

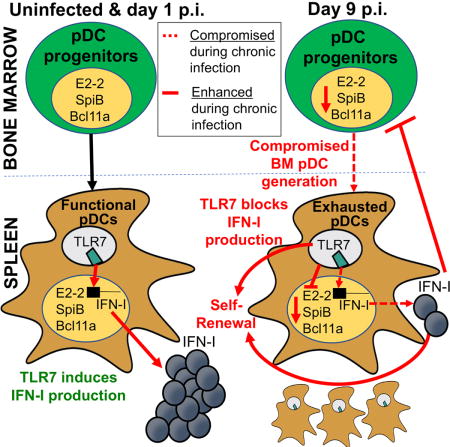
Summary
While characterization of T cell exhaustion has unlocked powerful immunotherapies, the mechanisms sustaining adaptations of short-lived innate cells to chronic inflammatory settings remain unknown. During murine chronic viral infection, we found that concerted events in bone marrow and spleen mediated by type-I-interferon (IFN-I) and Toll-like receptor 7 (TLR7) maintained a pool of functionally exhausted plasmacytoid dendritic cells (pDCs). In the bone marrow, IFN-I compromised the number and the developmental capacity of pDC progenitors, which generated dysfunctional pDCs. Concurrently, exhausted pDCs in the periphery were maintained by self-renewal via IFN-I- and TLR7-induced proliferation of CD4− subsets. On the other hand, pDC functional loss was mediated by TLR7, leading to compromised IFN-I production and resistance to secondary infection. These findings unveil the mechanisms sustaining a self-perpetuating pool of functionally exhausted pDCs and provide a framework to decipher long-term exhaustion of other short-lived innate cells during chronic inflammation.
Keywords: Type I interferon, plasmacytoid dendritic cells, exhaustion, TLR, chronic viral infection, LCMV, HIV, cancer
eTOC Blurb
The mechanisms underlying the maintenance and dysfunction of exhausted pDCs during chronic viral infection are unclear. Macal and Jo et al. find that exhausted pDCs are maintained by IFN-I and TLR7 signaling via multiple mechanisms, including inhibition of bone marrow pDC generation, sustained proliferation of exhausted pDCs, and promotion of pDC functional-loss, leading to impaired host defense to secondary infection.
Introduction
Chronic infections, including Human Immunodeficiency Virus (HIV) infection, mycobacterium tuberculosis and malaria, represent a major health burden. Immunosuppression is a hallmark of chronic infections, limiting the immune response to the ongoing pathogen as well as unrelated microbes, cancer and vaccination (Stelekati and Wherry, 2012; Virgin et al., 2009; Zuniga et al., 2015). While detrimental for host defense, such immunosuppression can be seen as a host adaptation to enable long-term survival and co-existence with the pathogen. Indeed, hosts with genetic ablation of core immunosuppressors often die after infection (despite enhanced microbial control) (Virgin et al., 2009; Zuniga et al., 2015). Nevertheless, short-term therapeutic relief of some immunosuppressors during infection can be tolerated and beneficial in clearing the persistent pathogen and/or secondary insults (Attanasio and Wherry, 2016; Zuniga et al., 2015). While host adaptations have been extensively studied in T and B cells, which undergo functional exhaustion during chronic infections and cancer (Attanasio and Wherry, 2016; Moir and Fauci, 2014), much less is known about the mechanisms mediating long-term adaptations in the innate immune system. In particular, it is unclear how innate immune exhaustion can be sustained long-term given that innate cells are generally short-lived.
Type I interferons (IFN-I) provide critical resistance to infections and tumors by limiting the spread of the infectious agent or malignant transformation and by activating multiple innate and adaptive immune cells (Garcia-Sastre and Biron, 2006; McNab et al., 2015; Zitvogel et al., 2015). Plasmacytoid dendritic cells (pDCs), the most potent IFN-I producing cells, rapidly respond to murine and human persistent viruses by releasing copious amounts of these cytokines (Borrow, 2011; Lee et al., 2009; Reizis et al., 2011; Swiecki and Colonna, 2015; Zuniga et al., 2015). Consistently, mice deficient in pDCs fail to control several acute and chronic viral infections (Reizis et al., 2011; Swiecki and Colonna, 2015). We and others, however, have reported sustained hypo-functionality of pDCs at later stages of chronic infections (Duan et al., 2004; Feldman et al., 2001; Kanto et al., 2004; Lee et al., 2009; Zuniga et al., 2008); hereafter referred to as pDC exhaustion. Such exhausted pDCs fail to produce IFN-I in response to the ongoing infection, synthetic toll-like receptor (TLR) ligands, and/or unrelated viruses, and this is accompanied by compromised innate responses and control of secondary opportunistic infections (Feldman et al., 2001; Lee et al., 2009; Zuniga et al., 2008). Furthermore, although systemic IFN-I is often elevated early after chronic infection, it is rapidly and profoundly attenuated afterwards, with only low levels of IFN-I and/or an “interferon signature” persisting in tissues (Borrow, 2011; Bruel et al., 2014; Ng et al., 2016; Snell and Brooks, 2015; Stacey et al., 2009, Zuniga et al., 2015). Prophylactic pDC stimulation, delayed IFN-I down-regulation, or IFN-I inoculation before or after infection with a chronic virus enhances T cell responses and host resistance, indicating that the rapid and profound IFN-I attenuation during chronic viral infection promotes T cell exhaustion and viral persistence (Blasius et al., 2012; Lee et al., 2009; Sandler et al., 2014; Vanderford et al., 2012; Wang et al., 2012). IFN-I exhaustion has also been reported in tumor-associated pDCs, and targeting of pDCs to restore their IFN-I production capacity has been proposed as a mean to overcome cancer immune suppression (Hartmann et al., 2003; Labidi-Galy et al., 2011; Sisirak et al., 2012). However, the mechanisms maintaining the numbers of exhausted pDCs and their functional loss in chronic settings remain unknown.
In this study, we used persistent lymphocytic choriomeningitis virus (LCMV) clone 13 (Cl13) infection in its natural murine host and found that long-term maintenance of exhausted pDCs involved both compromised numbers and pDC generative capacity of BM progenitors, as well as self-renewal of exhausted pDCs in the periphery. While both IFN-I and TLR7 signaling contributed to peripheral pDC proliferation, only IFN-I was responsible for reduced pDC progenitors. On the other hand, TLR7 mediated pDC functional impairment, compromising IFN-I elevation and resistance upon secondary infection. Together, these findings provide an anatomical, cellular and molecular explanation for pDC exhaustion during chronic inflammation.
Results
Both Spleen and BM pDCs exhibit sustained functional exhaustion during chronic LCMV infection
We previously reported that spleen pDCs, identified by co-expression of B220 and CD11c, exhibited long-lasting functional exhaustion during chronic LCMV infection (Zuniga et al., 2008). To further validate these findings and investigate if pDC exhaustion also occurred in the BM, we adopted a more rigorous pDC gating strategy that included the pDC marker BST2 (Asselin-Paturel et al., 2003; Blasius et al., 2006) (Fig. 1A). Consistent with our previous report (Zuniga et al., 2008), splenic pDCs from both LCMV Armstrong (ARM)- and Cl13-infected mice showed reduced IFN-I production in response to a TLR9 agonist CpG compared to uninfected controls at days 5 and 10 p.i., which was sustained only in pDCs from Cl13-infected mice at day 30 p.i. (Fig. 1B). Sustained suppression of IFN-I production was also observed in BM pDCs from Cl13 (but not ARM)-infected mice (Fig. 1C). Furthermore, the more rigorous pDC gate revealed reduced tumor necrosis factor (TNF)-α production by splenic and BM pDCs isolated at day 5 or 10 after either ARM or Cl13 infection (Fig. S1A). This defect was, however, fully or partially recovered 30 days after both infections. Finally, CD86 expression was upregulated upon CpG stimulation in splenic and BM pDCs from ARM or Cl13 infected mice (Fig. S1B), indicating that exhausted pDCs were still responsive to TLR stimulation. These data indicated that both splenic and BM pDCs acquired functional defects upon infection with either acute or persistent LCMV. While pDCs from acutely infected mice recovered few weeks after infection, pDC exhaustion was sustained in the chronic setting.
Figure 1. BM pDC progenitors are reduced in number and have compromised capacity to generate functional pDCs long-term after chronic LCMV infection. See also Figures S1 and S2.
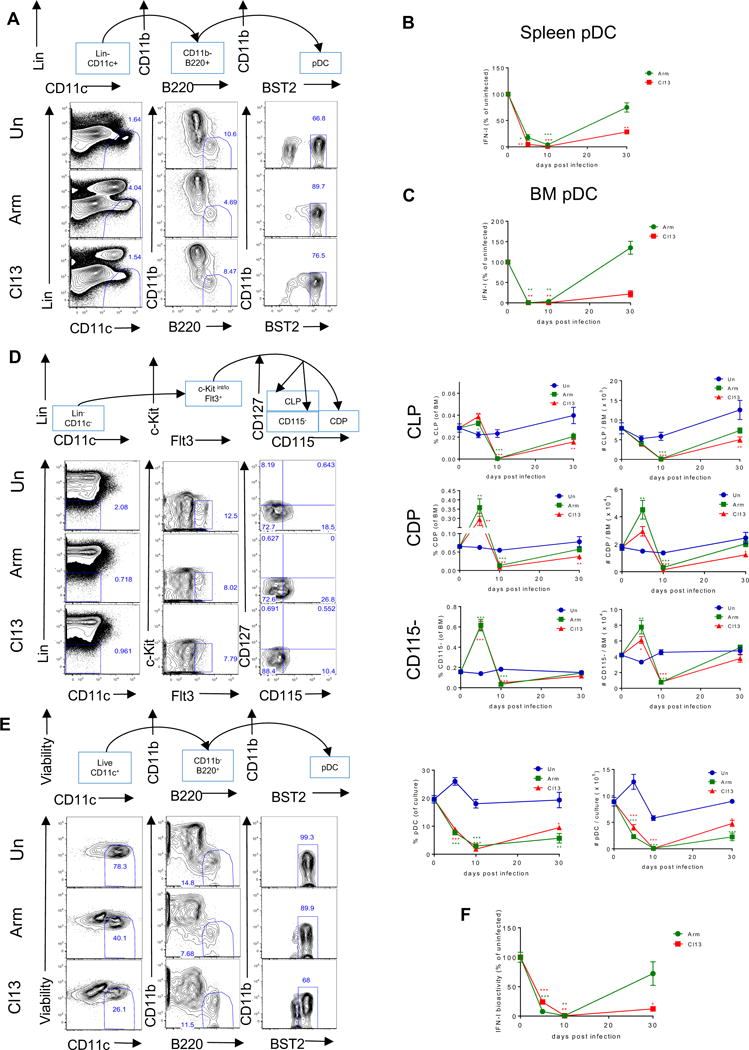
Wild-type (WT) mice were infected with LCMV ARM (green) or Cl13 (red) or left uninfected (blue) and sacrificed at days 5, 10, and 30 p.i. (A) Representative flow cytometric analysis of pDCs as Lin−CD11c+CD11b−B220+BST2+ in spleen at day 10 p.i. Lineage-negative gating (Lin) includes markers for Thy1.2, CD19, and NK1.1. (B-C) FACS-purified splenic pDCs (B) or BM pDCs (C) were stimulated with CpG-B for 15 hours and IFN-I activity in the supernatant was quantified. Graphs depict the percentage change in IFN-I activity normalized to the uninfected mice processed in parallel at each time point. (D) Representative flow cytometric analysis of BM pDC progenitors at day 10 p.i. where CLPs were identified as Lin−c-kitint/loFlt3+CD115−CD127+, CDPs as Lin−c-kitint/loFlt3+CD115+CD127−, and CD115− as Lin−c-kitint/loFlt3+CD115−CD127−. Lin includes markers for Thy1.2, CD19, NK1.1, CD3, CD4, CD8, B220, CD11b, Gr-1, and Ter119. Graphs depict the percentage (left) and the absolute number (right) of indicated progenitors in BM at each time point. (E) BM cells were cultured with Flt3L and pDCs were identified as CD11c+CD11b−B220+BST2high at day 8 post-culture. Representative FACS plots for BM-Flt3L-derived pDCs and graphs depicting their percentage (left) and absolute number (right) in the culture are shown. (F) FACS-purified BM-Flt3L-derived pDCs were stimulated with CpG-A for 15 hours and IFN-I activity in the supernatant was quantified. Graph depicts the percentage change in IFN-I activity normalized to the culture from uninfected mice processed in parallel at each time point. Graphs depict mean ± SEM. Data are representative of 2-3 independent experiments with 3-10 mice/group. *p<0.05, **p<0.01, ***p<0.001 (one-way Anova to compare ARM-infected (green asterisks) and Cl13-infected (red asterisks) groups to uninfected group processed in parallel at each time point).
BM pDC progenitor numbers are reduced long-term and fail to generate functional pDCs after chronic LCMV infection
Given that pDCs are short-lived (Liu et al., 2007; Zhan et al., 2016), we reasoned that sustained pDC exhaustion during chronic LCMV infection (Zuniga et al., 2008) might involve long-term adaptations occurring at the level of pDC progenitors. To evaluate this possibility, we first assessed the effect of acute and chronic LCMV infection on the numbers of BM progenitors that have been reported to generate pDCs: Common lymphoid progenitors (CLP), common DC progenitors (CDP), and CD115− CDP-like pDC progenitors (reviewed in (Swiecki and Colonna, 2015)). As shown in Fig. 1D, the percentage and number of CLPs were reduced by day 10 p.i. and numbers remained significantly decreased only in Cl13-infected mice by day 30 p.i.. The numbers of CDPs and CD115− progenitors tended to increase at day 5 p.i. but were also reduced by day 10 p.i. in both ARM- and Cl13-infected mice compared to uninfected controls. At day 30 p.i., CD115− progenitor numbers were restored while CDP numbers remained reduced in Cl13 (but not ARM)-infected mice. Similarly, the numbers of common myeloid progenitors (CMP), which can give rise to pDCs via CDP (Sathe et al., 2013), were also reduced in both ARM- and Cl13-infected mice at day 10 p.i. while such reduction was sustained only in the latter group at day 30 p.i. (Fig. S2A&B). In contrast, the numbers of granulocyte-monocyte progenitors (GMP) were not significantly reduced in neither acute nor chronic LCMV infections at any of the time points studied (Fig. S2A&C). When BM was cultured in the presence of Flt3 ligand (Flt3L) to induce DC development in vitro, pDC generation was significantly compromised in cultures derived from Cl13 or ARM-infected mice at day 5, 10 and 30 p.i. (Fig. 1E). To validate these findings in vivo, we transferred Lin−c-kitint/loFlt3+ cells from uninfected or Cl13-infected mice at day 30 p.i. into uninfected or infection-matched recipient mice, respectively, and quantified donor-derived pDCs after 10 days. In line with the BM-Flt3L culture (Fig. 1E), we observed a decrease in the number of donor-derived pDCs when recipients had received BM progenitors from Cl13-infected vs. uninfected donors (Fig. S2D&E).
To further understand BM progenitor adaptations after infection, we evaluated the function of pDCs derived from BM progenitors. pDCs from Flt3L-cultures of BM from ARM- or Cl13-infected mice at days 5 and/or 10 p.i. showed ablated IFN-I production in response to TLR stimulation (Fig. 1F). However, compromised BM capacity to generate functional pDCs was sustained in Cl13-infected mice by day 30 p.i., whereas BM-derived pDCs from ARM-infected mice exhibited restored IFN-I production upon CpG-A stimulation (Fig. 1F), although we continued observing reduced response to CpG-B at day 30 after ARM infection (not shown).
Together, these results indicated that both acute and chronic infections compromised long-term the ability of BM to generate pDCs. In addition, our results showed sustained ablation of CLPs, CMPs and CDPs and lasting functional exhaustion of BM-derived pDCs in chronically infected hosts.
Splenic and BM pDCs proliferate and expand CD4− subsets throughout chronic LCMV infection
We next investigated whether loss of pDC progenitors and diminished BM pDC generation affected pDC numbers. In agreement with apoptosis of pDCs early during infection (Swiecki et al., 2011) and the long-term compromise capacity of BM to generate pDCs after ARM and Cl13 infections (Fig. 1E), spleen pDC numbers were significantly reduced at days 5 and 30 after both infections (Fig. 2A, (Zuniga et al., 2008)). However, numbers of BM pDCs were mostly unchanged at all the time points studied (Fig. S3A) and spleen pDC numbers were restored or even enhanced at day 10 (Fig. 2A) after ARM and Cl13 infection. To determine how pDC numbers were maintained despite the reduced generation of pDCs from BM, we investigated the presence of proliferating cells within spleen and BM pDCs over the course of ARM and Cl13 infections. Consistent with previous studies (Liu et al., 2007; Zhan et al., 2016) we detected <2% proliferating pDCs in the spleen of uninfected mice, as indicated by the nuclear antigen Ki67 expression and in vivo bromodeoxyuridine (BrdU) incorporation (Fig. 2B&C). In contrast, we observed a substantial number of proliferating pDCs in spleen (Fig. 2B&C) and BM (Fig. S3B&C) from both ARM and Cl13 infected mice at day 5 p.i. and this was sustained at day 10 and 30 after chronic (but not acute) LCMV infection. BrdU was also incorporated by pDCs from Cl13-infected, but not uninfected, mice when splenocytes were cultured ex vivo, further supporting the proliferative capacity of splenic pDCs during Cl13 infection (Fig. S3D).
Figure 2. Splenic pDCs proliferate and CD4- subsets expand throughout chronic LCMV infection. See also Figure S3.
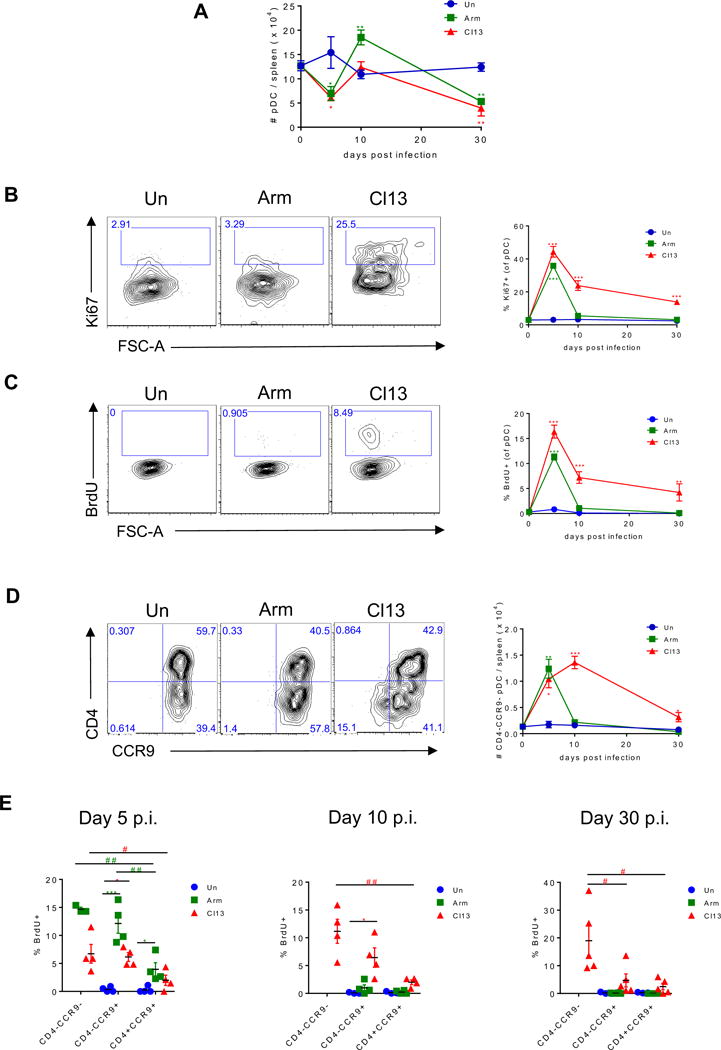
WT mice were infected with LCMV ARM (green) or Cl13 (red) or left uninfected (blue) and sacrificed at day 5, 10, and 30 p.i. Splenic pDCs were analyzed to determine their absolute number (A), Ki67 expression (B), BrdU incorporation in vivo (C), absolute number of CD4−CCR9− pDC subset (D) and BrdU incorporation by CD4+CCR9+, CD4−CCR9+, and CD4−CCR9- subsets (E). (B-D) Representative FACS plots indicate spleen pDCs from day 10 p.i. (A-E) Graphs depict mean ± SEM. Data are representative of 2-3 independent experiments with 3-5 mice/group. (A) Data obtained at day 10 after Cl13 infection are representative of 11 independent repeats with 7 and 4 experiments showing enhanced or unchanged pDC numbers vs. uninfected controls, respectively. (E) Symbols represent individual mice. *, # p<0.05, **, ## p<0.01, *** p<0.001 (one-way Anova to compare ARM-infected (green asterisks) and Cl13-infected group (red asterisks) to uninfected group processed in parallel at each time point (A-E) or to compare CD4+CCR9+, CD4−CCR9+, and CD4−CCR9− subsets in ARM-infected (green pounds) or Cl13-infected group (red pounds) (E)).
Proliferating CD4−CCR9− and CD4−CCR9+ pDC subsets have been reported in the BM of uninfected mice as immature pDCs or immediate precursors of CD4+CCR9+ pDCs (Schlitzer et al., 2012; Schlitzer et al., 2011; Zhan et al., 2016). On the other hand, almost all splenic pDCs express chemokine receptor CCR9 and do not proliferate under steady-state conditions (Schlitzer et al., 2012; Schlitzer et al., 2011; Zhan et al., 2016). In contrast to almost undetectable CD4−CCR9− pDCs in uninfected mice, we detected an increased number of CD4−CCR9− cells within splenic pDCs from both ARM- and Cl13-infected mice at day 5 p.i., a phenotype that was sustained at day 10 and 30 after chronic (but not acute) infection (Fig. 2D). Moreover, while none of the spleen pDC subsets proliferated in uninfected mice, both CD4−CCR9+ and CD4−CCR9− pDC subsets contained a significant proportion of proliferating cells at day 5 after either ARM or Cl13 infection, and this was only sustained in Cl13 infection at days 10 and 30 p.i. (Fig. 2E). A similar pattern was observed in the BM at day 5, 10 and 30 after ARM and Cl13 infection (Fig. S3E&F) and in both compartments the CD4−CCR9− pDC subset showed the highest proliferation in ARM-infected mice at day 5 p.i. and throughout Cl13 infection (Fig. 2E and S3F). Altogether, these data indicated that while BM pDC generation was compromised, CD4− subsets within differentiated BM and spleen pDCs significantly expanded to replenish functionally compromised pDCs early after acute and chronic LCMV infections and this was sustained for long-term in the chronic infection.
E2-2, SPIB and BCL11A are down-regulated in pDCs and their progenitors from LCMV Cl13-infected mice as well as in pDCs from HIV infected patients
Given that the aforementioned phenotypes in BM progenitors and differentiated pDCs were all present at day 9 after Cl13 infection (Fig. 1–2), we focused on this time-point and infection model to characterize their adaptations at the transcriptional level and the mechanisms involved. Because the class I helix-loop-helix TF E2-2 is a master regulator of pDC development, lineage commitment and function (Cisse et al., 2008), we first compared E2-2 expression in BM progenitors and pDCs from uninfected and Cl13-infected mice. We observed a significant down-regulation in the transcript levels of E2-2 (encoded by Tcf4) in FACS-purified BM progenitors and splenic pDCs isolated at day 9 p.i. from Cl13-infected vs. uninfected mice (Fig. 3A&B). The down-regulation of Tcf4 transcripts in Cl13-infected mice appeared of functional significance as transcripts of downstream target TFs, SPIB and BCL11A, also important in pDC development and/or function (Cisse et al., 2008; Murphy et al., 2016; Sasaki et al., 2012), were similarly reduced in both BM progenitors and splenic pDCs from Cl13-infected vs. uninfected mice (Fig. 3A&B), although Bcl11a reduction in splenic pDCs only reached significance in 2 out of 4 independent experiments (not shown). Furthermore, pDCs derived from BM of Cl13-infected mice also exhibited a significant decrease in transcripts of Tcf4, Spib and Bcl11a (Fig. 3C). Finally, to investigate whether such TF down-regulation extended to human chronic infections, we purified pDCs from HIV-infected patients and compared them to pDCs from HIV-negative controls (Table S1). pDC purity ranged from 77-99% and was not different between groups (Fig. S4). TCF4 expression was lower in viremic patients (Fig. 3D), where pDC IFN-I production capacity is significantly reduced (Feldman et al., 2001) with respect to healthy controls. Similarly, SPIB and BCL11A were significantly decreased in viremic patients compared to healthy controls (Fig. 3E&F).
Figure 3. E2-2, SPIB and BCL11A are down-regulated in pDCs and their progenitors from LCMV Cl13-infected mice as well as in pDCs from HIV infected patients. See also Figure S4 and Table S1.
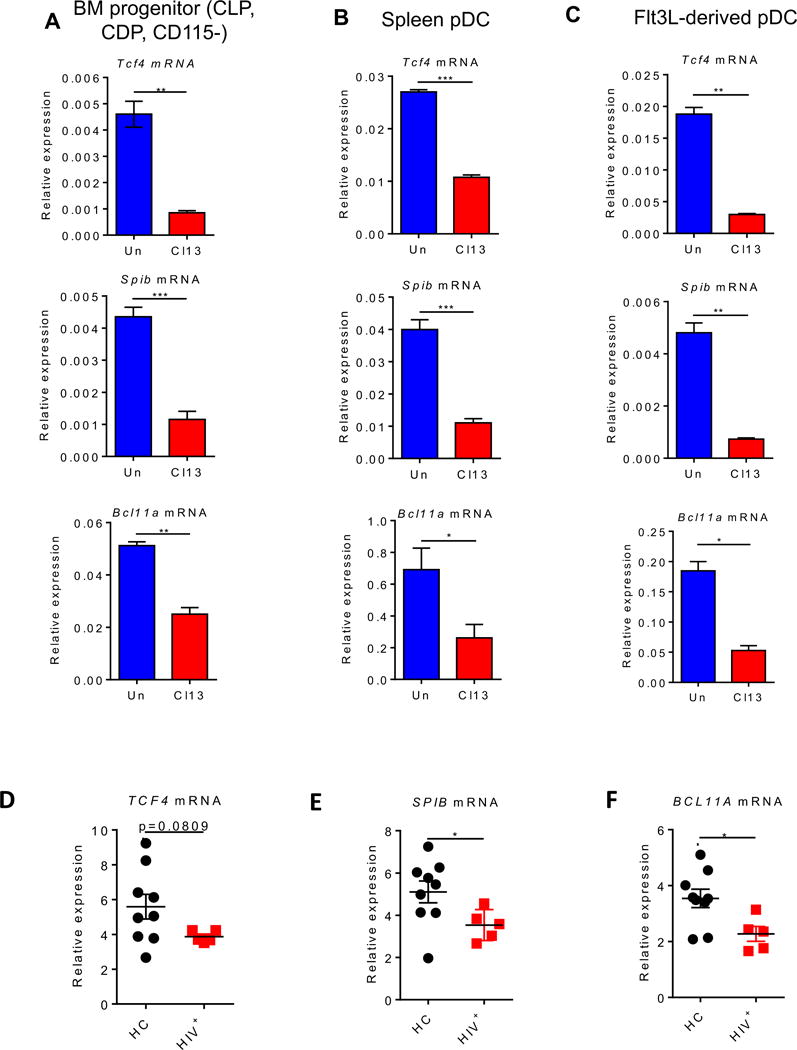
(A-C) WT mice were infected with LCMV Cl13 (red) or left uninfected (blue) and sacrificed at day 9 p.i. FACS-purified BM progenitors (Lin−c-kitint/loFlt3+) (A) and splenic pDCs (B) were evaluated for Tcf4 (top), Spib (middle), and Bcl11a (bottom) transcripts relative to Gapdh. Lin includes markers for Thy1.2, CD19, NK1.1, CD3, CD4, CD8, B220, CD11b, Gr-1, and Ter119. (C) BM cells were cultured with Flt3L for 8 days and FACS-purified BM-Flt3L-derived pDCs were evaluated for Tcf4 (top), Spib (middle), and Bcl11a (bottom) transcripts relative to Gapdh. (D-F) Enriched pDCs from peripheral blood of HIV-negative healthy controls (HC; black) and chronically HIV-infected patients (HIV+; red) were evaluated for TCF4 (D), SPIB (E), and BCL11A (F) transcripts relative to GAPDH. (A-C) Data are representative of 2-3 independent experiments with 3-4 mice/group. (D-F) Symbols represent individual blood donors. Bars depict mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 (unpaired, two-tailed t-test (A-C) or Mann-Whitney test (D-F)).
Altogether these results demonstrated down-regulation of Tcf4 and target TFs Spib and Bcl11a in BM progenitors and pDCs from chronically infected mice, a phenotype that appeared extendible to pDCs from HIV-viremic patients.
IFNAR-signaling reduces pDC progenitor numbers, their E2-2 expression, and BM pDC generation during chronic LCMV infection
Next, we investigated the mechanisms underlying the numerical and developmental adaptations in BM pDC progenitors. Previous studies demonstrated that IFN-I inhibited development of conventional dendritic cells during chronic LCMV infection (Hahm et al., 2005; Sevilla et al., 2004), whereas recombinant IFN-α treatment enhanced BM pDC generation in vitro and in vivo (Li et al., 2011). To test whether IFN-I induced in the context of Cl13 infection modulated BM pDC progenitors, we treated mice with anti-IFNAR neutralizing (n) Ab before and throughout the infection and monitored the number of BM progenitors and their potential to generate pDCs at day 9 p.i. We observed a restored or enhanced number of BM pDC progenitors in Cl13-infected mice treated with anti-IFNAR nAb in comparison to the isotype-treated group (Fig. 4A). Furthermore, significantly increased numbers of pDCs were generated in BM-Flt3L cultures from Cl13-infected mice that received anti-IFNAR nAb (Fig. 4B). Consistently, Tcf4 expression was restored in pooled BM progenitors from anti-IFNAR nAb-treated Cl13-infected mice (Fig. 4C). To investigate if IFNAR signaling regulated BM pDC progenitors in a cell-intrinsic manner, we next analyzed WT:Ifnar−/− mixed BM chimeras after Cl13 infection. In these chimeras, we observed a slight increase in the Ifnar−/− (CD45.2+) vs. WT (CD45.1+) BM pDC progenitors (Fig. 4D), suggesting that IFN-I regulation of BM progenitors was partly cell-intrinsic. In contrast, no difference was detected in Tcf4 expression in BM progenitors from WT vs. Ifnar−/− compartments (Fig. 4E), indicating that IFN-I also regulated BM progenitors in a cell-extrinsic manner.
Figure 4. IFN-I signaling suppresses BM pDC progenitors during chronic LCMV infection.
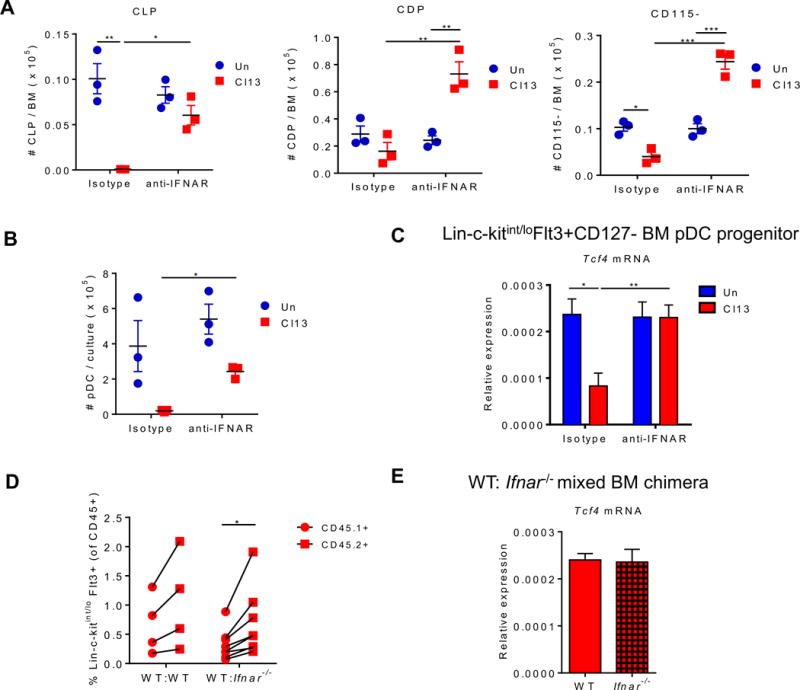
(A-C) WT mice were treated with isotype or anti-IFNAR nAb beginning 1 day before and throughout LCMV Cl13 infection and sacrificed at day 9 p.i. Absolute number of CLPs, CDPs, and CD115− (A) and pDCs derived from BM-Flt3L-cultures (continuing treatment with isotype or anti-IFNAR nAb in vitro) at day 8 post-culture (B) were quantified. (C) FACS-purified BM pDC progenitors (Lin-c-kitint/loFlt3+CD127−) were evaluated for Tcf4 expression relative to Gapdh. Lin includes markers for Thy1.2, CD19, NK1.1, CD3, CD4, CD8, B220, CD11b, Gr-1, and Ter119. (D-E) Mixed BM chimeras were generated using BM from WT (CD45.1+) and WT or Ifnar−/− (CD45.2+) mice and infected with LCMV Cl13 for 9 days. (D) Percentages of Lin−c-kitint/loFlt3+ pDC progenitors within CD45.1+ or CD45.2+ cells in BM of mixed chimeras. Connected symbols indicate values from the same mouse. (E) FACS-purified Lin−c-kitint/loFlt3+ BM pDC progenitors from WT and Ifnar−/− compartment were evaluated for Tcf4 transcripts relative to Gapdh. Bars depict mean ± SEM and symbols represent individual mice. Data are representative of 2-3 independent experiments with 3-4 mice/group (A-C and E) or pooled from 2 independent experiments with 2-4 mice/group (D). *p<0.05, **p<0.01 (two-way Anova (A-C), paired t-test (D), or unpaired, two-tailed t-test (E)).
Altogether, these results demonstrated that IFN-I regulated BM pDC progenitors via cell-intrinsic and -extrinsic pathways, contributing to the numerical reduction of BM progenitors, their diminished E2-2 expression, and their limited capacity to generate pDCs.
IFNAR signaling promotes splenic pDC proliferation and expansion of CD4− pDC subsets during chronic LCMV infection
IFN-I signaling has been shown to induce pDC apoptosis after viral infections (Swiecki et al., 2011). Thus, we reasoned that by preventing pDC apoptosis and restoring BM pDC generation, IFNAR blockade should increase the number of pDCs in periphery. However, LCMV Cl13-infected mice that received anti-IFNAR nAb exhibited reduced numbers of splenic pDCs compared with their isotype-treated counterparts at day 9 p.i. (Fig. 5A). Because the number of splenic pDCs during Cl13 infection would be impacted by pDC proliferation, we next investigated whether anti-IFNAR nAb treatment modulated proliferating pDCs. We observed that IFNAR blockade during Cl13 infection diminished pDC proliferation, as indicated by reduced Ki67+ and BrdU+ pDCs compared with isotype treatment in infected mice (Fig. 5B&C). Consistently, the numbers of CD4−CCR9− and CD4−CCR9+ splenic pDC subsets were reduced in infected mice that received anti-IFNAR nAb, while the number of CD4+CCR9+ pDCs was not significantly changed (Fig. 5D). To investigate if the IFN-I induction of pDC proliferation was cell-intrinsic, we again analyzed WT:Ifnar−/− mixed BM chimeras. We observed that Ki67 expression and in vivo BrdU incorporation were significantly reduced in Ifnar−/− vs. WT pDCs from Cl13-infected chimeras (Fig 5E&F), indicating that IFN-I promoted pDC proliferation in a cell-intrinsic manner.
Figure 5. IFN-I signaling promotes splenic pDC proliferation and increase in CD4- pDC subsets during chronic LCMV infection.
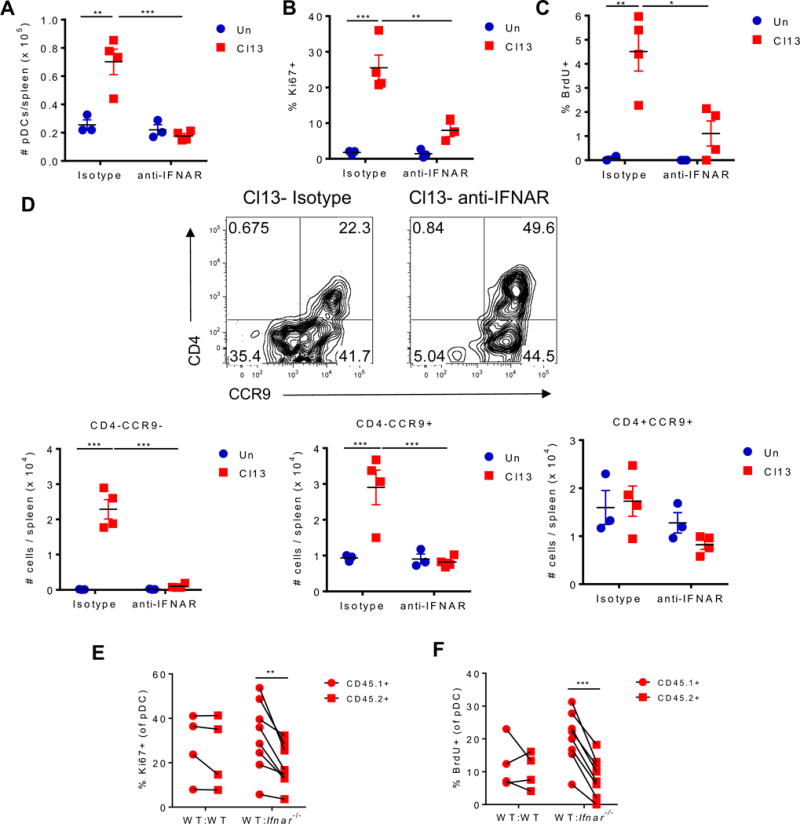
(A-D) WT mice were treated with isotype or anti-IFNAR nAb beginning 1 day before and throughout LCMV Cl13 infection and sacrificed at day 9 p.i. Splenic pDCs were evaluated for their absolute numbers (A), Ki67 expression (B), BrdU incorporation (C), and numbers of CD4+CCR9+, CD4−CCR9+, and CD4−CCR9− subsets (D). (D) Representative FACS plots indicate frequency of CD4+CCR9+, CD4−CCR9+, and CD4−CCR9− subsets in spleen pDCs. (E-F) Mixed BM chimeras were generated using BM from WT (CD45.1+) and WT or Ifnar−/− (CD45.2+) mice and infected with LCMV Cl13 for 9 days. Splenic pDCs from the CD45.1+ or CD45.2+ compartment were analyzed to determine their Ki67 expression (E) and BrdU incorporation in vivo (F). Data are representative of 1 (C) or 2 (A, B, and D) independent experiments with 3-4 mice/group or pooled from 2 independent experiments with 2-4 mice/group (E-F). Bars depict mean ± SEM. Symbols represent individual mice. *p<0.05, **p<0.01, ***p<0.001 (two-way Anova (A-D) or paired t-test (E-F)).
These data demonstrated that IFN-I induced the expansion of highly proliferative pDC subsets and promoted self-renewal of the exhausted pDC pool.
TLR7 signaling induces pDC proliferation and suppresses their IFN-I production after chronic LCMV infection
We next investigated the effect of IFN-I on the pDC functional-loss during chronic LCMV infection. We observed that both BM-derived and splenic pDCs from Cl13-infected mice treated with anti-IFNAR nAb or isotype control showed similarly impaired IFN-I production after ex vivo TLR stimulation (Fig. S5A&B). These data suggested that the loss of pDC IFN-I-production capacity occurred via an IFNAR-independent pathway. Given that arenavirus nucleoprotein (NP) can suppress IFN-I induction (Martinez-Sobrido et al., 2006) and that pDCs are productively infected by LCMV Cl13 (Macal et al., 2012), we explored the possibility that the suppression of pDC IFN-I production could be explained by overwhelming viral dissemination and NP expression in pDCs. Flow cytometric analysis of LCMV NP, however, demonstrated that the majority of pDCs did not express detectable NP at day 9 after Cl13 infection (Fig. S5C), suggesting that loss of pDC IFN-I production capacity may be, at least in part, a consequence of cellular adaptations to chronic viral infection and could not be fully explained by an anti-IFN-I effects of viral products in infected pDCs.
Considering that T cell exhaustion in chronic infection is driven, in great part, by persistent TCR stimulation (Attanasio and Wherry, 2016) and that uninfected pDCs can sense LCMV via TLR7 (Macal et al., 2012), we hypothesized that persistent TLR7 stimulation may contribute to hypo-functionality of pDCs. We first evaluated the numbers of pDC progenitors and their developmental potential, but we did not detect any difference between Cl13-infected WT and Tlr7−/− mice (Fig. S6A&B). Next, we analyzed spleen pDCs from WT and Tlr7−/− mice at day 9 after Cl13 infection. As we described at day 1 p.i. (Macal et al., 2012), expression of maturation markers was reduced in pDCs from Tlr7−/− vs. WT mice (Fig. S6C&D), while pDC numbers remained unchanged (Fig. S6E). This result indicated that TLR7 signaling continued exerting a positive effect on pDC maturation at later stages of infection. However, while pDCs from Cl13-infected WT mice were unable to produce IFN-I in response to ex vivo CpG stimulation, pDCs from Tlr7−/− Cl13-infected mice secreted significantly higher levels of IFN-I (Fig. 6A). The restored pDC response in Tlr7−/− mice occurred despite higher viral burden in Tlr7−/− vs. WT pDCs, indicated by LCMV glycoprotein (gp) transcript levels (Fig. S6F), in line with an antiviral effect of TLR7 signaling in pDCs and again arguing against functional defects of pDCs solely explained by a direct effect of a viral product (Martinez-Sobrido et al., 2006).
Figure 6. TLR7 suppresses pDC IFN-I production and promotes their proliferation during chronic LCMV infection. See also Figures S5 and S6.
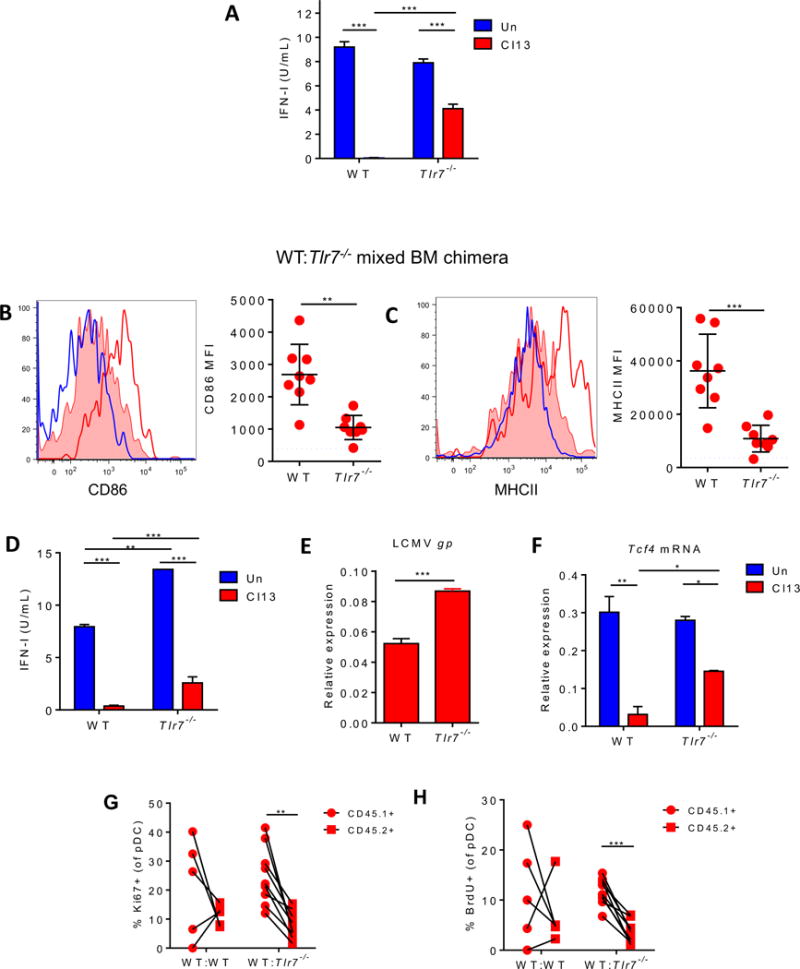
(A) WT and Tlr7−/− mice were left uninfected (blue) or infected with LCMV Cl13 for 9 days (red). FACS-purified splenic pDCs were stimulated with CpG-B and IFN-I activity in the supernatant was quantified. (B-H) Mixed BM chimeras were generated using BM from WT (CD45.1+) and Tlr7−/− (CD45.2+) mice and infected with LCMV Cl13 for 9 days. Representative histograms of CD86 (B) and MHCII (C) expression on splenic pDCs from WT uninfected mice (blue open) and WT (red open) and Tlr7−/− compartments (red filled) of Cl13-infected chimeric mice are shown. Graphs depict CD86 (B) and MHCII (C) MFI in pDCs from infected chimeric mice. Dotted line indicates the average MFI in WT uninfected mice. FACS-purified splenic pDCs were stimulated with CpG-B and IFN-I activity in the supernatant was quantified (D). Splenic pDCs were evaluated for LCMV gp (E) and Tcf4 (F) transcripts relative to Gapdh, Ki67 expression (G) and BrdU incorporation in vivo (H). Bars depict mean ± SEM. Symbols represent individual mice. Data are representative of 2-3 independent experiments with 3-5 mice/group (A-F) or are pooled from 2 independent experiments with 2-5 mice/group (G-H). *p<0.05, **p<0.01, ***p<0.001 (two-way Anova (A, D, and F), unpaired, two-tailed t-test (B, C, and E) or paired t-test (G-H)).
To discern whether TLR7 signaling induced pDC hypo-responsiveness in a cell-intrinsic manner, we generated WT:Tlr7−/− mixed BM chimeras and infected them with LCMV Cl13. Again, we observed reduced expression of CD86 and MHCII in Tlr7−/− vs. WT pDCs (Fig. 6B&C), indicating that the positive effects of TLR7 signaling on pDC maturation were cell-intrinsic. Tlr7−/− splenic pDCs also produced significant amounts of IFN-I (Fig. 6D) and exhibited significant Tnfα expression (Fig. S6G) in response to ex vivo CpG stimulation whereas their WT counterparts remained exhausted (Fig. 6D&S6G). Such increased pDC function in Tlr7−/− pDCs was again observed despite increased LCMV gp transcript levels in Tlr7−/− vs. WT pDCs from Cl13-infected chimeric mice (Fig. 6E). Furthermore, pDC functional restoration coincided with enhanced level of Tcf4 in splenic Tlr7−/− vs. WT pDCs (Fig. 6F), indicating that cell-intrinsic TLR7 signaling was, at least partly, responsible for the decreased E2-2 levels in splenic pDCs. We next evaluated pDC proliferation in WT:Tlr7−/− mixed BM chimeras upon Cl13 infection. We observed that both Ki67 expression and in vivo BrdU incorporation were significantly reduced in pDCs from the Tlr7−/− vs. WT compartments (Fig. 6G&H) indicating that direct TLR7 signaling also contributed to pDC proliferation after chronic LCMV infection.
Overall, our results indicated that the same TLR7 signaling that induces pDC maturation and IFN-I production at day 1 after LCMV Cl13 infection (Macal et al., 2012) continued to positively regulate pDC maturation and viral control, but instead of IFN-I induction, promoted functional loss and self-renewal of exhausted pDCs at later stages of infection.
TLR7 signaling compromises IFN-I production and pathogen control upon a secondary infection
We have previously reported that pDC IFN-I suppression during chronic viral infection coincided with enhanced susceptibility to secondary infections (Zuniga et al., 2008). To get more insight on the biological relevance of TLR7-mediated pDC exhaustion in the context of a secondary infection, we challenged WT and Tlr7−/− Cl13 infected mice with MCMV. At the infection dose used in this study, the role of pDCs in promoting MCMV control is well established (Dalod et al., 2003; Krug et al., 2004; Swiecki et al., 2010). We observed that pDC numbers were decreased in LCMV-MCMV co-infection vs. MCMV single infection, but this was not altered by the absence of TLR7 signaling (Fig. S7). However, in contrast to WT mice, Cl13-infected Tlr7−/− mice were able to produce systemic IFN-I in response to secondary MCMV infection, albeit to a lesser extent than mice receiving MCMV alone (Fig. 7A). Consistently, FACS-purified splenic pDCs isolated from Tlr7−/− (but not WT) Cl13-infected mice exhibited significant Ifnα and Ifnβ expression at 36hr post-MCMV infection (Fig. 7B&C), indicating that, in the absence of TLR7 signaling, pDCs are, at least partially, capable of responding to the secondary viral infection. MCMV viral clearance is impaired in LCMV co-infected mice (Fig. 7D & (Zuniga et al., 2008)) and the absence of TLR7 partially restored this impaired control, with MCMV titers ~10 times lower in Tlr7−/− vs. WT co-infected mice (Fig. 7D), consistent with higher systemic IFN-I and pDC functional restoration in the Tlr7−/− dually-infected group. Together, these results indicated that TLR7 signaling during LCMV Cl13 infection compromised pDC IFN-I response to an unrelated pathogen, which likely contributed to the limited hosts’ capacity to contain this unrelated infection.
Figure 7. TLR7 enhances susceptibility to secondary infection during chronic LCMV infection. See also Figure S7.
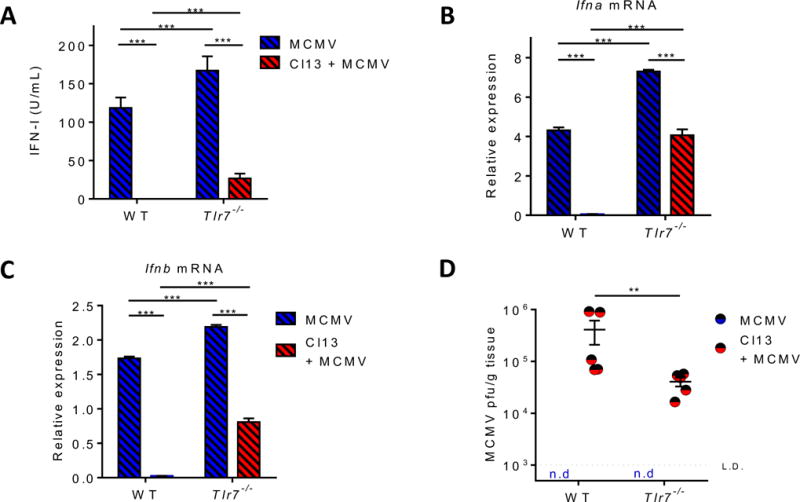
Uninfected and LCMV Cl13-infected WT and Tlr7−/− mice were infected with MCMV at day 20 p.i. 36 hours after MCMV infection, serum IFN-I activity was quantified (A) and FACS-purified splenic pDCs were evaluated for Ifnα (B) and Ifnβ (C) transcripts relative to Gapdh. MCMV titers in the liver were determined 3.5 days after MCMV infection (D). Dotted line represents the limit of detection. Bars depict mean ± SEM. Symbols represent individual mice. Data are representative of 2-3 independent experiments with 3-5 mice/group. **p<0.01, ***p<0.001 (two-way Anova).
Discussion
Production of soluble mediators, including IFN-I, after recognition of conserved pathogen-associated molecular patterns (PAMPs) represents a cornerstone of innate immune responses (Iwasaki and Medzhitov, 2015). Therefore, sustained silencing of systemic IFN-I (in the face of copious levels of PAMPs) during chronic viral infections (Borrow, 2011; Bruel et al., 2014; Stacey et al., 2009; Zuniga et al., 2008; Zuniga et al., 2015) is highly intriguing and suggests long-term innate adaptations to the chronic infectious environment, although the underlying mechanisms remained to be determined. Here we studied pDC adaptation to chronic infection and revealed that the maintenance of an exhausted pDC pool was achieved via two mechanisms: IFN-I signaling limited de novo pDC generation from BM progenitors while promoting (together with TLR7) self-renewal of exhausted pDCs in the periphery, whereas TLR7 signaling curtailed pDC IFN-I-production capacity.
All progenitor and pDC adaptations that we observed during chronic LCMV infection were also detected early after acute LCMV infection, supporting the view that they represent general responses to microbial exposure. However, most of these adaptations were sustained long-term only during chronic LCMV infection, suggesting that a persisting infectious environment is necessary to perpetuate them. It is conceivable that down-regulation of E2-2 and its targets SPIB and BCL11A, which are critical for pDC development, lineage commitment and/or function (Cisse et al., 2008; Ghosh et al., 2010; Murphy et al., 2016; Sasaki et al., 2012), contributed to the limited generation of functional pDCs from BM and/or the functional loss of exhausted pDCs during chronic LCMV infection in mice, and this may extend to HIV-infected patients. Similarly, as we observed in LCMV chronically infected mice, it is possible that an initial IFN-I peak and/or a persistently low IFN-I signature (Borrow, 2011; Feldman et al., 2001; Snell and Brooks, 2015; Stacey et al., 2009; Zuniga et al., 2015) could suppress BM pDC generation in chronically infected patients, explaining the decreased numbers of circulating pDCs (Duan et al., 2004; Feldman et al., 2001; Kanto et al., 2004). Although this may appear at odds with a previous report showing that (under non-infectious conditions) recombinant IFN-α enhances BM pDC generation (Li et al., 2011), it is likely that factors in the infectious environment may modulate the cell-intrinsic or -extrinsic effects of IFN-I signaling on BM progenitors.
Increased BrdU incorporation and Ki67 expression in pDCs have been shown in SIV-infected macaques (Brown et al., 2009; Bruel et al., 2014; Li et al., 2015). Ex vivo BrdU incorporation was, however, not detected in the original SIV infection study (Brown et al., 2009), leading to the interpretation that Ki67+ or BrdU+ pDCs represent cells recently egressed from BM (Brown et al., 2009; Bruel et al., 2014). In our study, ex vivo BrdU was incorporated by splenic pDCs from chronically infected mice, supporting peripheral pDC proliferation. Thus, we propose that initial TLR7 signaling and IFN-I elevation may induce expansion of CD4− pDC subsets after both acute and chronic LCMV infection while continuous TLR stimulation and low IFN-I levels perpetuate peripheral self-renewal of exhausted pDCs during chronic infection. The initial, more robust, pDC proliferation may allow restoration of pDC numbers after their initial ablation (Swiecki et al., 2011) despite limited BM pDC generation and result in pDC accumulation in lymphoid organs that we observed at day 10 after LCMV infection and that others have reported in HIV-infected patients (Dillon et al., 2008; Foussat et al., 2001; Nascimbeni et al., 2009; Tilton et al., 2008). pDC proliferation was, however, subsequently attenuated, explaining the decline in pDC numbers at later stages after LCMV, HIV and SIV infections (Biancotto et al., 2007; Brown et al., 2009; Zuniga et al., 2008).
We also showed that pDC functional loss involved TLR7 signaling which coincidentally down-regulated E2-2 levels. pDC exhaustion due to sustained TLR stimulation may be particularly relevant for persistent viral infections but may also extend to tumor-associated pDCs (Hartmann et al., 2003; Labidi-Galy et al., 2011; Sisirak et al., 2012), which may receive continuous TLR signaling by nucleic acids from dying cells (Fuertes et al., 2013). Our results are in line with in vitro studies showing that CpG stimulation down-regulates E2-2 expression in murine pDCs and a human pDC cell line (Cisse et al., 2008) and that pDCs are unable to induce secondary responses after TLR stimulation (Ito et al., 2006). However, TLR7-deficiency only partially protected pDC IFN-I production capacity, indicating that other mechanisms contributed to pDC exhaustion, as previously proposed (Schwartz et al., 2017; Vincent et al., 2011; Zuniga et al., 2015). Together with these reports, our data highlight involvement of host innate regulatory mechanism analogous to T and B cell exhaustion in IFN-I suppression during chronic infection. Finally, our findings suggest that prolonged therapeutic administration of TLR ligands (Iribarren et al., 2016) may cause pDC exhaustion and enhanced susceptibility to secondary infections.
It is tempting to speculate that the evolutionary driver of the rapidly triggered pDC exhaustion in both acute and chronic infections may have been the need for limiting IFN-I-induced immunopathology, which can be lethal after some infections (McNab et al., 2015; Ng et al., 2016). Furthermore, maintenance of an exhausted pDC pool via local self-renewal may provide an infected host with greater plasticity to tailor pDC functions according to pathogen load or other cues in the nearby environment. Sustained IFN-I exhaustion comes, however, with a cost for the infected host, who becomes compromised to mount optimal innate responses and control certain secondary infections and tumors (Feldman et al., 2001; Jarrett, 2006; Kohler et al., 1990; Zuniga et al., 2008). Nonetheless, increased IFN-I levels and enhanced resistance to a secondary pathogen upon preventing pDC exhaustion in our study opens the possibility that transient recovery from innate exhaustion could be combined with existing treatments targeting adaptive immune cells to enable more effective control of chronic infections and associated opportunistic pathogens and/or tumors.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Elina Zuniga (eizuniga@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Female C57BL/6 mice (6-8 weeks old) were purchased from The Jackson Laboratory. Tlr7−/− mice were obtained from Dr. Maripat Corr (University of California, San Diego) (Lund et al., 2004). Ifnar−/− mice were obtained from Dr. Charles Surh (La Jolla Institute for Allergy and Immunology). All mice were housed under specific-pathogen-free conditions at the University of California, San Diego. Mice were bred and maintained in a closed breeding facility and mouse handling and experiments conformed to the requirements of the National Institute of Health and the Institutional Animal Care and Use Guidelines of UC San Diego. Unless stated otherwise, experiments were initiated in mice (female and male) at 7-12 weeks of age.
Virus strains
Mice were infected with 2 × 106 plaque-forming units (pfu) of LCMV Arm (ARM) or LCMV clone 13 (Cl13) intravenously (i.v.) via tail vein. Viruses were propagated on BHK cells and quantified by plaque assay performed on Vero cells (Ahmed et al., 1984). Briefly, Vero cell monolayers were infected with 500 μL of serially diluted viral stock and incubated for 60 minutes at 37°C in 5% CO2 with gentle shaking every 15 minutes. Agarose overlay was added to infected cells and placed in incubator for 6 days at 37°C in 5% CO2. Cells were fixed with formaldehyde and stained with crystal violet for 5 minutes at room temperature and plaques were counted.
Mice were infected with 1 × 104 PFU of MCMV Smith intraperitoneally (i.p.). Viruses were propagated in vivo in 3-week-old BALB/c mice infected with 1 × 104 pfu by an i.p. route and quantified by plaque assay as described below (Tabeta et al., 2004).
Human Samples
All human subject studies were approved by the Institutional Review Board (IRB) at Rutgers, the State University of New Jersey, New Jersey Medical School. Blood samples from HIV-negative and HIV-infected volunteers were obtained with consent according to institutional guidelines and the Declaration of Helsinki. Age and sex of the subjects are provided in table S1 of the manuscript.
Cell lines
ISRE-L929 reporter cells (male origin, provided by Dr, Bruce Beutler, University of Texas South Western Medical Center) and NIH-3T3 cells (sex information not available) were cultured in DMEM containing 10% FBS, penicillin-streptomycin, and supplements of L-glutamine. 1×106 cells were passaged every 3-4 days. Cells were washed in PBS pH 7.4 then lifted from culture flasks with 0.05% Trypsin-EDTA. Trypsin was deactivated by addition of serum containing media.
Primary cell culture
BMDCs were differentiated at the concentration of 2x106 cells/ml for 8 days in 5ml of RPMI-1640 supplemented with 10% (vol/vol) fetal bovine serum, L-glutamine, penicillin-streptomycin, and HEPES pH 7.2) supplemented with 100ng/mL Flt3L and 50μM β-mercaptoethanol. Half of the medium was replaced after 5 days with fresh cytokines added.
METHOD DETAILS
Mouse Cell Purification
Spleens were incubated with 1mg/mL collagenase D for 20 min at 37°C and passed through a 100μm strainer to achieve a single cell suspension. For purification of spleen or BM pDCs, splenocytes or BM cells were enriched with PanDC microbeads using an Automacs system (Miltenyi) or by using EasySep™ Mouse Streptavidin RapidSpheres™ Isolation Kit (Stemcell Technologies), biotin-conjugated anti-Thy1.2 (53-2.1), and anti-CD19 (6D5). PanDC+ fractions or fractions unbound to streptavidin beads were stained with propidium iodide (PI) and FACS-purified using a BD ARIA II (BD Biosciences) for pDCs (PI−CD11cintermediate/dimCD11b−B220+BST2+) after B (CD19), T (Thy1.2) and NK (Nk1.1) cell exclusion. Plasma cells (CD138) were also excluded for purification of BM pDCs. BM pDC progenitors were enriched by using EasySep™ Mouse Streptavidin RapidSpheres™ Isolation Kit (Stemcell Technologies,), biotin-conjugated anti-Thy1.2 (53-2.1), anti-B220 (RA3-6B2), anti-CD11b (M1/70), anti-CD4 (RM4-5), anti-CD19 (6D5) and anti-CD8 (536.7). Fractions unbound to streptavidin beads were stained with PI and FACS-purified using a BD ARIA II (BD) for progenitors (PI−CD11c−c-Kitintermediate/dimFlt3+) after B (CD19, B220), T (Thy1.2, CD3, CD4, and CD8), NK (Nk1.1), red blood cell (Ter119), granulocyte (Gr-1), and monocyte (CD11b) exclusion. pDCs derived from culture of BM cells with Flt3L were stained and sorted as PI−CD11c+CD11b−B220+BST2high. Purity of the cells was > 95% (data not shown).
Human pDC Purification
Peripheral blood mononuclear cells were separated from freshly collected blood using Lymphocyte Separation Media (Mediatech, Inc). pDCs were further purified by using a diamond pDC isolation kit (Miltenyi) following the manufacturer’s instructions. Purity was evaluated by flow cytometry.
Flow Cytometry
The following antibodies were used to stain single cell suspensions prepared from murine BM or spleens: Thy 1.2 (53-2.1), CD19 (eBio1D3), NK 1.1 (PK136), Gr-1 (RB6-8C5), CD11c (N418), CD11b (M1/70), B220 (RA36B2), BST2 (eBio927), CD45.2 (104), CD4 (RM4-5), CD8 (53-6.7), Ter119 (TER-119), CD3 (145-2C11), CD45.1 (A20), MHC class II (I-A/I-E) (M5/114.15.2), CCR9 (eBioCW-1.2), Ki67 (SolA15), c-kit (2B8), CD115 (AFS98), Flt3 (A2F10), Streptavidin, CD127 (eBioRDR5), CD86 (GL1), CD34 (RAM34), CD16/32 (93), and CD138 (281-2). PI or Ghost dye (Tonbo Biosciences, San Diego, CA) was used to exclude dead cells. Cells were pre-incubated with CD16/CD32 Fc block (BD Pharmingen) prior to surface staining. Staining for Ki67 (SolA15) was performed as directed using the Foxp3/Transcription Factor Staining kit. To measure in vivo BrdU incorporation in pDCs, mice were injected with 2mg BrdU (Sigma Aldrich). 16 hours later, spleens were harvested and splenocytes were stained using the BrdU Flow kit (BD Biosciences) following manufacturer instructions. To measure BrdU incorporation ex vivo, splenocytes were cultured at a concentration of 2 × 106 cells/ml with 10mM BrdU. 7 hours later, cells were harvested and stained using the BrdU Flow kit (BD Biosciences) following manufacturer instructions. Cells were acquired with a LSRII flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Treestar, Inc.).
Cytokine Measurements
For cytokine measurement in culture supernatants, spleen or BM cells from individual mice in the same group were pooled prior to FACS purification. FACS-purified pDCs (3 × 104 cells/well) were incubated in 120ul RPMI-1640 supplemented with 10% (vol/vol) fetal bovine serum, L-glutamine, penicillin-streptomycin, and HEPES pH 7.2) supplemented with 50μM β-mercaptoethanol in the presence or absence of CpG-B 1668 (Integrated DNA Technologies) at 0.1μM (Flt3L culture-derived pDC) or 1μM (splenic and BM pDC) or CpG-A 1585 (InvivoGen) at 0.5uM for 6 or 15 hours at 37°C. Supernatants were collected and stored at −80°C until further analysis. IFN-I bioactivity was measured with reference to a recombinant mouse IFNβ standard (Sigma-Aldrich) using ISRE-L929 reporter cells (L-929 cell line transfected with an interferon-sensitive luciferase) (Jiang et al., 2005). IFNα and IFNβ were measured by Lumikine mIFNα and Lumikine Xpress mIFNβ ELISA (InvivoGen), respectively, following manufacturer instructions. TNFα was measured by Mouse TNF alpha ELISA Ready-SET-Go!® (eBioscience) following manufacturer instructions. Graphs depicting cytokine measurements in this paper represent individual wells of stimulation.
Quantitative Real-time RT-PCR (qRT-PCR) Analysis
For qRT-PCR analysis, spleen or BM cells from individual mice in the same group were pooled prior to FACS purification. Total RNA was extracted using RNeasy kits (Qiagen), digested with DNase I (RNase-free DNase set; Qiagen) and reverse-transcribed into cDNA using Superscript III RT (Invitrogen). The expression of various genes was quantified using Fast SYBR Green Master Mix (Thermo Fisher Scientific) or TaqMan™ Fast Universal PCR Master Mix with probe sets from the Universal Probe Library (Roche) and a Real-Time PCR Detection System (Applied Biosystems) or CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Primers for the test genes and probe sets are listed in Table S2. The relative transcript levels were normalized against murine Gapdh or human GAPDH as described previously (Macal et al., 2016). Graphs depicting qRT-PCR analysis of murine genes in this paper represent technical triplicates from one representative experiment.
In vivo IFNAR Blockade
WT mice were treated with 500μg/mouse intraperitoneally (i.p.) with neutralizing antibody (nAb) to IFNAR (clone MAR1-5A3; BioXCell) or Anti-Mouse IgG1 Isotype Control (clone MOPC-21; BioXCell) on days -1 and 0 post-LCMV Cl13-infection. Mice were then treated with 250μg/mouse anti-IFNAR nAb or isotype control i.p. on days 2, 4, and 6 post-LCMV Cl13 infection as previously described (Wilson et al., 2013). Uninfected control mice were similarly treated in parallel.
Generation of Mixed BM Chimeras
WT CD45.1+ C57BL/6 recipient mice were sub-lethally irradiated with 1000 rads and reconstituted with a 1:1 mix of BM cells from CD45.1+ WT mice and CD45.2+ Tlr7−/− or Ifnar −/− mice (or CD45.2+ WT mice for WT:WT controls). 5-10×106 total mixed BM cells were transferred i.v. into the irradiated recipient mice, which were treated with antibiotics (Trimethoprim 8 mg/ml and Sulfamethoxazole 40 mg/ml supplied in drinking water; Akorn) for 3 weeks to prevent infection and allow immune reconstitution. Reconstitution was analyzed 8 weeks after BM transfer by determining the ratio of CD45.1+ to CD45.2+ immune subsets in blood by flow cytometry.
MCMV Plaque Assays
Livers (or salivary glands for MCMV viral stock titration) were homogenized 3.5 days post-MCMV infection and serially diluted samples were incubated on NIH 3T3 monolayer with rotation for 2hr at 37°C. Samples were removed and monolayers were overlayed with DMEM-Carboxymethylcellulose 5 days at 37°C. Cells were fixed with formaldehyde and stained with crystal violet for 5 minutes at room temperature and plaques were counted (Orange et al., 1995).
In vivo Progenitor Transfer Assay
FACS-purified Lin−c-kitint/loFlt3+ progenitors (1.5 × 104 cells) from CD45.2+ uninfected or Cl13-infected donor mice at day 30 p.i. were injected i.v. into CD45.1+ non-irradiated uninfected or infection-matched recipient mice, respectively. Mice were sacrificed at day 10, and spleen cells were analyzed by flow cytometry after splenocytes were enriched by using EasySep™ Mouse Streptavidin RapidSpheres™ Isolation Kit (Stemcell Technologies), biotin-conjugated anti-Thy1.2 (53-2.1), and anti-CD19 (6D5).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical parameters are described in figure legends. Student’s t tests (two-tailed, unpaired, or where indicated paired), one-way Anova, two-way Anova, Mann-Whitney tests and multiple comparisons were performed by using GraphPad Prism 6.0 (GraphPad). Significance was defined as p ≤ 0.05. In all figures, error bars indicate S.E.M.
Supplementary Material
Highlights.
-
-
IFN-I down-regulates E2-2 in BM progenitors and compromises BM pDC generation
-
-
IFN-I and TLR7 induce pDC proliferation and sustain exhausted pDC numbers
-
-
TLR7 down-regulates E2-2 and mediates loss of function in exhausted pDCs
-
-
TLR7 deficiency enhances IFN-I elevation and resistance upon secondary MCMV infection
Acknowledgments
We thank Christie Lyn Costanza and Marta Paez-Quinde for help with human subject recruitment. This study was supported by a scholar grant from Lupus Research Institute and a scholar research grant from American Cancer Society as well as NIH grants AI081923 and AI113923 (to E.I.Z.) and R01AI106125 (to P.F.B). S.D. was a visiting graduate student from University of Milan, Italy, and was supported by a scholarship from Ministero dell’Istruzione, dell’Università e della Ricerca. E.I.Z. was a Leukemia and Lymphoma Society Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
M.M. designed, performed, and analyzed experiments related to BM progenitors and BM-derived and splenic pDCs at day 9 time-point, IFNAR blockade, Tlr7−/− mice, Tcf4 and SpiB expression, and MCMV experiments; Y.J. designed, performed and analyzed experiments related to BM pDCs, LCMV ARM infection, day 5 and 30 time-points, pDC proliferation, SpiB, Bcl11a and NP expression, and WT:Ifnar−/− mixed BM chimera. S.D., J.D., and P.F.B. designed, performed, and analyzed human studies. A.Y.C. helped design, perform and interpret TLR7 studies in BM. S.S. helped recruited human subjects. E.I.Z. conceived the study and supervised the design, analysis and interpretation of all experiments. E.W. contributed to data interpretation and manuscript editing. M.M., Y.J., and E.I.Z. wrote manuscript.
Declaration of Interests
The authors declare no competing interests.
References
- Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44:1052–1068. doi: 10.1016/j.immuni.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, Debernardo R, Garate K, Rodriguez B, Margolis LB, Lederman MM. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood. 2007;109:4272–4279. doi: 10.1182/blood-2006-11-055764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Krebs P, Sullivan BM, Oldstone MB, Popkin DL. Slc15a4, a gene required for pDC sensing of TLR ligands, is required to control persistent viral infection. PLoS Pathog. 2012;8:e1002915. doi: 10.1371/journal.ppat.1002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P. Innate immunity in acute HIV-1 infection. Curr Opin HIV AIDS. 2011;6:353–363. doi: 10.1097/COH.0b013e3283495996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5:e1000413. doi: 10.1371/journal.ppat.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel T, Dupuy S, Demoulins T, Rogez-Kreuz C, Dutrieux J, Corneau A, Cosma A, Cheynier R, Dereuddre-Bosquet N, Le Grand R, Vaslin B. Plasmacytoid dendritic cell dynamics tune interferon-alfa production in SIV-infected cynomolgus macaques. PLoS Pathog. 2014;10:e1003915. doi: 10.1371/journal.ppat.1003915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SM, Robertson KB, Pan SC, Mawhinney S, Meditz AL, Folkvord JM, Connick E, McCarter MD, Wilson CC. Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J Acquir Immune Defic Syndr. 2008;48:1–12. doi: 10.1097/QAI.0b013e3181664b60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan XZ, Wang M, Li HW, Zhuang H, Xu D, Wang FS. Decreased frequency and function of circulating plasmocytoid dendritic cells (pDC) in hepatitis B virus infected humans. J Clin Immunol. 2004;24:637–646. doi: 10.1007/s10875-004-6249-y. [DOI] [PubMed] [Google Scholar]
- Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, Sun Y, Megjugorac N, Fitzgerald-Bocarsly P. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol. 2001;101:201–210. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- Foussat A, Bouchet-Delbos L, Berrebi D, Durand-Gasselin I, Coulomb-L'Hermine A, Krzysiek R, Galanaud P, Levy Y, Emilie D. Deregulation of the expression of the fractalkine/fractalkine receptor complex in HIV-1-infected patients. Blood. 2001;98:1678–1686. doi: 10.1182/blood.v98.6.1678. [DOI] [PubMed] [Google Scholar]
- Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34:67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity. 2010;33:905–916. doi: 10.1016/j.immuni.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, Giese T, Gires O, Endres S, Hartmann G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003;63:6478–6487. [PubMed] [Google Scholar]
- Iribarren K, Bloy N, Buque A, Cremer I, Eggermont A, Fridman WH, Fucikova J, Galon J, Spisek R, Zitvogel L, et al. Trial Watch: Immunostimulation with Toll-like receptor agonists in cancer therapy. Oncoimmunology. 2016;5:e1088631. doi: 10.1080/2162402X.2015.1088631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. 2006;107:2423–2431. doi: 10.1182/blood-2005-07-2709. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett RF. Viruses and lymphoma/leukaemia. J Pathol. 2006;208:176–186. doi: 10.1002/path.1905. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- Kanto T, Inoue M, Miyatake H, Sato A, Sakakibara M, Yakushijin T, Oki C, Itose I, Hiramatsu N, Takehara T, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190:1919–1926. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- Kohler M, Ruttner B, Cooper S, Hengartner H, Zinkernagel RM. Enhanced tumor susceptibility of immunocompetent mice infected with lymphocytic choriomeningitis virus. Cancer Immunol Immunother. 1990;32:117–124. doi: 10.1007/BF01754208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, Combes JD, Faget J, Mithieux F, Cassignol A, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011;71:5423–5434. doi: 10.1158/0008-5472.CAN-11-0367. [DOI] [PubMed] [Google Scholar]
- Lee LN, Burke S, Montoya M, Borrow P. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J Immunol. 2009;182:7178–7189. doi: 10.4049/jimmunol.0802526. [DOI] [PubMed] [Google Scholar]
- Li H, Evans TI, Gillis J, Connole M, Reeves RK. Bone marrow-imprinted gut-homing of plasmacytoid dendritic cells (pDCs) in acute simian immunodeficiency virus infection results in massive accumulation of hyperfunctional CD4+ pDCs in the mucosae. J Infect Dis. 2015;211:1717–1725. doi: 10.1093/infdis/jiu671. [DOI] [PubMed] [Google Scholar]
- Li HS, Gelbard A, Martinez GJ, Esashi E, Zhang H, Nguyen-Jackson H, Liu YJ, Overwijk WW, Watowich SS. Cell-intrinsic role for IFN-alpha-STAT1 signals in regulating murine Peyer patch plasmacytoid dendritic cells and conditioning an inflammatory response. Blood. 2011;118:3879–3889. doi: 10.1182/blood-2011-04-349761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol. 2007;8:578–583. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, Zuniga EI. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe. 2012;11:617–630. doi: 10.1016/j.chom.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macal M, Tam MA, Hesser C, Di Domizio J, Leger P, Gilliet M, Zuniga EI. CD28 Deficiency Enhances Type I IFN Production by Murine Plasmacytoid Dendritic Cells. J Immunol. 2016;196:1900–1909. doi: 10.4049/jimmunol.1501658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol. 2006;80:9192–9199. doi: 10.1128/JVI.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Fauci AS. B-cell exhaustion in HIV infection: the role of immune activation. Curr Opin HIV AIDS. 2014;9:472–477. doi: 10.1097/COH.0000000000000092. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, Kretzer NM, Durai V, Murphy KM. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol. 2016;34:93–119. doi: 10.1146/annurev-immunol-032713-120204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni M, Perie L, Chorro L, Diocou S, Kreitmann L, Louis S, Garderet L, Fabiani B, Berger A, Schmitz J, et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood. 2009;113:6112–6119. doi: 10.1182/blood-2008-07-170803. [DOI] [PubMed] [Google Scholar]
- Ng CT, Mendoza JL, Garcia KC, Oldstone MB. Alpha and Beta Type 1 Interferon Signaling: Passage for Diverse Biologic Outcomes. Cell. 2016;164:349–352. doi: 10.1016/j.cell.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Wang B, Terhorst C, Biron CA. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med. 1995;182:1045–1056. doi: 10.1084/jem.182.4.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki I, Hoshino K, Sugiyama T, Yamazaki C, Yano T, Iizuka A, Hemmi H, Tanaka T, Saito M, Sugiyama M, et al. Spi-B is critical for plasmacytoid dendritic cell function and development. Blood. 2012;120:4733–4743. doi: 10.1182/blood-2012-06-436527. [DOI] [PubMed] [Google Scholar]
- Sathe P, Vremec D, Wu L, Corcoran L, Shortman K. Convergent differentiation: myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood. 2013;121:11–19. doi: 10.1182/blood-2012-02-413336. [DOI] [PubMed] [Google Scholar]
- Schlitzer A, Heiseke AF, Einwachter H, Reindl W, Schiemann M, Manta CP, See P, Niess JH, Suter T, Ginhoux F, Krug AB. Tissue-specific differentiation of a circulating CCR9- pDC-like common dendritic cell precursor. Blood. 2012;119:6063–6071. doi: 10.1182/blood-2012-03-418400. [DOI] [PubMed] [Google Scholar]
- Schlitzer A, Loschko J, Mair K, Vogelmann R, Henkel L, Einwachter H, Schiemann M, Niess JH, Reindl W, Krug A. Identification of CCR9- murine plasmacytoid DC precursors with plasticity to differentiate into conventional DCs. Blood. 2011;117:6562–6570. doi: 10.1182/blood-2010-12-326678. [DOI] [PubMed] [Google Scholar]
- Schwartz JA, Clayton KL, Mujib S, Zhang H, Rahman AK, Liu J, Yue FY, Benko E, Kovacs C, Ostrowski MA. Tim-3 is a Marker of Plasmacytoid Dendritic Cell Dysfunction during HIV Infection and Is Associated with the Recruitment of IRF7 and p85 into Lysosomes and with the Submembrane Displacement of TLR9. J Immunol. 2017;198:3181–3194. doi: 10.4049/jimmunol.1601298. [DOI] [PubMed] [Google Scholar]
- Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest. 2004;113:737–745. doi: 10.1172/JCI20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, Renaudineau S, Poyet G, Labidi-Galy SI, Goddard-Leon S, et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012;72:5188–5197. doi: 10.1158/0008-5472.CAN-11-3468. [DOI] [PubMed] [Google Scholar]
- Snell LM, Brooks DG. New insights into type I interferon and the immunopathogenesis of persistent viral infections. Curr Opin Immunol. 2015;34:91–98. doi: 10.1016/j.coi.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelekati E, Wherry EJ. Chronic bystander infections and immunity to unrelated antigens. Cell Host Microbe. 2012;12:458–469. doi: 10.1016/j.chom.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15:471–485. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. 2010;33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med. 2011;208:2367–2374. doi: 10.1084/jem.20110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton JC, Manion MM, Luskin MR, Johnson AJ, Patamawenu AA, Hallahan CW, Cogliano-Shutta NA, Mican JM, Davey RT, Jr, Kottilil S, et al. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished alpha interferon production in vitro. J Virol. 2008;82:3997–4006. doi: 10.1128/JVI.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderford TH, Slichter C, Rogers KA, Lawson BO, Obaede R, Else J, Villinger F, Bosinger SE, Silvestri G. Treatment of SIV-infected sooty mangabeys with a type-I IFN agonist results in decreased virus replication without inducing hyperimmune activation. Blood. 2012;119:5750–5757. doi: 10.1182/blood-2012-02-411496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P, Zoulim F, Tommasino M, Trepo C, Hasan U, Chemin I. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One. 2011;6:e26315. doi: 10.1371/journal.pone.0026315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, Colonna M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe. 2012;11:631–642. doi: 10.1016/j.chom.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Chow KV, Soo P, Xu Z, Brady JL, Lawlor KE, Masters SL, O’Keeffe M, Shortman K, Zhang JG, Lew AM. Plasmacytoid dendritic cells are short-lived: reappraising the influence of migration, genetic factors and activation on estimation of lifespan. Sci Rep. 2016;6:25060. doi: 10.1038/srep25060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4:374–386. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga EI, Macal M, Lewis GM, Harker JA. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu Rev Virol. 2015;2:573–597. doi: 10.1146/annurev-virology-100114-055226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.