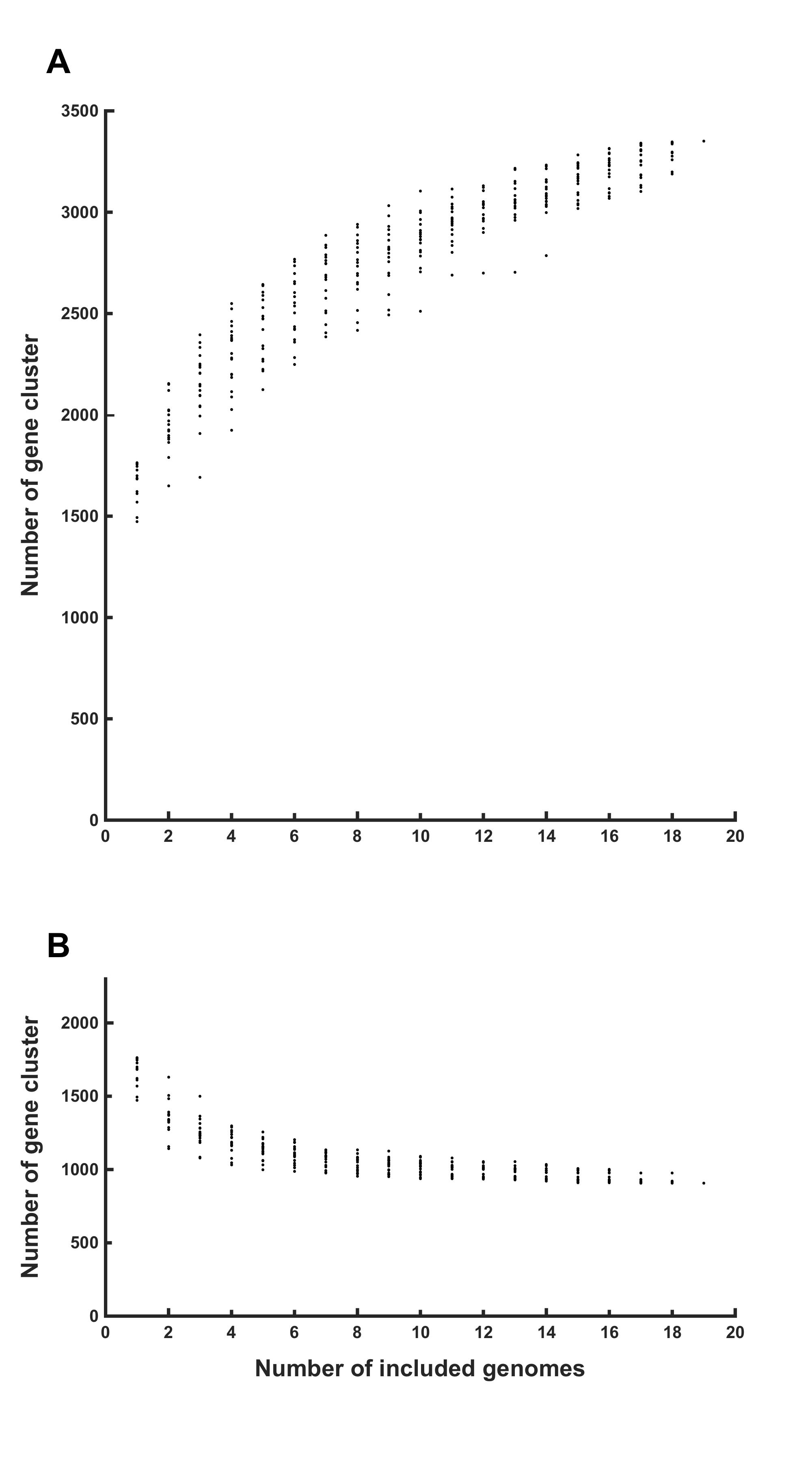
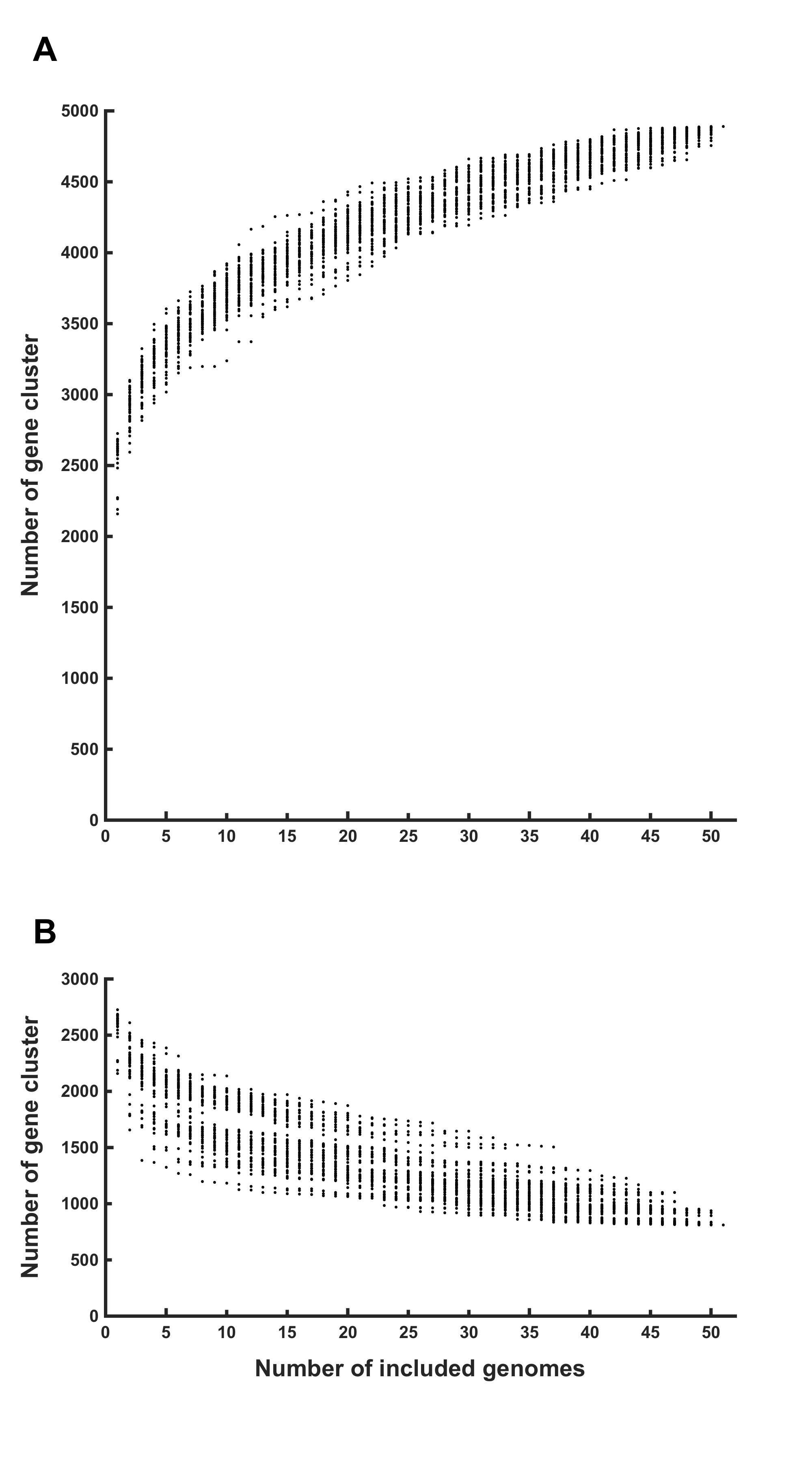
To obtain a general view of Lactobacillus genome contents, the core- and pan-genome for 98 completely assembled Lactobacillus genomes were calculated. The pan-genome for the Lactobacillus genus still increased with approximately 50 genes after addition of a 98th genome and thus can be considered as open (Fig. 1a). The core-genome rapidly decreased with the first set of genomes, but stabilizes after the 70th is added, showing it’s closed (Fig. 1b). The core-genome contained 266 genes and the pan-genome 20′800 genes (Table 1; Additional files 3 and 4). A core-genome based clustering revealed 4 major clades: (A), a reuteri-fermentum-salivarius clade, (B), a plantarum-paraplantarum clade, (C) a casei-paracasei-rhamnosus clade and (D) a helveticus-delbrueckii-johnsonii clade (Fig. 2). The softcore- and pan-genome were also clustered and the 4 clades appeared again as separate clusters and contained the same isolates (Fig. 3). The highly similar pan- and core-genome clusters shows that evolutionary relationship appears already in the core genome. In general, species clustered together. However, some strains from the species L. casei / L. paracasei and L. helveticus / L. gallinarum did not.
Fig. 1.
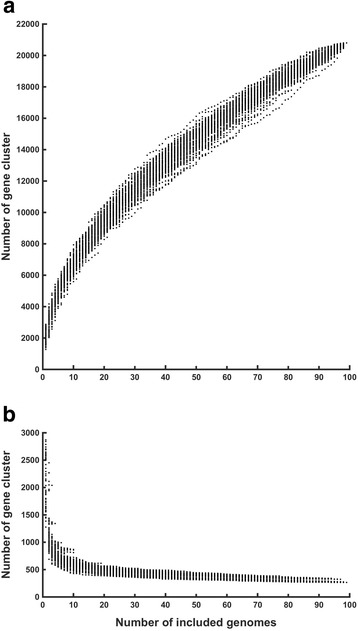
Pan- and core-genome evolution of Lactobacillus. a For every included genome the size of the pan-genome increases. b Evolution of core-genome of 98 complete Lactobacillus genomes. After 70 genomes, the size of the core-genome is only decreasing by a few genes per included genome. Order of calculation was randomized for 98 sets, each represented by a single point
Table 1.
Core- and pan-genome of the genus Lactobacillus and of 5 Lactobacillus spec
| Genus | Species | N genomes | Genome size (Mbp) | NGenes | core | softcore | shell | cloud | pan |
|---|---|---|---|---|---|---|---|---|---|
| Lactobacillus | – | 98 | 2.47 ± 0.55 | 2274 ± 528 | 266 | 594 | 7249 | 12,957 | 20,800 |
| Lactobacillus | helveticus | 19 | 2.02 ± 0.13 | 2050 ± 164 | 908 | 1062 | 1133 | 1155 | 3350 |
| Lactobacillus | reuteri | 25 | 2.10 ± 0.12 | 2050 ± 117 | 897 | 1306 | 1364 | 1290 | 3960 |
| Lactobacillus | rhamnosus | 51 | 2.97 ± 0.08 | 2788 ± 71 | 811 | 1920 | 1736 | 1233 | 4889 |
| Lactobacillus | plantarum | 122 | 3.27 ± 0.13 | 3075 ± 140 | 1037 | 2144 | 2826 | 2640 | 7610 |
| Lactobacillus | delbrueckii | 29 | 1.88 ± 0.13 | 1873 ± 93 | 756 | 1042 | 1336 | 1082 | 3460 |
Fig. 2.
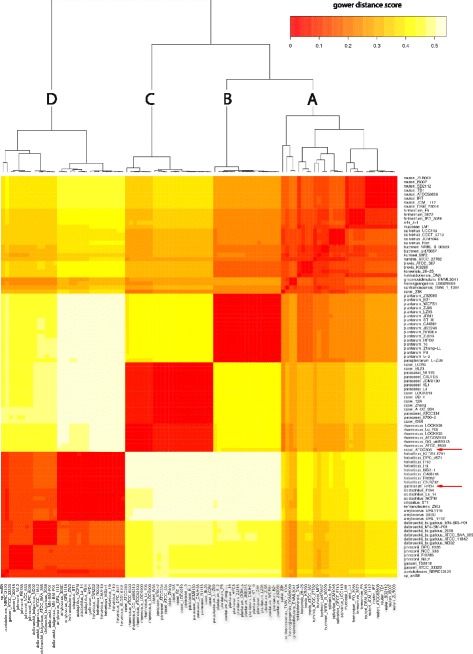
Core-genome clustering of genus Lactobacillus. Heatmap clustering according to 266 core genes from 98 Lactobacillus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, Outliers are marked with red arrows. a reuteri-fermentum-salivarius clade, (b) plantarum clade, (c) casei-paracasei-rhamnosus clade and (d) helveticus-delbrueckii-johnsonii clade
Fig. 3.
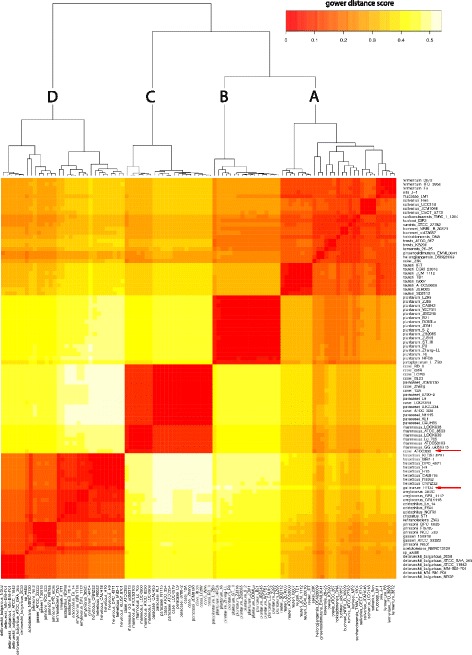
Pan- genome clustering of genus Lactobacillus. Heatmap clustering according to 20′800 pan-genome genes from 98 Lactobacillus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, Outliers are marked with red arrows. a reuteri-fermentum-salivarius clade, (b) plantarum clade, (c) casei-paracasei-rhamnosus clade and (d) helveticus-delbrueckii-johnsonii clade
Detailed analysis of strains with unexpected clustering
The L. casei type strain ATCC 393 did not clusters with other L. casei strains, but with 6 L. rhamnosus strains (Figs. 2 and 3). The genome of strain ATCC 393 contains 213 KEGG orthology (KO) assignments that are not present in any other of the 21 L. casei, L. paracasei and L. rhamnosus genomes. 11 of these 213 KOs are related to carbohydrate metabolism, 7 to environmental information processing and all other to hypothetical functions (Table 2). From 27 annotated KOs, 22 describe functions that are present in other L. casei, L. paracasei and L. rhamnosus isolates but are encoded by isogenes. However, L. casei ATCC 393 contains 5 KOs with a unique function, including one catalase (Table 3).
Table 2.
Unique genes in L. casei ATCC393, L. gallinarum HDF4 and in L. delbrueckii strains as identified with GhostKOALA functional categories
| KEGG orthology | L. casei ATCC393 | L. gallinarum HDF4 | L. delbruecki bulgaricus clade | L. delbrueckii diverse clade |
|---|---|---|---|---|
| Carbohydrate metabolism | 11 | 5 | 7 | 1 |
| Energy metabolism | 0 | 0 | 2 | 1 |
| Lipid metabolism | 0 | 2 | 3 | 0 |
| Nucleotide metabolism | 1 | 0 | 0 | 1 |
| Amino acid metabolism | 1 | 3 | 3 | 3 |
| Metabolism of other amino acid | 1 | 0 | 0 | 0 |
| Glycan biosynthesis and metabolism | 2 | 0 | 0 | 0 |
| Metabolism of cofactors and vitamins | 0 | 3 | 0 | 1 |
| Metabolism of terpenoids and polyketides | 0 | 1 | 0 | 0 |
| Biosynthesis of other secondary metabolites | 0 | 1 | 0 | 0 |
| Xenobiotics biodegradation and metabolism | 0 | 0 | 3 | 0 |
| Enzyme families | 2 | 1 | 0 | 0 |
| Genetic Information Processing | 2 | 11 | 1 | 1 |
| Environmental Information Processing | 7 | 9 | 6 | 0 |
| Cellular Processes | 5 | 6 | 3 | 0 |
| Organismal Systems | 1 | 0 | 0 | 0 |
| Human Diseases | 1 | 2 | 2 | 0 |
| Unclassified | 5 | 9 | 0 | 1 |
| Annotated KEGG orthologous | 27a | 38a | 12a | 5 |
| Hypothetical function | 186 | 143 | 30 | 5 |
| Query dataset | 213 | 181 | 42 | 10 |
aKEGG orthologous can be present in single or multiple categories
Table 3.
Unique genes of Lactobacillus strains from Table 2 with no isoenzymes in the pan-genome that they were compared to, which would comply the same function. K-number according to KEGG database
| Present in isolate | K-number | EC-number | Function |
|---|---|---|---|
| L. casei ATCC393 | K01788 | 5.1.3.9 | N-acylglucosamine-6-phosphate 2-epimerase |
| K03781 | 1.11.1.6 | Catalase | |
| K00681 | 2.3.2.2 | gamma-glutamyltranspeptidase | |
| K00681 | 3.4.19.13 | glutathione hydrolase | |
| K20997 | polysaccharide biosynthesis protein (pslA) | ||
| L. gallinarium HFD4 | K00278 | 1.4.3.16 | L-aspartate oxidase (nadB) |
| K00558 | 2.1.1.37 | DNA (cytosine-5)-methyltransferase 1 | |
| K03517 | 2.5.1.72 | quinolinate synthase (nadA) | |
| K18231 | Macrolide transporter symstem ATP-binding/permease protein (msrA) | ||
| L. delbrueckii diverse cluster | K00135 | 1.2.1.16 | Succinate semialdehyde dehydrogenase |
| K00926 | 2.7.2.2 | carbamate kinase | |
| K00611 | 2.1.3.3 | ortnithine carbamoyltransferase | |
| K02970 | small subunit ribosomal protein S21 |
L. zeae was not included in the pan/core-genome analyses because a closed genome is not available for the species. However, if the incomplete genome of L. zeae DSM 20178 is included, its clusters together with L. casei ATCC 393 and next to the rhamnosus clade (Additional file 5).
L. gallinarum HFD4 clusters in the core-genome within the helveticus clade (Fig. 2 and Additional file 6). In the pan-genome, however, it clusters outside of the helveticus clade (Fig. 3). Analyses of the 16S rRNA gene sequence search of HDF4 revealed over 99% identity with the 16S rRNA gene sequence of various L. helveticus strains. L. gallinarum HFD4 contains 181 KOs that are not present in the 8 L. helveticus strains. Beside 135 hypotheticals, 10 KOs are associated with genetic information processing and 9 KOs with environmental information processing. Isolate HFD4 possesses an L-aspartate oxidase, an enzyme that converts L-aspartate to oxaloacetate and a DNA (cytoseine-5)-methyltransferase 1, which catalyzes the conversion from L-aspartate-4-semialdehyde to L-homoserine. However, these two KOs do not allow the strain to produce additional amino acids compared to the 8 L. helveticus strains. Additionally, isolate HFD4 contains macrolide transport system ATP-binding/permease protein (Table 3).
Analysis of core- and pan-genome of the genus Lactobacillus
The metabolic capacity of the core- and pan-genome of the genus Lactobacillus was analyzed by using Brite protein family enrichment and pathway reconstruction in GhostKOALA. Reconstruction of protein families revealed an average increase of 6.1 fold from core- to pan-genome. A significant lower increase of 2.8-fold was observed in the class “genetic information processing” from core- to pan-genome and a significant higher increase of 17.9-fold in the “signaling and cellular processes” class (Table 4). The pathway reconstruction analysis revealed a 7.1-fold increase core- to pan-genome, a significant lower increase of 2.4-fold of genes in “genetic information processing” and a significant higher 24.9-fold increase for “Environmental information processing”, paralleling the observation in the protein family enrichment analysis.
Table 4.
Reconstruction of core-, softcore- and pan-genome of the genus Lactobacillus with Brite and Pathway algorithm of GhostKOALA
| Brite Reconstruction Result | n-fold increase | ||||
| Core | Softcore | Pan | Core-pan | Softcore-pan | |
| Orthologs and modules | 237 | 471 | 1650 | 7.0 | 3.5* |
| Protein families: metabolism | 180 | 320 | 1093 | 6.1_ | 3.4_ |
| Protein families: genetic information processing | 165 | 292 | 458 | 2.8* | 1.6* |
| Protein families: signaling and cellular processes | 27 | 73 | 484 | 17.9* | 6.6* |
| Total | 609 | 1156 | 3685 | 6.1_ | 3.2_ |
| Pathway Reconstruction Result | n-fold increase | ||||
| core | softcore | pan | core to pan | softcore-pan | |
| Metabolism | 303 | 502 | 2298 | 7.6* | 4.6* |
| Genetic Information Processing | 84 | 156 | 199 | 2.4* | 1.3* |
| Environmental Information Processing | 10 | 28 | 249 | 24.9* | 8.9* |
| Cellular Processes | 11 | 20 | 135 | 12.3_ | 6.8_ |
| Organismal Systems | 8 | 9 | 83 | 10.4_ | 9.2_ |
| Human Diseases | 18 | 33 | 109 | 6.1_ | 3.3_ |
| Total | 434 | 748 | 3073 | 7.1_ | 4.1_ |
* indicates p-value < 0.01
Core- and pan-genome of the type species Lactobacillus delbrueckii
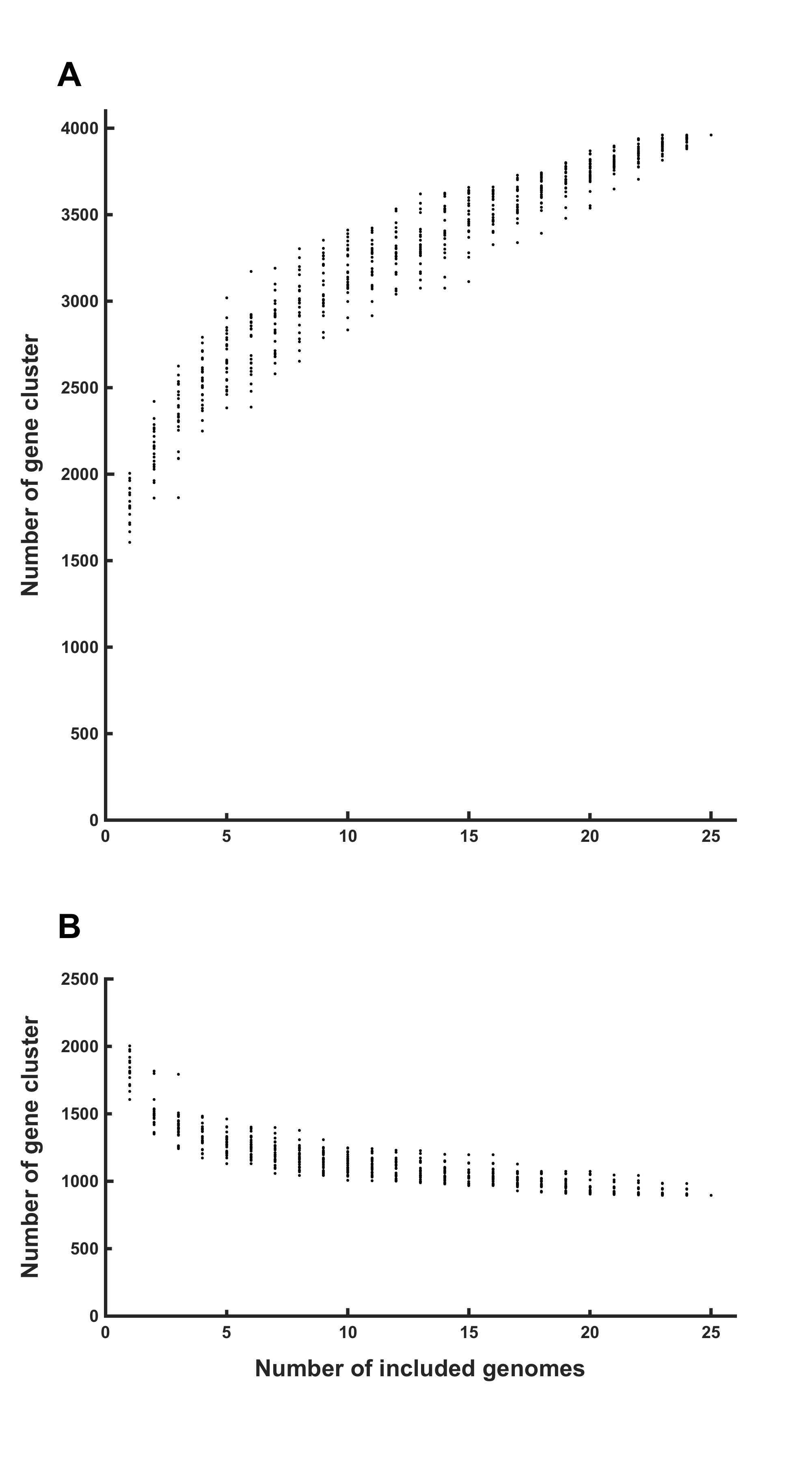
To gain insight in the core- and pan-genome of a Lactobacillus species, similar analyses as for the genus were performed with the type species of the genus: Lactobacillus delbrueckii (Additional files 7 and 8). The L. delbrueckii core-genome contained 756 genes, the softcore-genome 1042 genes and the pan-genome 3460 genes. The average genome size was 1873 ± 93 genes (Table 1). The pan-genome of L. delbrueckii is gaining only 4–5 genes per genome after 26 included genomes and can be considered as closed (Fig. 4).
Fig. 4.
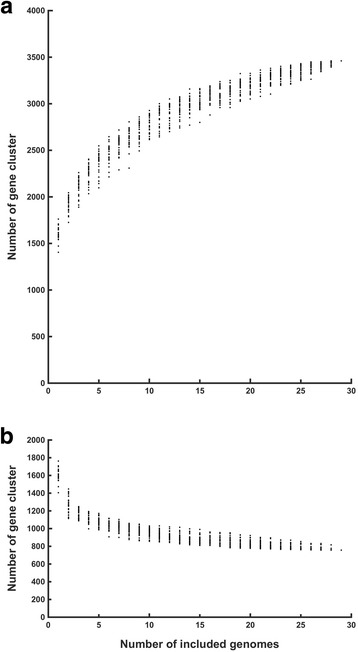
Pan- and core-genome evolution of Lactobacillus delbrueckii. a Evolution of the pan-genome for L. delbrueckii. After 20 included genomes, the pan-genome is closed. b Evolution of the core-genome for L. delbrueckii. Order of calculation was randomized for 29 sets, each represented with a single point
If L. delbrueckii MN-BM-F01, formerly L. acidophilus MN-BM-F01 [37], was excluded from the analyses, the core-genome increased only by 4 genes. This supports strongly the new classification of MN-BM-F01 from L. acidophilus to L. delbrueckii.
The quality criterion for genomes was set that the gene number should be within a range of ± 2σ around the average gene and protein number. If genomes that do not match this criterion were included in the analyses, e.g. the genomes of L. delbrueckii JCM1002, L. delbrueckii JCM1012 and L. delbrueckii CRL871, the core-genome dropped dramatically from 756 to 302 core genes, showing clearly the sensibility of the core genome for low quality sequenced genomes.
The 29 L. delbrueckii strains are separated in 2 clades in both the softcore- and pan-genome tree (Fig. 5). In the core-genome a small third clade containing the 3 strains PB2003_004-T3–4, ND02 and JCM17838 occurs. One clade in the pan-genome tree contains 13 strains that all belong to Lactobacillus delbrueckii subsp. bulgaricus and was therefore designated “bulgaricus” clade. The second clade, contains 16 isolates of the subspecies Lactobacillus delbrueckii subsp. delbrueckii, −lactis, −indicus, −sunkii, −jakobsenii and -bulgaricus, was designated “diverse” clade.
Fig. 5.
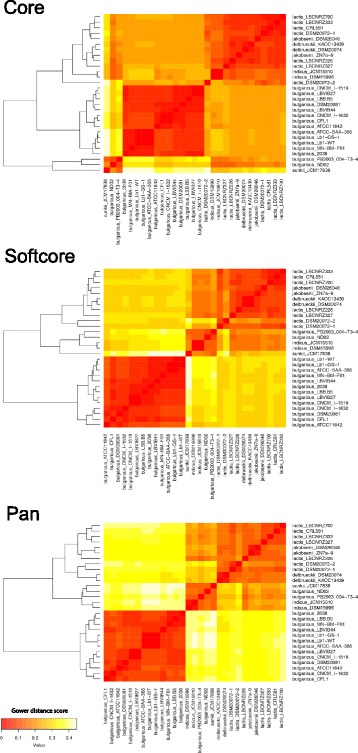
Heatmap of core-, softcore- and pan-genome for L. delbrueckii. In the core-genome heatmap the bulgaricus clade is embedded within the diverse clade. In the softcore- and pan-genome heatmap is the bulgaricus and the diverse clade are separated. Gower distance score based on ANI: Red = more similar, white = less similar
The average ANI over all L. delbrueckii genomes was 96.58 ± 0.93%. The average ANI in the bulgaricus clade was 98.05 ± 0.23% and in the diverse clade 96.23 ± 0.93%.
Core-genomes of both clades were constructed and the core genes were categorized with GhostKOALA. The bulgaricus clade core-genome contains 42 KOs that are not found in the diverse core-genome, of which 30 are hypothetical KOs. The 12 functionally annotated KOs are associated with carbon metabolism and environmental information processing (Table 2), including a complete sucrose-specific type II PTS system. There were, however, no functional differences between the two clades. This shows that evolutionary distinct genes with predicted identical functions are conserved in the strains in the clades. This is illustrated by the different aspartate kinases found in the 2 core genomes. Asparte kinase connects the glycine, serine and threonine metabolism with a number of other amino acid synthesis pathways. The enzymes in the bulgaricus clades are more than 79% identical to each other and the enzymes the diverse clades more than 95%. The two types of enzymes; however, have only an identity percentage of 34% or less and thus seem evolutionary distinct. This suggests that the L. delbrueckii subsp. bulgaricus is evolving differently in aspartate metabolism compared to the non-bulgaricus strains.
The diverse clade core-genome contains 9 KOs that are not present in the bulgaricus clade. Of these KOs, 5 encoded for hypothetical KOs, 3 for amino acid metabolism KOs and one small subunit ribosomal protein S21 (Table 3). Further, an α-glucoside transport system is uniquely present in the diverse cluster. This ABC transporter transports, amongst others, maltose.
Analysis of core- and pan-genome of Lactobacillus delbrueckii
The metabolic capacity of the core- and pan-genome of L. delbrueckii was analyzed using protein family enrichment and pathway reconstruction in GhostKOALA. An increase of 1.8-fold from core- to pan-genome was measured with a significant lower increase of 1.4-fold in the class “genetic information processing” from core- to pan-genome and a significant higher 2.6 -fold increase in the “signaling and cellular processes” class (Additional file 9). In the pathway reconstruction, a significant lower increase of genes in “genetic information processing” and a higher increase for “Environmental information processing” was measured. These findings parallel the previous analysis the core- and pan-genome of the genus Lactobacillus (Table 4).
The reconstruction according to manually defined functional units, KEGG modules, revealed that short pathways are completely present in the core-genome whereas 1 or 2 enzymes were missing in many longer pathways (Additional file 10). However, many of such longer pathways such as the glycolysis, purine ribonucleotide biosynthesis, RNA polymerase, aminoacyl-tRNA biosynthesis and the ribosome protein complex are complete in the softcore. Taken together, fundamental processes in the cell are conserved in the softcore-genome and processes involved in interactions with the environment are only complete in the pan-genome.
Analyses of core- genes of L. delbrueckii
To determine whether EcoSNPs in the core genes are responsible for the occurrence of the two clades in the core-genome tree, the consensus sequence for all 756 genes in the L. delbrueckii core-genome was calculated and SNPs in all 29 strains analyzed. In total, 53′583 SNPs were detected in all core genes. However, no cluster specific SNPs were detected, showing that the formation of 2 clades in the clustering is not dependent on a small set of EcoSNPs.
To analyze if all genes in the core genome had a similar phylogenetic tree, the tree of every gene in the core-genome was compared to the tree based on the core-genome (Fig. 5). The top 5% (n = 38) of genes with trees most similar to the core-genome tree had an average nodal distance score of 2.10 ± 0.13 and an average gene size of 1424 bp. The consensus sequences of the genes had an SNP density of 75.3 SNPs/kb. Of these 38 genes, 9 genes were interacting with DNA or RNA and there were no hypothetical genes (Additional file 11). The genes with trees least similar to the core-genome tree had an average nodal distance score of 6.47 ± 0.78, an average gene size of 407 bp. Of the 38 genes, 16 are either annotated as 30S or 50S ribosomal proteins. The consensus sequences of the 38 genes had an SNP density of 26.54 SNPs/kb, a density that is clearly lower than the average SNP density of 70.25 SNPs/kb. The 38 genes are thus highly homologous. This shows that highly conserved genes have a different phylogenetic tree than moderate conserved genes and such genes are not useful for phylogenetic reconstruction at species level.
Potential HGT in Lactobacillus delbrueckii
To detect whether gene transfer appeared between the two clades in L. delbrueckii, we screened for potential HGT-genes within the two clades. In the L. delbrueckii pan-genome, a total of 57 genes were detected that were present in a subset of strains in both clades and are therefore potentially involved in HGT. 42 of those 57 genes encode for hypothetical proteins or are associated with phages or transposons (Additional file 12). Phages and transposons are commonly associated with HGT and their occurrence shows that our simple algorithm can detect HGT related genes.
Core- and pan-genome of other Lactobacillus species
To determine if the type species L. delbrueckii is representative for other Lactobacillus species, we calculated the core- and pan-genome for four other species, one from each of the four clades observed in the core-genome clustering; L. helveticus, L. rhamnosus, L. reuteri and L. plantarum (Fig. 1). L. helveticus has a core-genome of 908 and pan-genome of 3350 genes with an average genome size of 2050 ± 164 genes (Table 1, Additional files 13 and 14). A similar ratio of core-genome to average genome size was calculated for L. reuteri with 897 core genes and 3960 pan genes on an average genome size of 2050 ± 117 genes (Table 1, Additional files 15 and 16). A lower ratio of core-genome to average genome size was calculated for L. rhamnosus with 811 core genes and 4889 pan genes on an average genome size of 2788 ± 71 genes (Table 1, Additional files 17 and 18). The core- and pan-genome for those three species are all closed (Additional files 19, 20 and 21). The biggest core-genome was calculated for the species L. plantarum with 1037 core-genes which is around 34% of the average genes in a L. plantarum genome (Table 1, Additional files 22 and 23). Neither the core- nor the pan-genome of L. plantarum were closed even after 122 genomes were included (Additional file 24).
Clustering of core- and pan-genome of other Lactobacillus species
L. rhamnosus and L. plantarum clustered in two clades (Additional files 25 and 26), L. reuteri and L. helveticus in a number of minor clades (Additional files 27 and 28). The smaller L plantarum clade contained the type strain L. plantarum subsp. argentoratensis DSM 16365 and was designated the argentoratensis clade whereas the bigger clade contained the type strain L. plantarum subsp. plantarum ATCC 14917 and was designated the plantarum clade.
Potential HGT in other Lactobacillus species
The species L. plantarum and L. rhamnosus cluster in 2 clearly separated clades and were used for HGT analyses. L. plantarum and L. rhamnosus possess 95 and 38 potential HGT genes in their pan-genome, respectively (Table 5). The majority of those genes encode hypothetical proteins. In L. helveticus only one gene was detected, a transposase, and in L. reuteri none.
Table 5.
Gene annotation for potential HGT genes in Lactobacillus species
| Genes related to | ||||||
|---|---|---|---|---|---|---|
| Organism | Clades | transferred genes | phage | transposon | hypothetical | others |
| L. delbrueckii | 2 | 57 | 0 | 5 | 26 | 26 |
| L. helveticus | 3 | 1 | 0 | 1 | 0 | 0 |
| L. plantarum | 2 | 95 | 5 | 2 | 44 | 44 |
| L. reuteri | 2 | 0 | 0 | 0 | 0 | 0 |
| L. rhamnosus | 2 | 38 | 6 | 5 | 11 | 16 |
HGT between clades in the genus Lactobacillus
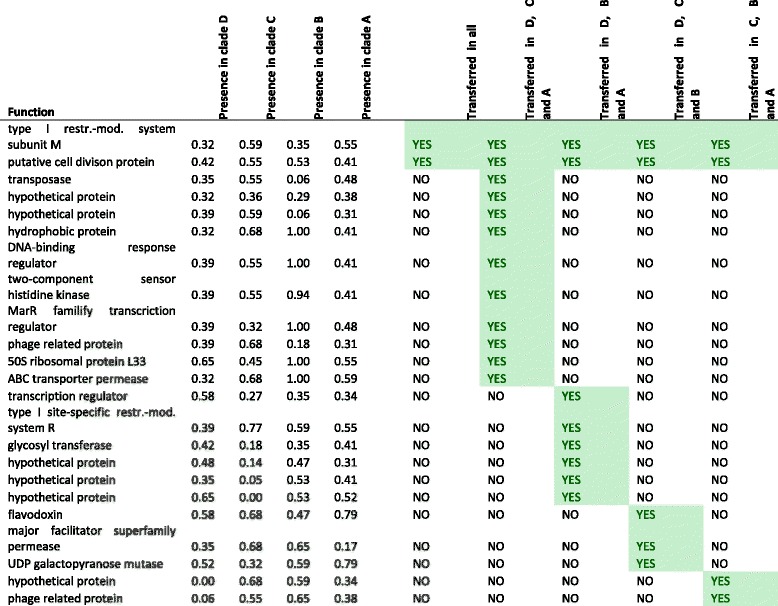
Since we detected in four out of five analyzed species potential genes related to HGT, also the pan-genome of Lactobacillus genus was analyzed for HGT. The 20′800 pan genes of the genus Lactobacillus contains 2 genes occuring in all 4 clades with a probability of 30–70% (Table 6). Gene 1 encodes a type I restriction-modification system subunit M (ID = YP_004888889 in L. plantarum WCFS1) with a length of 539 aa. Gene 2 encodes a putative cell division protein (ADY84228 in L. delbrueckii 2038) with a length of 659 aa. Therefore, HGT occurs even between Lactobacillus species.
Table 6.
Gene annotation of potential HGT genes in the genus Lactobacillus. Genes that were potentially horizontally transferred within clades are marked in green
Discussion
We clustered 98 complete sequenced genomes of 32 species of the genus Lactobacillus and calculated core- and pan-genome. The core-genome contained 266 genes. A core-genome of 175 Lactobacillus isolates and 26 strains from 8 Lactobacillus-related genera calculated with similar parameters presented a core-genome of only 73 genes [38]. The lower amount of core genes in the latter study is likely due to the higher number of genomes in the dataset, the integration of genomes from other genera, and including draft genomes in the analysis [18]. Especially incomplete or poorly assembled genomes have a large impact on the core-genome, as shown for core-genome of L. delbrueckii in this work. Since the core-genome is very sensitive to heterogeneous datasets and low sequence quality, a prior quality selection is necessary [16, 39]. The minimum standards for submitting a prokaryotic genome to Genbank are, amongst others, at least one copy of 5S, 16S and 23S rRNA-operon, a tRNA gene for each amino acid, and a ratio of genes to genome length close to 1 [40]. However, we showed that those standards are not restrictive enough for core-genome analysis and an additional selection of 2-fold the standard deviation of genes number was therefore used.
Another study using closed genomes revealed that the core-genome of 67 Lactobacillus strains from 25 species contained 311 genes [39]. The core-genome of Lactobacillus in our study was however, not closed after 67 genomes and between 290 and 406 genes (Fig. 1). The difference in the core-genomes is therefore likely due to the lower number of genomes in the previous study. The pan-genome of the Lactobacillus genus based on 67 strains contained 11′047 genes, clearly less than the pan-genome calculated in this study: 16148–18,318 genes for 67 genomes and 20′800 genes for 98 genomes. The larger pan-genome in our study is likely due to the more heterogenic dataset containing 32 species. Remarkably, the pan-genome of Lactobacillus is 4 times larger than the combined pan-genome of the narrow range genera Staphylococcus and Macrococcus. This exemplifies the wide habitat range and versatility of the Lactobacillus genus compared to Staphylococcus and Macrococcus [39, 41]. Moreover, the pan-genome of Lactobacillus was not closed after 98 genomes (Fig. 1). A closed pan-genome is rapidly reached in species that occur in a few habitats only or have a low capacity to acquire genes, such as Bacillus anthracis [42], and, in this study, L. delbrueckii. Non-closed pan-genomes are typical for heterogeneous datasets, like the Lactobacillus dataset in this study, for species with diverse habitats, like L. plantarum in this study, and in species with high acquisition of genes, such as natural competent streptococci [16, 21]. Acquisition of genes occurs in lactobacilli occurs via HGT, which parallels observations in another genus frequently associated with the human gut: Bifidobacteria [43].
We analyzed the Lactobacillus type species L. delbrueckii, and the species L. helveticus, L. reuteri, L. rhamnosus and L. plantarum in more detail. The relative core-genome to average genome size was similar for all 5 species. In general, species with more genomes included in pan-genome analyses and higher genomic diversity, such as L. plantarum, have a smaller core-genome compared to the average genome size than species with less included genomes and a lower genomic diversity such as L. delbrueckii [44, 45]. A previous study revealed a core-genome of 2164 genes for 40 L. rhamnosus genomes [46] which is much higher than the 811 genes in our core-genome, yet close to soft-core genome of 1920 genes from 51 isolates (Table 1). Other studies revealed a L. plantarum core-genome of 1957 genes from 54 genomes and a L. rhamnosus core-genome of 2419 genes from 100 included genomes [47, 48], again much higher compared to the core-genomes in this study (Table 1). These core-genomes were; however, based on conserved function and not on sequence identity. The homologous-based comparison in this study is preferable, because it is based on true evolutionary events. This is clearly illustrated by the two clades in the core-genome of L. delbrueckii. The clades are clearly different from evolutionary view, but possess identical functional capacity (Table 3).
Analysis of the core- and pan-genome content revealed that fundamental processes like processing of genetic information and key metabolic pathways were conserved in the core-genome of L. delbrueckii, whereas environmental genes were not. These results are similar with compositions found in S. aureus [21] and P. aeruginosa [49], and parallels previous finding in lactobacilli [39].
In general, clustering of core- and pan-genome resulted in highly similar trees. Since the core genome contains the same genes for all isolates, the phylogenetic trees have to be based on information in the core-genome sequences. The strains of Lactobacillus clustered in species specific clusters (Fig. 1), with the exception of two strains: L. casei ATCC 393 and L. gallinarum HFD4. Differences of type strain L. casei ATCC 393 with other strains of L. casei are well documented [50–59]. The clustering in this study shows that ATCC 393 is most closely related to L. zeae DSM 20178 (Additional file 5), which confirms previous studies [53, 60]. However, a reclassification of type strain ATCC 393 as L. zeae was rejected by the Judicial Commission of the International Committee on Systematics of Bacteria [61]. Strain L. gallinarum HFD4 clustered different in core- and pan-genome clustering (Figs. 2 and 3). Genotypic differentiation for L. gallinarum and L. helveticus based on 16S rRNA sequence is not evident [62]. Initially, L. gallinarum and L. helveticus were differentiated based on their sugar fermentation pattern; L. gallinarum ferments amygdalin, cellobiose, salicin and sucrose, L. helveticus not [63]. However, none of the 181 KOs uniquely present in HDF4 encodes for any of these carbon sources and the phylogenetic differentiation between L. helveticus and L. gallinarum remains therefore unclear.
A separation in subspecies in the clustering of L. delbrueckii was already detected in a previous study based on MLST [64]. The separation is also visible in the ANI values within the clades, which were higher between members of the bulgaricus clade than between members of the mixed clade. Nevertheless, the ANI values were still above the cutoff value of 94% for different species [65] and all the analyses strains belong therefore to the same species.
Separation of populations into groups and further to species has been explained with several models. The infinitely many genes (IMG) model relates evolution and separation to all non-core-genome genes [66]. Since the bulgaricus clade separation already appears in the core-genome, the IMG model does not fit the evolution of L. delbrueckii. The ecotype model relates a mutation, identifiable as an ecoSNP, within a population to evolve into two subpopulations [67, 68]. EcoSNPs were not found in the L. delbrueckii analyses. However, ecoSNPs are only visible in recently diverged populations [12] whereas the division of L. delbrueckii into subspecies might not be recent. Convergent evolution was suggested in the genus Lactobacillus [69]. The example of two distinct aspartate kinases in the two clades of L. delbrueckii suggests convergent-like evolution. The aspartate kinase activity is; however, only an annotated function and it is possible that the two enzymes have different functions or activities in the cell, which would speak against convergent evolution.
The enrichment of environmental function in the accessory genome suggests that a L. delbrueckii population occupies a novel niche and then adapts via gene gain. In addition, gene exchange between the L. delbrueckii subpopulations occurred (Table 5). L. delbrueckii evolved therefore into subspecies with a mechanism that resembles the parapatric model used for specification in sexually reproducing organisms: a novel niche is occupied by a subpopulation that differentiates, but gene exchange with its original population is still possible.
The detection of HGT in L. plantarum and L. rhamnosus suggests they evolved similarly. Remarkably, L. plantarum, and L. rhamnosus were both considered as nomadic in a recent study [70] and such lifestyle provides opportunities for parapatric specification. L. reuteri and L. helveticus were not considered as nomadic [70] and indeed no evidence for parapatric differentiation in these species was found in our analyses.
Conclusion
The sequenced based core- and pan-genome analyses of Lactobacillus and are useful to cluster and classify lactobacilli. The core- and pan-genome clustering yield similar trees. However, core-genomes clustering does not respect environmental adaptations, specific evolution or horizontal gene transfer. Pan-genome clustering was therefore necessary to show that L. delbrueckii evolved into subspecies via a parapatric-like model.
Our data provide novel insight how lactobacilli evolve and are related. This knowledge is useful for rational selection of strains for use in food fermentation.
Additional files
All strains used for core- and pan-genome analysis study. (XLSX 54 kb)
Home-made scripts used in this study. (XLSX 39 kb)
Core-genome fasta for genus Lactobacillus (Fasta). (XLSX 59 kb)
Pan-genome fasta for genus Lactobacillus (Fasta). (XLSX 48 kb)
Pan-genome clustering of genus Lactobacillus. Heatmap clustering according to 20,969 pan-genome genes from 99 Lactobacillus genomes including the non-complete genome of L. zeae DSM 20178. Gower distance score based on ANI: Red = more similar, white = less similar. L. zeae DSM 20178 marked with a red arrow. (FASTA 305 kb)
Softcore-genome clustering of genus Lactobacillus. Heatmap clustering according to 594 softcore genes from 98 Lactobacillus genomes. Gower distance score based on ANI: Red = more similar, white = less similar. (FASTA 944 kb)
Core-genome fasta for Lactobacillus delbrueckii (Fasta). (FASTA 278 kb)
Pan-genome fasta for Lactobacillus delbrueckii (Fasta). (FASTA 1226 kb)
Reconstruction of core, softcore- and pan-genome of Lactobacillus delbrueckii species with the Brite and pathway algorithm of GhostKOALA. (FASTA 219 kb)
Reconstruction of core, softcore- and pan-genome of Lactobacillus delbrueckii species with the Module algorithm of GhostKOALA. Pathways are fractured in blocks (number). Green = pathway complete; yellow = 1 block is missing in the pathway; red = 2 or more blocks are missing in the pathway. (FASTA 1473 kb)
{kind=link}
TOPD/FMTS nodal distance scores and SNPs evaluation. Genes are sorted according to the nodal distance scores compared with the pan-genome tree and the 5% and 95% quantile is listed. Sum SNP – The sum of all SNP according to the consensus sequence of the 29 homologous sequences; SNP/base – sum of SNP divided by length of consensus sequence in nucleotide; Sum polyvariable positions (SPP) – sum of all positions with 3 or more different nucleotides a specific position; Length in bp – length of consensus sequence (JPEG 206 kb)
Potential HGT in Lactobacillus delbrueckii. Clade B = bulgaricus clade, clade D = diverse clade, Number indicates copies of gene within the genome, presence = possibility of occurrence within the clade in percent. (TXT 2464 kb)
{kind=link}
Core-genome fasta for Lactobacillus helveticus (Fasta). (JPEG 215 kb)
{kind=link}
Pan-genome fasta for Lactobacillus helveticus (Fasta). (JPEG 289 kb)
Core-genome fasta for Lactobacillus reuteri (Fasta). (FASTA 312 kb)
Pan-genome fasta for Lactobacillus reuteri (Fasta). (FASTA 2283 kb)
{kind=link}
Core-genome fasta for Lactobacillus rhamnosus (Fasta). (JPEG 302 kb)
Pan-genome fasta for Lactobacillus rhamnosus (Fasta). (PDF 3325 kb)
Pan- and core-genome evolution of L. helveticus. A Evolution of the pan-genome for L. helveticus. After 14 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. helveticus. Order of calculation was randomized for 19 sets, each represented with a single point. (PDF 5105 kb)
Pan- and core-genome evolution of L. reuteri. A Evolution of the pan-genome for L. reuteri. After 20 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. reuteri. Order of calculation was randomized for 25 sets, each represented with a single point. (PDF 2010 kb)
Pan- and core-genome evolution of L. rhamnosus. A Evolution of the pan-genome for L. rhamnosus. After 20 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. rhamnosus. Order of calculation was randomized for 51 sets, each represented with a single point. (PDF 1978 kb)
Core-genome fasta for Lactobacillus plantarum (Fasta). (FASTA 193 kb)
Pan-genome fasta for Lactobacillus plantarum (Fasta). (FASTA 5115 kb)
Pan- and core-genome evolution of L. plantarum. A Evolution of the pan-genome for L. plantarum. The pan-genome remains open even after 122 genomes were included. B Evolution of the core-genome for L. plantarum. Order of calculation was randomized for 122 sets, each represented with a single point. (PDF 6945 kb)
Pan-genome heatmap of L. rhamnosus. Heatmap clustering according to 4889 pan-genome genes from 51 Lactobacillus rhamnosus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (PDF 7940 kb)
Pan-genome heatmap of L. plantarum. Heatmap clustering according to 7610 pan-genome genes from 122 Lactobacillus plantarum genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (FASTA 469 kb)
Pan-genome heatmap of L. reuteri. Heatmap clustering according to 3960 pan-genome genes from 25 Lactobacillus reuteri genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (FASTA 1001 kb)
Pan-genome heatmap of L. helveticus. Heatmap clustering according to 3350 pan-genome genes from 19 Lactobacillus helveticus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (XLSX 36 kb)
Acknowledgements
We acknowledge Bruno Contreras-Moreira, CSIC – Estación Experimental de Aula Dei (EEAD) / Fundación ARAID, Spain for his bioinformatics support with the program GET_HOMOLOGUES.
Funding
This project was financed by the Swiss National Science Foundation with the National Research Program 69, project number 145214.
Availability of data and materials
All data generated or analysed in this study are included in this published article and its supplementary information files.
Abbreviations
- ANI
Average nucleotide identity
- DDH
DNA-DNA hybridization
- HGT
Horizontal gene transfer
- KO
KEGG orthology
- NGS
Next generation sequencing
- OMCL
Ortho Markov Cluster algorithm
- WGS
Whole genome sequencing
Authors’ contributions
RI, LM, and MS designed the study. RI and MS performed analyses. MS and LM supervised the study. RI, LM, and MS wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4601-5) contains supplementary material, which is available to authorized users.
Contributor Information
Raffael C. Inglin, Email: raffael.inglin@hest.ethz.ch
Leo Meile, Email: leo.meile@hest.ethz.ch.
Marc J. A. Stevens, Email: marc.stevens@hest.ethz.ch
References
- 1.Ekblom R, Wolf JBW. A field guide to whole-genome sequencing, assembly and annotation. Evol Appl. 2014;7:1026–1042. doi: 10.1111/eva.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet. 2015;16:275–284. doi: 10.1038/nrg3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leekitcharoenphon P, Nielsen EM, Kaas RS, Lund O, Aarestrup FM. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One. 2014;9 10.1371/journal.pone.0087991. [DOI] [PMC free article] [PubMed]
- 5.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;12:427–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Vandamme P, Peeters C. Time to revisit polyphasic taxonomy. Antonie Van Leeuwenhoek. 2014;106:57–65. doi: 10.1007/s10482-014-0148-x. [DOI] [PubMed] [Google Scholar]
- 7.Colwell RR. Polyphasic taxonomy of the genus Vibrio: numerical taxonomy of Vibrio cholerae, Vibrio parahaemolyticus, and related Vibrio species. J Bacteriol. 1970;104(I):410–433. doi: 10.1128/jb.104.1.410-433.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray RGE, Brenner DJ, Colwell RR, de Vos P, Goodfellow M, Grimont PAD, et al. Report of the ad hoc committee on approaches to taxonomy within the Proteobacteria. Int J Syst Bacteriol. 1990:213–5.
- 9.Vandamme P, Pot B, Gillis M, de Vos P, Kersters K, Swings J. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol Rev. 1996;60:407–438. doi: 10.1128/mr.60.2.407-438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stackebrandt E, Ebers J. Taxonomic parameters revisited: tarnished gold standards. Microbiol Today. 2006;33:152–155. [Google Scholar]
- 11.Wayne LG, Brenner DJ, Colwell RR, Grimont P a. D, Kandler O, Krichevsky MI, et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 1987;37:463–464. doi:10.1099/00207713-37-4-463.
- 12.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabo G, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 15.Deloger M, El Karoui M, Petit MAA. Genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J Bacteriol. 2009;91:91–99. doi: 10.1128/JB.01202-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Naomi L, et al. Genome analysis of multiple pathogenic isolates of for Streptococcus agalactiae: implications for the microbial “pan-genome”. PNAS. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaas RS, Friis C, Ussery DW, Aarestrup FM. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics. 2012;13:577. doi: 10.1186/1471-2164-13-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefébure T, Bitar PDP, Suzuki H, Stanhope MJ. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol. 2010;2:646–655. doi:10.1093/gbe/evq048. [DOI] [PMC free article] [PubMed]
- 19.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Georgiades K, Raoult D. Defining pathogenic bacterial species in the genomic era. Front Microbiol 2011;1:1–13 doi:10.3389/fmicb2010.00151. [DOI] [PMC free article] [PubMed]
- 21.Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BØ. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci. 2016;113:3801–3809. doi: 10.1073/pnas.1523199113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bezuidt OK, Pierneef R, Gomri AM, Adesioye F, Makhalanyane TP, Kharroub K, et al. The Geobacillus pan-genome: implications for the evolution of the genus. Front Microbiol. 2016;7:1–9. doi: 10.3389/fmicb.2016.00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein EJC, Tyrrell KL, Citron DM. Lactobacillus species: taxonomic complexity and controversial susceptibilities. Clin Infect Dis. 2015;60(Suppl 2):98–107. doi: 10.1093/cid/civ072. [DOI] [PubMed] [Google Scholar]
- 24.Claesson MJ, Van Sinderen D, O’Toole PW. The genus Lactobacillus - a genomic basis for understanding its diversity. FEMS Microbiol Lett. 2007;269:22–28. doi: 10.1111/j.1574-6968.2006.00596.x. [DOI] [PubMed] [Google Scholar]
- 25.Panel EFSA-NDA. Scientific opinion on the substantiation of a health claim related to glucosamine and maintenance of joints pursuant to article 13(5) of regulation (EC) no 1924/2006. EFSA J. 2015;13:3951. doi: 10.2903/j.efsa.2015.3951. [DOI] [Google Scholar]
- 26.Saito T. Selection of useful probiotic lactic acid bacteria from the Lactobacillus acidophilus group and their applications to functional foods. Anim Sci J. 2004;75:1–13. doi: 10.1111/j.1740-0929.2004.00148.x. [DOI] [Google Scholar]
- 27.Goh Y-J, Klaenhammer TR. Genomic features of Lactobacillus species. Front Biosci. 2009;(14):1362–86. [DOI] [PubMed]
- 28.NCBI Resource Coordinators Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2016;44:D7–19. doi: 10.1093/nar/gkv1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins MD, Rodrigues U, Ash C, Aguirre M, Farrow JAE, Martinez-Murcia A, et al. Phylogenetic analysis of the genus Lactobacillus and related lactic acid bacteria as determined by reverse transcriptase sequencing of 16S rRNA. FEMS Microbiol Lett. 1991;77:5–12. doi: 10.1111/j.1574-6968.1991.tb04313.x. [DOI] [Google Scholar]
- 30.Felis GE, Dellaglio F. Taxonomy of lactobacilli and bifidobacteria. Curr Issues Intest Microbiol. 2007;8:44–61. [PubMed] [Google Scholar]
- 31.Salvetti E, Torriani S, Felis GE. The genus Lactobacillus: a taxonomic update. Probiotics Antimicrob Proteins. 2012;4:217–226. doi: 10.1007/s12602-012-9117-8. [DOI] [PubMed] [Google Scholar]
- 32.Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Stoeckert CJ, Jr, Roos DS. Ortho MCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gower JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966;53(3/4):325–338. doi: 10.2307/2333639. [DOI] [Google Scholar]
- 35.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–731. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Puigbò P, Garcia-Vallvé S, McInerney JO. TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics. 2007;23:1556–1558. doi: 10.1093/bioinformatics/btm135. [DOI] [PubMed] [Google Scholar]
- 37.Yang L, Yun C, Zhiwei L, Yudong S, Zhouyong L, Zhao X. Correction for Yang et al., complete genome sequence of Lactobacillus delbrueckii subsp. bulgaricus MN-BM-F01. Genome Announc. 2016;4:2016. doi: 10.1128/genomeA.00420-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Z, Harris HMB, McCann A, Guo C, Argimon S, Zhang W, et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun. 2015;6 10.1038/ncomms9322. [DOI] [PMC free article] [PubMed]
- 39.Mendes-Soares H, Suzuki H, Hickey RJ, Forneya LJ. Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. J Bacteriol. 2014;196:1458–1470. doi: 10.1128/JB.01439-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genome Annotation Coordinators NCBI. NCBI prokaryotic genome annotation standards. 2017. [Google Scholar]
- 41.Suzuki H, Lefébure T, Bitar P, Stanhope MJ. Comparative genomic analysis of the genus Staphylococcus including Staphylococcus aureus and its newly described sister species Staphylococcus simiae. BMC Genomics. 2012;13:38. doi: 10.1186/1471-2164-13-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouli L, MBengue M, Robert C, Ndiaye M, La Scola B, Raoult D. Genomic analysis of three African strains of Bacillus anthracis demonstrates that they are part of the clonal expansion of an exclusively pathogenic bacterium. New Microbes New Infect. 2014;2:161–169. doi: 10.1002/nmi2.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vazquez-Gutierrez P, Stevens MJA, Gehrig P, Barkow-Oesterreicher S, Lacroix C, Chassard C. The extracellular proteome of two Bifidobacterium species reveals different adaptation strategies to low iron conditions. BMC Genomics. 2017;18:41. doi: 10.1186/s12864-016-3472-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siezen RJ, Tzeneva V a., Castioni A, Wels M, Phan HTK, Rademaker JLW, et al. Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ Microbiol 2010;12:758–773. doi:10.1111/j.1462-2920.2009.02119.x. [DOI] [PubMed]
- 45.Song Y, Sun Z, Guo C, Wu Y, Liu W, Yu J, et al. Genetic diversity and population structure of Lactobacillus delbrueckii subspecies bulgaricus isolated from naturally fermented dairy foods. Sci Rep. 2016;6:22704. doi: 10.1038/srep22704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceapa C, Davids M, Ritari J, Lambert J, Wels M, Douillard FP, et al. The variable regions of Lactobacillus rhamnosus genomes reveal the dynamic evolution of metabolic and host-adaptation repertoires. Genome Biol Evol. 2016;8:1889–1905. doi: 10.1093/gbe/evw123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martino ME, Bayjanov JR, Caffrey BE, Wels M, Hughes S, Gillet B, et al. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ Microbiol. 2016;18:1–41. doi: 10.1111/1462-2920.13455. [DOI] [PubMed] [Google Scholar]
- 48.Douillard FP, Ribbera A, Kant R, Pietilä TE, Järvinen HM, Messing M, et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013;9 10.1371/journal.pgen.1003683. [DOI] [PMC free article] [PubMed]
- 49.Ozer EA, Allen JP, Hauser AR. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools spine and AGEnt. BMC Genomics. 2014;15:1–17. doi: 10.1186/1471-2164-15-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mills CK, Lessel EF. Lactobacterium zeae Kuznetsov, a later subjective synonym of Lactobacillus casei (Orla-Jensen) Hansen and Lessel. Int J Syst Bacteriol. 1973;23:430–432. doi: 10.1099/00207713-23-4-430. [DOI] [Google Scholar]
- 51.Dellaglio F, Bottazzi V, Vescovo M. Deoxyribonucleic acid homology among Lactobacillus species of the subgenus Streptobacterium Orla-Jensen. Int J Syst Bacteriol. 1975;25:160–172. doi: 10.1099/00207713-25-2-160. [DOI] [Google Scholar]
- 52.Dellaglio F, Dicks L, du Toit M, Torriani S. Designation of ATCC 334 in place of ATCC 393 (NCDO 161) as the neotype strain of Lactobacillus casei subsp. casei and rejection of the name Lactobacillus paracasei (Collins et al., 1989) Int J Syst Bacteriol. 1991;41:340–342. doi: 10.1099/00207713-41-2-340. [DOI] [Google Scholar]
- 53.Dicks LMT, Du Plessis EM, Dellaglio F, Lauer E. Reclassification of Lactobacillus casei subsp. casei ATCC 393 and Lactobacillus rhamnosus ATCC 15820 as Lactobacillus zeae nom. Rev., designation of ATCC 334 as the neotype of L. casei subsp. casei, and rejection. Int J Syst Bacteriol. 1996;46:337–340. doi: 10.1099/00207713-46-1-337. [DOI] [PubMed] [Google Scholar]
- 54.Collins MD, Phillips BA, Zanoni P. Deoxyribonucleic acid homology studies of Lactobacillus casei, Lactobacillus paracasei sp. nov., subsp. paracasei and subsp. tolerans, and Lactobacillus rhamnosus sp. nov., comb. nov. Int J Syst Bacteriol. 1989;39:105–108. doi: 10.1099/00207713-39-2-105. [DOI] [Google Scholar]
- 55.Ferrero M, Cesena C, Morelli L, Scolari G, Vescovo M. Molecular characterization of Lactobacillus casei strains. FEMS Microbiol Lett. 1996;140:215–219. doi: 10.1111/j.1574-6968.1996.tb08339.x. [DOI] [Google Scholar]
- 56.Mori K, Yamazaki K, Ishiyama T, Katsumata M, Kobayashi K, Kawai Y, et al. Comparative sequence analyses of the genes coding for 16S rRNA of Lactobacillus casei-related taxa. Int J Syst Bacteriol. 1997;47:54–57. doi: 10.1099/00207713-47-1-54. [DOI] [PubMed] [Google Scholar]
- 57.Chen H, Lim CK, Lee YK, Chan YN. Comparative analysis of the genes encoding 23S–5S rRNA intergenic spacer regions of Lactobacillus casei-related strains. Int J Syst Evol Microbiol. 2000;50:471–478. doi: 10.1099/00207713-50-2-471. [DOI] [PubMed] [Google Scholar]
- 58.Felis GE, Dellaglio F, Mizzi L, Torriani S. Comparative sequence analysis of a recA gene fragment brings new evidence for a change in the taxonomy of the lactobacillus casei group. Int J Syst Evol Microbiol. 2001;51:2113–2117. doi: 10.1099/00207713-51-6-2113. [DOI] [PubMed] [Google Scholar]
- 59.Acedo-Félix E, Pérez-Martínez G. Significant differences between Lactobacillus casei subsp. casei ATCC 393T and a commonly used plasmid-cured derivative revealed by a polyphasic study. Int J Syst Evol Microbiol. 2003;53:67–75. doi: 10.1099/ijs.0.02325-0. [DOI] [PubMed] [Google Scholar]
- 60.Toh H, Oshima K, Nakano A, Takahata M, Murakami M, Takaki T, et al. Genomic adaptation of the Lactobacillus casei group. PLoS One. 2013;8 10.1371/journal.pone.0075073. [DOI] [PMC free article] [PubMed]
- 61.Tindall BJ. The type strain of lactobacillus casei is ATCC 393, ATCC 334 cannot serve as the type because it represents a different taxon, the name lactobacillus paracasei and its subspecies names are not rejected and the revival of the name ‘lactobacillus. Int J Syst Evol Microbiol. 2008;58:1764–1765. doi: 10.1099/ijs.0.2008/005330-0. [DOI] [PubMed] [Google Scholar]
- 62.Jebava I, Chuat V, Lortal S, Valence F. Peptidoglycan hydrolases as species-specific markers to differentiate Lactobacillus helveticus from Lactobacillus gallinarum and other closely related homofermentative lactobacilli. Curr Microbiol. 2014;68:551–557. doi: 10.1007/s00284-013-0512-5. [DOI] [PubMed] [Google Scholar]
- 63.Hammes WP, Hertel C. Genus I. Lactobacillus Beijerink 1901. In: Bergey’s manual of systematic bacteriology: volume 3, 2nd. New York: Springer New York; 2009. p. 465–510.
- 64.Tanigawa K, Watanabe K. Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiology. 2011;157:727–738. doi: 10.1099/mic.0.043240-0. [DOI] [PubMed] [Google Scholar]
- 65.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baumdicker F, Hess WR, Pfaffelhuber P. The infinitely many genes model for the distributed genome of bacteria. Genome Biol Evol. 2012;4:443–456. doi: 10.1093/gbe/evs016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: ecological diversity. Science. 2009;323:741–746. doi: 10.1126/science.1159388. [DOI] [PubMed] [Google Scholar]
- 68.Cohan FM, Perry EB. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol. 2007;17:373–386. doi: 10.1016/j.cub.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 69.Makarova KS, Koonin EV. Evolutionary genomics of lactic acid bacteria. J Bacteriol. 2007;189:1199–1208. doi: 10.1128/JB.01351-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duar RM, Lin XB, Zheng J, Martino ME, Grenier T, Pérez-Muñoz ME, et al. Lifestyles in transition: evolution and natural history of the genus Lactobacillus. FEMS Microbiol Rev. 2017;41(Supp_1):S27–S48. doi: 10.1093/femsre/fux030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All strains used for core- and pan-genome analysis study. (XLSX 54 kb)
Home-made scripts used in this study. (XLSX 39 kb)
Core-genome fasta for genus Lactobacillus (Fasta). (XLSX 59 kb)
Pan-genome fasta for genus Lactobacillus (Fasta). (XLSX 48 kb)
Pan-genome clustering of genus Lactobacillus. Heatmap clustering according to 20,969 pan-genome genes from 99 Lactobacillus genomes including the non-complete genome of L. zeae DSM 20178. Gower distance score based on ANI: Red = more similar, white = less similar. L. zeae DSM 20178 marked with a red arrow. (FASTA 305 kb)
Softcore-genome clustering of genus Lactobacillus. Heatmap clustering according to 594 softcore genes from 98 Lactobacillus genomes. Gower distance score based on ANI: Red = more similar, white = less similar. (FASTA 944 kb)
Core-genome fasta for Lactobacillus delbrueckii (Fasta). (FASTA 278 kb)
Pan-genome fasta for Lactobacillus delbrueckii (Fasta). (FASTA 1226 kb)
Reconstruction of core, softcore- and pan-genome of Lactobacillus delbrueckii species with the Brite and pathway algorithm of GhostKOALA. (FASTA 219 kb)
Reconstruction of core, softcore- and pan-genome of Lactobacillus delbrueckii species with the Module algorithm of GhostKOALA. Pathways are fractured in blocks (number). Green = pathway complete; yellow = 1 block is missing in the pathway; red = 2 or more blocks are missing in the pathway. (FASTA 1473 kb)
TOPD/FMTS nodal distance scores and SNPs evaluation. Genes are sorted according to the nodal distance scores compared with the pan-genome tree and the 5% and 95% quantile is listed. Sum SNP – The sum of all SNP according to the consensus sequence of the 29 homologous sequences; SNP/base – sum of SNP divided by length of consensus sequence in nucleotide; Sum polyvariable positions (SPP) – sum of all positions with 3 or more different nucleotides a specific position; Length in bp – length of consensus sequence (JPEG 206 kb)
Potential HGT in Lactobacillus delbrueckii. Clade B = bulgaricus clade, clade D = diverse clade, Number indicates copies of gene within the genome, presence = possibility of occurrence within the clade in percent. (TXT 2464 kb)
Core-genome fasta for Lactobacillus helveticus (Fasta). (JPEG 215 kb)
Pan-genome fasta for Lactobacillus helveticus (Fasta). (JPEG 289 kb)
Core-genome fasta for Lactobacillus reuteri (Fasta). (FASTA 312 kb)
Pan-genome fasta for Lactobacillus reuteri (Fasta). (FASTA 2283 kb)
Core-genome fasta for Lactobacillus rhamnosus (Fasta). (JPEG 302 kb)
Pan-genome fasta for Lactobacillus rhamnosus (Fasta). (PDF 3325 kb)
Pan- and core-genome evolution of L. helveticus. A Evolution of the pan-genome for L. helveticus. After 14 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. helveticus. Order of calculation was randomized for 19 sets, each represented with a single point. (PDF 5105 kb)
Pan- and core-genome evolution of L. reuteri. A Evolution of the pan-genome for L. reuteri. After 20 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. reuteri. Order of calculation was randomized for 25 sets, each represented with a single point. (PDF 2010 kb)
Pan- and core-genome evolution of L. rhamnosus. A Evolution of the pan-genome for L. rhamnosus. After 20 included genomes, the pan-genome is closed. B Evolution of the core-genome for L. rhamnosus. Order of calculation was randomized for 51 sets, each represented with a single point. (PDF 1978 kb)
Core-genome fasta for Lactobacillus plantarum (Fasta). (FASTA 193 kb)
Pan-genome fasta for Lactobacillus plantarum (Fasta). (FASTA 5115 kb)
Pan- and core-genome evolution of L. plantarum. A Evolution of the pan-genome for L. plantarum. The pan-genome remains open even after 122 genomes were included. B Evolution of the core-genome for L. plantarum. Order of calculation was randomized for 122 sets, each represented with a single point. (PDF 6945 kb)
Pan-genome heatmap of L. rhamnosus. Heatmap clustering according to 4889 pan-genome genes from 51 Lactobacillus rhamnosus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (PDF 7940 kb)
Pan-genome heatmap of L. plantarum. Heatmap clustering according to 7610 pan-genome genes from 122 Lactobacillus plantarum genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (FASTA 469 kb)
Pan-genome heatmap of L. reuteri. Heatmap clustering according to 3960 pan-genome genes from 25 Lactobacillus reuteri genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (FASTA 1001 kb)
Pan-genome heatmap of L. helveticus. Heatmap clustering according to 3350 pan-genome genes from 19 Lactobacillus helveticus genomes. Gower distance score based on ANI: Red = more similar, white = less similar, strains with deviating cluster behavior marked with red arrows. (XLSX 36 kb)
Data Availability Statement
All data generated or analysed in this study are included in this published article and its supplementary information files.