

Abstract
Pulmonary hypertension (PH) is caused by a group of pulmonary vascular disorders. A recent study has provided evidence that pulmonary vascular remodeling and PH can be observed in lung cancer, and this may be associated with tumor cell–immune cell inflammatory cross-talk. These findings highlight the pressing need to better understand and manage pulmonary vascular comorbidities in lung cancer.
Keywords: Pulmonary Hypertension, Lung Cancer, Inflammatory, Disease Comorbidity, Cardio-Oncology
Lung cancer is a major public health problem worldwide and is accounted for more than one-quarter (26%) of all U.S. cancer deaths in 2017 [1]. Furthermore, the five-year survival rate of lung cancer is only 20% after diagnosis [1]. Including cardiovascular and pulmonary comorbidities, the contribution of tumor heterogeneity and disease comorbidities to high mortality in lung cancer patients is poorly understood. A recent study published in Science Translational Medicine reported new findings suggesting that pulmonary vascular remodeling and pulmonary hypertension (PH) that can lead to right ventricular dysfunction, heart failure, and high mortality, were observed in both human patients and three genetic mouse models of lung cancer (LCC1, cRaf-BxB, and KRasLA2) which could be linked to tumor cell–immune cell inflammatory cross-talk [2]. Pullamsetti and colleagues found that 48% (250/519) of lung cancer patients presented a mean pulmonary artery diameter of more than 28 mm, suggestive of PH, as assessed from histopathological data analysis. Furthermore, they showed that 46% of patients exhibited a pulmonary artery systolic pressure (PASP) above the upper limit of normal value of 34 mmHg, as evidenced from the available echocardiographic data for 70 individuals. Histological analysis of tumors from the patients and mice revealed extensive pulmonary vascular remodeling (assessing pulmonary vessel wall thickness) relative to non-tumor sections. These complementary data suggested that lung cancer might be able to trigger PH [2].
Pullamsetti et al. further established in vitro models by exposing pulmonary arterial smooth muscle cells (PASMCs) or pulmonary arterial adventitial fibroblasts (PAAFs) isolated from normal human donor lung tissue to conditioned medium derived from adenocarcinoma cell/macrophage co-cultures. They showed that co-culturing human lung tumor cells with tumor-associated macrophages and lymphocytes led to the release of several cytokines [e.g., including interleukin-8 (IL-8), interleukin-1 receptor antagonist (IL-1RA), granulocyte macrophage colony stimulating factor (GM-CSF), and other chemokines], promoting pulmonary vascular cell proliferation in vitro [2]. Phosphodiesterase-5 (PDE5), a known downstream protein in PH pathogenesis, was significantly upregulated in the pulmonary vasculature of human and mouse lung tumor samples. Furthermore, treatment with sildenafil, a known PDE5 inhibitor, significantly improved pulmonary vascular remodeling (e.g., based on echocardiographic, hemodynamic, and morphometric measurements) in the LCC1 mouse lung cancer model. These findings are relevant in that they may lead to the development of novel candidate approaches for the management of cardiovascular and pulmonary comorbidities in lung cancer patients. Mechanistically, this proof-of-concept study reveals that an inflammatory microenvironment in lung cancer might constitute one potential driving force in tumor-associated PH development [2]; indeed, improving pulmonary function via vascular remodeling would certainly be beneficial for lung cancer patients. Thus, these findings may help identify novel treatment strategies for lung cancer-associated PH (Figure 1). Accordingly, all lung cancer patients with dyspnea syndromes (shortness of breath associated with lung or heart pathology) should undergo echocardiographic assessment in clinical practice to diagnose potential cardiovascular and pulmonary comorbidities. Combining existing tumor-targeted therapeutic agents with approved PH drugs (e.g., sildenafil) might also constitute useful adjunctive therapies for treating lung cancer and coexisting PH, aiming to improve both survival and quality of life of individuals with lung cancer (Figure 1).
Figure 1. Lung Cancer-associated Pulmonary Hypertension.
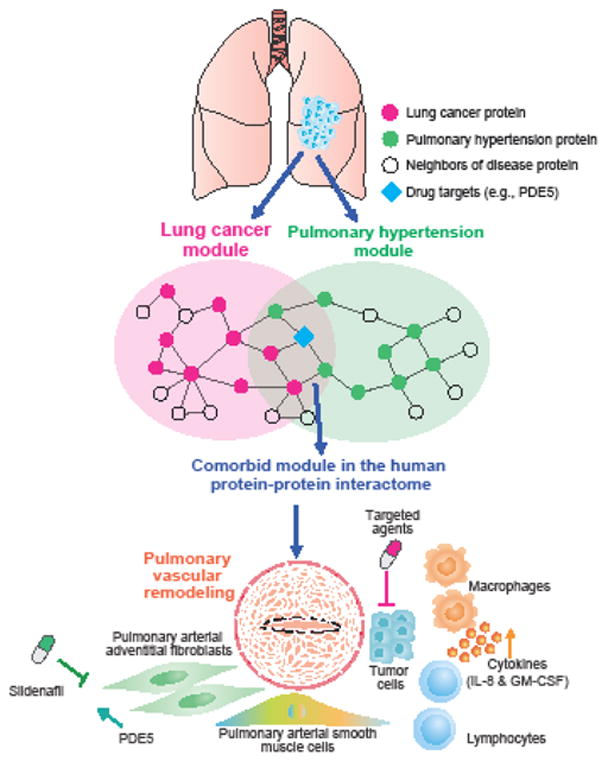
Diagram illustrating the biological hypothesis of lung cancer-associated pulmonary hypertension (PH). Lung cancer may trigger pulmonary comorbidities by tumor cell-immune cell inflammatory cross-talk (e.g. macrophages; lymphocytes), mediated in part, by the up-regulation of phosphodiesterase-5 (PDE5 [a well-known downstream protein involving in PH pathogenesis]) and the release of several cytokines and chemokines (e.g., interleukin-8 [IL-8] and granulocyte macrophage colony stimulating factor [GM-CSF]) that can promote vascular remodeling. This cross-talk is illustrated by the comorbid disease module between the lung cancer and pulmonary hypertension in the human protein-protein interactome. Potential actionable biomarkers for patient stratification and personalized treatment in patients with lung cancer and pulmonary comorbidities might be identified using the human interactome network. A combination of sildenafil (a known PDE5 inhibitor) and existing targeted cancer therapeutic agents might potentially improve vascular remodeling, and presumably, provide therapeutic benefit for lung cancer patients with PH comorbidities.
Beyond tumor cell cross-talk with inflammatory cells, several underlying molecular mechanisms might contribute to mediating the pathogenesis and development of lung cancer-associated PH, which were not explored in this study [2]. PH is characterized by vascular remodeling owing to cell proliferation and migration in the walls of resistant pulmonary arteries. Cell proliferation indicates various cancer-like profiles often driven by somatic genomic events in human cells. Several recent human genomic studies have suggested that somatic genomic events may contribute to atherosclerotic cardiovascular disease [3], secondary hypertension [4], and pulmonary arterial hypertension [5]. The unifying hypothesis of somatic genomic events-driven PH is that cancer cells and vascular cells or systems might share cross-talk pathways (e.g., including tumor cell-immune cells [2] or cell metabolic responses [6,7]) promoted by somatic events. Hence, investigating whether somatic mutations are involved in lung cancer-associated PH via next-generation sequencing technologies in both patients and genetic mouse models could offer novel mechanisms and candidate therapeutic targets for the treatment of both PH and lung cancer, although this remains to be robustly tested. For example, repurposing a generic drug that is already approved to treat one disease or condition (e.g., cancer) might offer novel treatment strategies in PH, as this generic drug might potentially targeta shared biological pathways (e.g., cell metabolic responses and inflammatory cross-talk) between cancer and PH. A recent study reported that the generic drug dichloroacetate (DCA), an inhibitor of the protein pyruvate dehydrogenase kinase – a well-known mitochondrial target in certain cancers and congenital mitochondrial disease – might harbor potential in the treatment of genetically susceptible PH patients [8]. Specifically, in a four-month, open-label study, DCA administration in PH patients was reported to lead to a significant reduction in mean pulmonary arterial pressure and pulmonary vascular resistance, presumably by improving mitochondrial function in these individuals [8].
Systematic identification of cross-talk or comorbid disease modules via network medicine approaches [9] (e.g., endophenotype network model [10]) may also offer putative personalized treatment strategies for cancer-associated cardiovascular and pulmonary comorbidities. For example, an integrative network medicine infrastructure that incorporates innovative genomic technologies, radiomics (e.g., echocardiographic and other imaging methods), systems biology, and modern network science approaches [9], might offer unexpected opportunities that may help identify actionable biomarkers for patient stratification and personalized therapies. Furthermore, early diagnosis and treatment of cardiovascular and pulmonary comorbidities associated with cancer via combination therapies or polypharmacy may potentially improve survival and quality of life during and after cancer treatments, which may facilitate taking a closer step towards achieving precision medicine approaches.
Acknowledgments
This work was supported by K99HL138272 (F.C.) from NHLBI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, et al. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Pullamsetti SS, et al. Lung cancer-associated pulmonary hypertension: role of microenvironmental inflammation based on tumor cell-immune cell crosstalk. Sci Transl Med. 2017;9:eaai9048. doi: 10.1126/scitranslmed.aai9048. [DOI] [PubMed] [Google Scholar]
- 3.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beuschlein F, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet. 2013;45:440–444. doi: 10.1038/ng.2550. [DOI] [PubMed] [Google Scholar]
- 5.Aldred MA, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:1153–1160. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metabolism. 2015;21:777–789. doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oldham WM, et al. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metabolism. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michelakis ED, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9:eaao4583. doi: 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- 9.Menche J, et al. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347:1257601. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghiassian SD, et al. Endophenotype network models: common core of complex diseases. Sci Rep. 2016;6:27414. doi: 10.1038/srep27414. [DOI] [PMC free article] [PubMed] [Google Scholar]