

Abstract
Cancer metabolism is significantly altered from normal cellular metabolism allowing cancer cells to adapt to changing microenvironments and maintain high rates of proliferation. In the past decade, stable-isotope tracing and network analysis have become powerful tools for uncovering metabolic pathways that are differentially activated in cancer cells. In particular, 13C metabolic flux analysis (13C-MFA) has emerged as the primary technique for quantifying intracellular fluxes in cancer cells. In this review, we provide a practical guide for investigators interested in getting started with 13C-MFA. We describe best practices in 13C-MFA, highlight potential pitfalls and alternative approaches, and conclude with new developments that can further enhance our understanding of cancer metabolism.
Cancer research: tagging to understand cancer metabolism
Tracing tagged molecules can help researchers understand the altered metabolism of cancer cells. The abilities of cancer cells to multiply rapidly and invade new tissues are supported by metabolic alterations, which can be investigated by feeding tagged molecules to cells and tracing how they are metabolized. These techniques, such as 13C metabolic flux analysis (13C-MFA), have been perceived as difficult to use, but recent advances are making them more accessible. Maciek Antoniewicz, University of Delaware, Newark, USA, has published a practical guide for researchers wanting to use 13C-MFA. The review includes best practices, pitfalls, alternative approaches, and new developments, especially new user-friendly software that allows researchers without extensive training in mathematics, statistics, or coding to perform 13C-MFA. Broadening access to tools for investigating altered metabolic pathways may spur development of new cancer therapies targeting these pathways.
Introduction
In the past decade, measuring intracellular metabolism has become an indispensable tool in biomedical research1,2. Cancer metabolism is an especially active area of research3–8. It has long been recognized that cancer cells exhibit rewired metabolism compared to normal cells. A century ago, Warburg9 described how cancer cells take up large amounts of glucose and preferentially convert it to lactate, even under aerobic conditions. This so-called Warburg effect, or aerobic glycolysis, is a major hallmark of cancer metabolism10–12. More recently, with the aid of stable-isotope tracers and network analysis, additional metabolic pathways were identified that are activated in cancer cells, including reductive metabolism of glutamine13, altered glycolysis14, serine and glycine metabolism15–17, one-carbon metabolism18,19, transketolase-like 1 (TKTL1) pathway20,21, and acetate metabolism22–25. The activities of these pathways allow cancer cells to extract cellular building blocks and energy from substrates and use them for cell growth. With the rapid progress in cancer research, an increasingly clearer picture is generated how cancer cells rewire their metabolism, adapt to and manipulate their microenvironment26–28, and maintain a continuous supply of anabolic precursors, reducing equivalents and energy to fuel the reproduction of more cancer cells5,29.
The complexities of mammalian metabolism require a systems-level analysis of the underlying networks and metabolic phenotypes30,31. Currently, 13C metabolic flux analysis (13C-MFA) is the preferred tool for quantitative characterization of metabolic phenotypes in microbial32–34 and mammalian cells3,4,35–38. The emergence of 13C-MFA as a primary research tool was made possible in large part due to several major advances in theoretical approaches for conducting 13C-MFA calculations39–41, and more recently, by the availability of dedicated and user-friendly software tools for 13C-MFA such as Metran and INCA42,43. However, 13C-MFA it is still not widely used by cancer biologists, outside of a few expert groups. This may be in part because 13C-MFA is sometimes perceived as unintuitive, obscure, demanding in terms of time and data, and costly in terms of initial capital investment and isotopic tracers. Moreover, few guidelines exist to help researchers get started with 13C-MFA44,45. The main objective of this review is to address these concerns by providing practical guidelines for cancer biologists interested in 13C-MFA. First, we describe the basics of 13C-MFA, discuss key assumptions that are inherent in 13C-MFA but may not always be explicitly stated, highlight best practices in 13C-MFA, and identify potential pitfalls as well as alternative approaches. Throughout, we emphasize key aspects that should be considered when planning tracer experiments and performing 13C-MFA calculations to ensure correct interpretation of data and results, and to increase insights obtained from these studies.
Basics of 13C-MFA
Cellular metabolism serves four important functions in proliferating cancer cells: (1) supply of anabolic building blocks for cell growth; (2) generation of metabolic energy in the form of ATP to drive thermodynamically unfavorable reactions; (3) generation of redox equivalents in the form of NADPH for anabolic processes such as fatty acid biosynthesis and to combat oxidative stress; and (4) maintaining redox homeostasis by oxidizing excess NADH generated in central metabolic pathways.
The first step in obtaining a quantitative picture of cellular metabolism is to measure the growth rate of the cells and quantify nutrient uptake and secretion rates such as glucose and glutamine uptake and lactate secretion46,47 (Fig. 1). These external rates provide important boundary constraints on intracellular pathway activities. However, due to redundancies in mammalian metabolic pathways, external rates alone do not allow detailed conclusions to be drawn about the relative contribution of specific metabolic pathways to overall metabolism46,48. To examine intracellular fluxes in detail, stable isotopes such as 13C are utilized. When a labeled substrate, e.g., [1,2-13C]glucose, is metabolized by cells, enzymatic reactions rearrange carbon atoms resulting in specific labeling patterns in downstream metabolites that can be measured with analytical techniques such as mass spectrometry (MS), or nuclear magnetic resonance. For a well-selected tracer, different metabolic pathways will produce distinctly different labeling patterns in the measured metabolites from which fluxes can be inferred49,50. However, in most cases, isotopic labeling data cannot be interpreted intuitively due to the highly complex nature of atom rearrangements in metabolic pathways51; instead, a formal model-based analysis approach is required to extract flux information from the labeling data. In the past 20 years, 13C-MFA has emerged as the primary approach used for converting isotopic labeling data into corresponding metabolic flux maps45.
Fig. 1. Glucose and glutamine are the two most highly consumed carbon substrates in cancer cells.
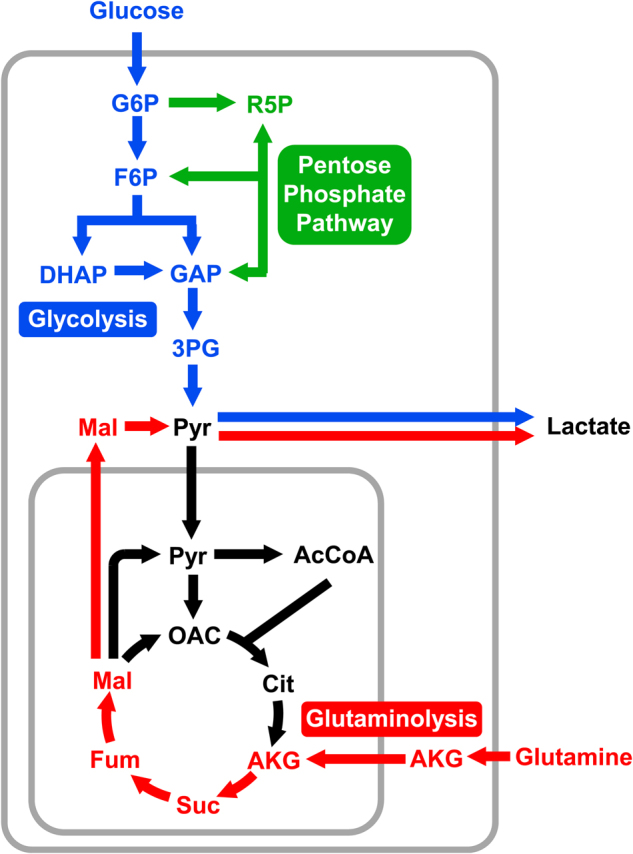
Both substrates can be converted to lactate via glycolysis and glutaminolysis, respectively. High lactate secretion, especially from glucose, is a major hallmark of cancer cells known as the Warburg effect, or aerobic glycolysis
The main objective of 13C-MFA is thus to generate a quantitative map of cellular metabolism by assigning flux values to the reactions in the network model and confidence intervals for each estimated flux (Fig. 2). At a high level, 13C-MFA is formulated as a least-squares parameter estimation problem, where fluxes are unknown model parameters that must be estimated by minimizing the difference between the measured labeling data and labeling patterns simulated by the model, subject to stoichiometric constraints resulting from mass balances for intracellular metabolites and metabolite labeling states, the so-called isotopomers40,52. When 13C-MFA first emerged in 1990s53, the main challenge was to develop efficient algorithms for solving large sets of isotopomer mass balances54. Eventually, the computational problems in 13C-MFA were resolved with the development of the elementary metabolite unit (EMU) framework that allows efficient simulation of isotopic labeling in any arbitrary biochemical network model39. The EMU framework was subsequently incorporated into user-friendly software tools for 13C-MFA, such as Metran and INCA42,43, that are freely available to the scientific community. These powerful tools have opened up 13C-MFA to a much wider scientific audience, including cancer biologists, that may not have extensive background in mathematics and statistics, which was required before these software packages became available. In the next sections, we describe in detail the three inputs that are required for performing 13C-MFA calculations: (i) external rates; (ii) isotopic labeling; and (iii) metabolic model (Fig. 2).
Fig. 2. 13C metabolic flux analysis (13C-MFA) is a powerful approach for quantifying intracellular metabolic fluxes in cancer cells.

The three inputs required for 13C-MFA are external uptake and secretion rates, isotopic labeling measurements, and a comprehensive compartmentalized model of cellular metabolism. User-friendly software tools for 13C-MFA, such as Metran and INCA, can be used to perform 13C-MFA calculations. These tools produce as outputs fluxes for all reactions in the model, confidence intervals for the estimated fluxes, and statistical analysis of the goodness-of-fit
Determination of external rates
To quantify intracellular metabolic fluxes, first, the cross talk between the cells and their environment must be quantified. Collectively referred to as external rates, this includes measuring influxes of nutrients such as glucose and glutamine, and secretion of metabolic by-products such as lactate and glutamate. In addition, the rate of cell growth must be determined. Assuming cells are continuously dividing, the cell number will increase exponentially according to:
| 1 |
Here Nx is the number of cells (typically expressed in millions of cells), and µ (1/h) is the growth rate. The growth rate is easily determined by plotting the natural logarithm of Nx vs time and determining the slope of the curve. If cells are counted only at two time points, then the growth rate is determined as follows:
| 2 |
The doubling time (td) is inversely related to the growth rate, according to:
| 3 |
External rates, i.e., nutrient uptake rates and waste product secretion rates, can be determined in a straightforward way by measuring changes in metabolite concentrations during the labeling experiment. For exponentially growing cells, external rates (ri, in units nmol/106 cells/h) can be calculated as follows:
| 4 |
Here ΔCi (mmol/L) is the change in concentration of a particular metabolite i between two sampling time points, ΔNx is the change in cell number (expressed in millions of cells) during the same time period, V (mL) is the culture volume, and µ (1/h) is the growth rate. Based on this expression, external rates have negative values for uptake rates and positive values for secretion rates. For non-proliferating cells, external rates are determined by a slightly different expression:
| 5 |
Because glutamine is an unstable molecule, i.e., it spontaneously degrades to pyroglutamate and ammonium under normal culture conditions, the calculated glutamine uptake rate must be corrected for glutamine degradation, i.e., the measured rate reflects both net uptake of glutamine by the cells and glutamine degradation. Glutamine degradation can be expressed as a first-order degradation process with a degradation constant of around 0.003/h55. After correcting for glutamine degradation55, the true net glutamine uptake rate is obtained. For long tracer experiments (e.g., >24 h), it may also be necessary to correct for evaporation effects. For this purpose, control experiments without cells are performed. By measuring the apparent increases in metabolite concentrations over time, the rate of evaporation can be estimated. The dynamics of glutamine degradation are also easily determined from these control experiments.
For 13C-MFA studies, external rates are often determined for glucose uptake, lactate secretion, and amino-acid uptake and secretion. For proliferating cancer cells, typical values are as follows: 100–400 nmol/106 cells/h for glucose uptake; 200–700 nmol/106 cells/h for lactate secretion; 30–100 nmol/106 cells/h for glutamine uptake; and 2–10 nmol/106 cells/h for uptake or secretion of other amino acids. Depending on the scope of the study, it may also be important to measure the rates of other metabolites such as ammonium, pyruvate, acetate, citrate, and any other significant nutrients or by-products that cancer cells exchange with their environment.
Measurement of isotopic labeling
When conducting 13C-tracer experiments, a labeled substrate is introduced to the culture medium that is then taken up by the cells and metabolized through various metabolic pathways. It takes a certain amount of time before intracellular metabolites reach a constant labeling state, which is referred to as isotopic steady state46 (Fig. 3). The time required to reach isotopic steady state depends on the turnover rate of metabolites in a pathway and the labeling dynamics of upstream metabolites that feed into the pathway. The turnover rate of a metabolite pool is roughly equivalent to the ratio of the metabolite pool size and the flux through that metabolite pool. For proliferating cells, isotopic steady state can be reached relatively quickly, i.e., within a few hours after the introduction of the isotopic tracer56. However, in some cases, due to exchange of intracellular and external metabolites, significantly slower labeling incorporation rates can be observed. In particular, external lactate often acts as a large buffer that slows down labeling of intracellular pyruvate and downstream metabolic pathways, e.g., tricarboxylic acid (TCA) cycle, when 13C-glucose tracers are used55. Slow labeling may be observed even if there is large net secretion of lactate, since external lactate readily exchanges with intracellular lactate, which in turn rapidly equilibrates with cytosolic pyruvate. The effective pool size of intracellular pyruvate thus becomes the combined pool of intracellular pyruvate, intracellular lactate, and external lactate. This buffering effect can be so extreme that certain metabolites may never reach isotopic steady state55. One strategy to reduce the buffering effect of lactate is to ensure that little or no lactate is present in the medium at the beginning of 13C-glucose tracer experiments.
Fig. 3. Parallel labeling experiments with different 13C-labeled substrates greatly enhance the resolution of metabolic fluxes in complex models.

The rate of labeling incorporation after the introduction of a 13C-tracer depends on the turnover rate of intracellular metabolites and exchanges between intracellular and extracellular metabolites. In particular, external lactate can slow down labeling of intracellular pyruvate and TCA cycle metabolites from 13C-glucose tracers. If isotopic steady state is reached then labeling data can be analyzed with 13C-MFA. However, if the system has not reached isotopic steady state, then the labeling data must be analyzed using isotopic non-stationary 13C-MFA (13C-NMFA)
An important inherent assumption of 13C-MFA calculations is that all metabolites are at isotopic steady state. It is thus critical to validate this assumption for all tracer experiments performed. To validate this, isotopic labeling is measured for at least two time points, e.g., 18 and 24 h, after the introduction of tracer. If isotopic labeling is identical for the two time points, then isotopic steady state is confirmed and the labeling data can be analyzed using classical 13C-MFA. However, if isotopic labeling is changing with time, then the data must be analyzed using a more advanced 13C-MFA approach called isotopic-non-stationary 13C-MFA, or 13C-NMFA41. Most software packages for 13C-MFA can only perform classical 13C-MFA calculations, i.e., assuming isotopic steady state, although a few software packages such as INCA can perform both 13C-MFA and 13C-NMFA calculations43.
MS is currently the preferred analytical technique used for measuring isotopic labeling of intracellular metabolites. With the technological advances in gas chromatography/MS (GC/MS) and liquid chromatography/MS (LC/MS) in the past two decades, it is now possible to measure mass isotopomer distributions for a large number of intracellular metabolites from as few as one million cells, including for intermediates of glycolysis pathway: fructose 6-phosphate (F6P), dihydroxyacetone phosphate, glycerol 3-phosphate, 3-phosphoglycerate (3PG), phosphoenolpyruvate, pyruvate, and lactate; intermediates of the pentose phosphate pathway (PPP; LC/MS mainly): xylulose 5-phosphate (X5P), ribose 5-phosphate (R5P), and sedoheptulose 7-phosphate; intermediates of the TCA cycle: citrate, α-ketoglutarate (AKG), succinate, fumarate, and malate; and most amino acids, including alanine, aspartate, glutamate, glutamine, proline, serine, and glycine.
Parallel labeling experiments
The selection of an isotopic tracer (or multiple tracers) is one of the most important considerations when designing 13C-MFA studies, since this ultimately determines the quality (i.e., precision and accuracy) of flux results that can be obtained50. It is now well-known that there is no single best tracer for 13C-MFA studies. Generally, 13C-glucose tracers are best for determining fluxes in upper metabolism (e.g., glycolysis and PPP), while 13C-glutamine tracers typically produce better resolution of fluxes in lower parts of metabolism (e.g., TCA cycle and reductive carboxylation)57,58 (Fig. 3). A powerful approach to achieve high resolution of multiple metabolic pathways is to perform parallel labeling experiments with different tracers and then integrate all data into a single comprehensive flux model59,60. For example, parallel labeling experiments with [1,2-13C]glucose and [U-13C]glutamine have been demonstrated to be particularly informative and complementary56,58,61. When conducting parallel labeling experiments, it is important that the only difference between the experiments is which metabolite is labeled, i.e., concentrations of all nutrients in the media must be the same for parallel labeling experiments62. With recent advances in 13C-MFA methodology it is now fairly straightforward to analyze isotopic labeling data from parallel labeling experiments45. The Metran software was the first tool that allowed comprehensive analysis of parallel labeling experiments for high-resolution 13C-MFA. Recently, other 13C-MFA software packages have also included this feature.
Metabolic model for 13C-MFA
All 13C-MFA calculations are based on a model of biochemical reactions within a specified metabolic network. Determining the scope of the model is an important decision in 13C-MFA studies. Unfortunately, there is only limited consensus in the literature on the optimal scope of metabolic models for flux analysis in cancer cells. This is in part due to the fact that the appropriate model complexity will depend to some degree on the specific choice of isotopic tracer (or tracers), how many parallel labeling experiments are performed, and how many and which labeling measurements are collected. In general, more comprehensive data sets, i.e., based on multiple parallel labeling experiments with different labeled substrates36,56,60,63, will permit the use of more complex models for 13C-MFA than smaller data sets obtained using a single tracer experiment.
Typically, 13C-MFA models will include all major metabolic pathways of central carbon metabolism such as glycolysis, PPP, TCA cycle, as well as any relevant reactions that connect these pathways (Fig. 4a). Compartmentalization of metabolites and metabolic reactions is an important feature of mammalian cells that must be captured in the model. Metabolites and reactions are therefore assigned to specific metabolic compartments such as cytosol or mitochondrion. Certain metabolites will be present in multiple compartments, for example, pyruvate, acetyl coenzyme A, citrate, malate, fumarate, oxaloacetate, and AKG. These metabolites are treated as separate entities in the model that can have different labeling states in different compartments. Transport reactions in the model allow specific metabolites to be transferred between cellular compartments. Compartment-specific isozymes, which can operate independently, must be included as separate reactions in the model (e.g., cytosolic and mitochondrial isocitrate dehydrogenases; and cytosolic and mitochondrial malic enzymes). Finally, 13C-MFA models will include a lumped biomass formation reaction that drains anabolic precursors from central metabolism (and extracellular medium, e.g., essential amino acids) for the biosynthesis cellular macromolecules55. The stoichiometric coefficients for this lumped biomass reaction are easily determined based on the macromolecular composition of cells (Fig. 4b). Recently, a number of GC/MS-based protocols have been developed that allow biomass compositions of cells to be determined easily and accurately64–66. Typical values for anabolic precursor effluxes for proliferating cancer cells are shown in Fig. 4b.
Fig. 4. 13C metabolic fluxes are estimated based on comprehensive compartmentalized models of cellular metabolism.

a The diagram shows important metabolic pathways in cancer metabolism, including glycolysis, pentose phosphate pathway, TCA cycle, reductive carboxylation of glutamine, and transketolase-like 1 (TKTL1) pathway. One of the key functions of cellular metabolism is to supply anabolic building blocks needed for cell growth, shown here as draining reactions from central metabolic pathways. b A typical macromolecular composition of cancer cells is shown. The macromolecular composition and the growth rate of cells determine the rates at which anabolic precursors must be produced to sustain cell growth. Typical values of anabolic precursor fluxes in proliferating cancer cells are shown
13C-MFA and statistical analysis
Current software tools for 13C-MFA such as Metran and INCA are designed so that users are not required to have any extensive background in mathematics, statistics, or writing computer code. All of the complex math associated with performing 13C-MFA computations is hidden from the user. These software tools accept as inputs: (1) a user-defined metabolic network model consisting of biochemical reactions and corresponding atom transitions; and (2) a set of measurements consisting of isotopic labeling data and external rates. As outputs, the software returns the following: (1) metabolic fluxes for the entire network; (2) confidence intervals for all estimated fluxes; and (3) statistical analysis of the goodness-of-fit (Fig. 2).
13C-MFA should be viewed as an iterative process that requires careful scrutiny of the analysis results. After the software returns a result, it is up to the user to determine how acceptable the result is, and this requires some level of experience. Generally, it is rare that the first result returned by the software will be the optimal solution. There are several important reasons for this. First, as mentioned in the introduction, in 13C-MFA a highly nonlinear multi-dimensional parameter estimation problem is solved40. Problems of this kind have many suboptimal local solutions, and there is no guarantee that the first solution returned by the software will be the global optimal solution. To address this concern, 13C-MFA is typically restarted many times with random initial values for all fluxes and the goodness-of-fit of these iterations is compared. The goodness-of-fit is expressed by the sum of squared weighted residuals, or the SSR value40 (Fig. 2). The lower the SSR value, the better the agreement between the measured data and the model fit. Assuming that the metabolic model is correct and data are without gross measurement errors, the minimized SSR is a stochastic variable with a χ2-distribution. Based on this property, it is possible to calculate a maximum statistically acceptable value for SSR, which is roughly equal to the number of fitted measurements (n) minus the number of estimated independent parameters (p). More technically, the acceptable range of SSR values is between χ2α/2(n − p) and χ21−α/2(n − p), where α is a certain chosen threshold value, for example, 0.05 for the 95% confidence interval.
The strategy for performing 13C-MFA is thus to restart flux estimation many times (typically at least 10 times, but more is preferred) and compare the SSR values. The solution with the lowest SSR value is then selected as the optimal solution. Often, multiple iterations will produce the same low SSR value, which increases the likelihood that the solution is indeed the global optimal solution. In practice, however, it is not uncommon that the lowest SSR value obtained in this way is still greater than the maximum statistically allowed SSR. Some common reasons for this are as follows:
Errors in the metabolic model. Mistakes in the user-specified metabolic model such as incorrect reaction stoichiometries or errors in atom transitions are generally easy to identify and correct.
Incomplete metabolic model. Omitting important reactions or pathways from the model will result in poor fits. Thus, depending on the quality of fit, the scope of the model may need to be adjusted. In some cases, it may be necessary to include hypothetical reactions in the model in order to achieve an acceptable fit. In this way, 13C-MFA can be used as a hypothesis generating tool that can eventually lead to the discovery of novel metabolic pathways or reactions67–72. As an example, the TKTL1 pathway was recently discovered in Chinese hamster ovary cells by this approach73.
Gross measurement errors. It is not uncommon that certain labeling data will contain gross measurement errors, for example, due to co-elution of metabolites in GC/MS and LC/MS analyses. Careful inspection of ion chromatograms can in most cases help to identify co-elution problems. In such cases, labeling data for the contaminated metabolite fragments should be excluded from flux analysis.
Incorrect assumptions about measurement errors. The SSR value is calculated by summing up the weighted squared differences between the measured and simulated values. The weighting factors are inverses of measurement standard deviations squared. The assumed measurement errors thus greatly influence the calculated SSR value. Typical measurement errors used in 13C-MFA studies are as follows: 0.004 (or 0.4 mol%) for GC/MS data; 0.01 (or 1 mol%) for LC/MS data; and 5–10% relative error for external rates. In cases when very high or very low SSR values are obtained, it may be necessary to reevaluate the assumptions regarding measurement errors. Moreover, inspection of weighted residuals can inform if correct measurement errors have been assigned. Assuming measurement errors are random, the weighted residuals should follow a normal distribution N(µ = 0, σ2 = 1), which can be easily tested40.
Isotopomer spectral analysis
Isotopomer spectral analysis (ISA) is a related and widely used analysis approach for analyzing de novo fatty acid biosynthesis74 (Fig. 5). ISA calculations can be performed with most current software tools for 13C-MFA. Initially developed in early 1990s (before the 13C-MFA approach was fully formalized), the ISA approach is based on a relatively simple two-parameter model for analyzing mass isotopomer distributions of fatty acids from tracer experiments with fully 13C-labeled substrates, e.g., [U-13C]glucose. In the classical ISA formulation, two model parameters are determined: the D-value and the g(t)-value74. The D-value quantifies the fractional contribution of the fully 13C-labeled metabolite to lipogenic AcCoA, and the g(t)-value quantifies the fraction of fatty acids that were newly synthesized during the labeling time t.
Fig. 5. The isotopomer spectral analysis (ISA) approach is used to quantify de novo fatty acid biosynthesis based on tracer experiments with fully 13C-labeled substrates.

In the classical ISA formulation, two model parameters are determined, the D-value and the g(t)-value. The ISA approach can be generalized and extended to include additional model parameters such as fM2
Typically, several parallel labeling experiments are performed with different fully 13C-labeled substrates, e.g., [U-13C]glucose and [U-13C]glutamine, and isotopic labeling is measured for multiple fatty acids is each experiment, e.g., C16:0, C16:1, C18:0, and C18:1, using GC/MS. In theory, for a given tracer the D-values should be identical for all fatty acids, since all fatty acids are derived from the same cytosolic AcCoA pool. In contrast, the g(t)-values may be different for each fatty acid since different fatty acids may be synthesized at different rates. However, g(t)-values for a particular fatty acid determined with different tracers, e.g., with [U-13C]glucose and [U-13C]glutamine, should be the same since the synthesis rate of a particular fatty acid should not depend on which substrate is labeled. The ISA approach can be generalized for analysis of odd-chain fatty acids, e.g., C15:0 and C17:0, as was recently demonstrated62. Moreover, ISA can be extended to include additional model parameters62 (Fig. 5). In the classical ISA model, it is assumed that fully labeled substrates, e.g., [U-13C]glutamine, will produce only fully labeled AcCoA (i.e., M + 2-labeled). However, this assumption may not always be valid. For example, metabolism of [U-13C]glutamine in the TCA cycle can result in some loss of 13C, which will produce a mixture of M + 1- and M + 2-labeled AcCoA. Moreover, catabolism of certain substrates such as [U-13C]leucine will always produce a mixture of M + 1- and M + 2-labeled AcCoA due to carbon exchange with unlabeled CO275. For example, for the case of [U-13C]leucine, 33% of AcCoA will be M + 1-labeled and 67% of AcCoA will be M + 2-labeled62. By including an additional fM2 parameter in the ISA model, losses of 13C atoms can be captured, which produces more accurate estimates of D- and g(t)-values.
As indicated above, ISA analysis is typically performed with different fully 13C-labeled substrates in parallel experiments. These studies provide important insights into the relative contributions of different nutrients for de novo lipogenesis13,76. The estimated g(t)-values are also informative, since they can be used to calculate absolute de novo biosynthesis rates of fatty acids (nmol/106 cells/h):
| 6 |
Here FA is the macromolecular content of a particular fatty acid in cancer cells (in units nmol/106 cells; a typical value for palmitate is 40 nmol/106 cells), and Δt (h) is the length of the tracer experiment. The fatty acid content of cancer cells is easily determined with GC-flame ionization detector, or using the protocols described by Long and Antoniewicz65.
Quantifying fluxes in upper metabolism
In the next two sections, we describe briefly common stable-isotope tracing strategies for determining fluxes in upper and lower parts of central carbon metabolism, respectively. When performing flux analysis in upper metabolism, the drain of metabolic precursors toward biomass synthesis such as glucose 6-phosphate (G6P) for carbohydrates, R5P for nucleotides, and glycerol 3-phosphate for lipids can be generally ignored, since the glucose uptake rate (~100–400 nmol/106 cells/h) is typically two orders of magnitude greater than the drain of anabolic precursors for cell growth (~2–3 nmol/106 cells/h; Fig. 4). However, when performing flux analysis in lower metabolism, the drain of AcCoA for lipogenesis (~28 nmol/106 cells/h) cannot be ignored since this flux is comparable in magnitude to other fluxes in lower metabolism.
At present, [1,2-13C]glucose is one of the most widely used tracers to quantify fluxes of glycolysis and PPP (Fig. 6a). With this tracer the two pathways produce distinctly different labeling patterns in downstream metabolites such as 3PG, which can be easily measured with GC/MS and LC/MS. Metabolism of glucose via glycolysis produces 3PG that is 50% M + 2-labeled and 50% unlabeled (i.e., M + 0), while metabolism of glucose via oxidative PPP (oxPPP) produces a mixture of M + 0-, M + 1-, and M + 2-labeled 3PG. For a single pass through oxPPP, the labeling of 3PG is 60% M + 0, 20% M + 1, and 20% M + 2. The ratio of M + 1/M + 2 mass isotopomers of 3PG thus roughly approximates the relative contribution of oxPPP to glucose metabolism. However, this approximation should be used with caution. Specifically, the reversible G6P isomerase reaction, which interconverts G6P and F6P, can reroute a significant fraction of F6P that is produced via PPP back to G6P to be metabolized via oxPPP a second time (and possibly a third time), which results in additional losses of 13C (Fig. 6a). Thus, depending on the equilibration of F6P and G6P, the M + 1 and M + 2 mass isotopomers of 3PG can be significantly <20% and the ratio M + 1/M + 2 may be different from unity. Thus, to obtain a reliable estimate of oxPPP flux, the 3PG labeling data should be analyzed formally with 13C-MFA.
Fig. 6. Two alternative 13C-glucose-tracing strategies for analysis of metabolic fluxes in upper metabolism based on mass isotopomer measurements of 3-phosphoglycerate (3PG).

a The [1,2-13C]glucose tracer allows good resolution of relative glycolysis and pentose phosphate pathway fluxes. b A mixture of 50% [2-13C]glucose and 50% [4,5,6-13C]glucose is an improved tracer approach that also allows precise quantification of the transketolase-like 1 (TKTL1) pathway flux
Recently, a third metabolic pathway was discovered in cancer cells by which glucose can be metabolized, the TKTL1 pathway, which converts X5P (an intermediate of PPP) to glyceraldehyde 3-phosphate and a two-carbon metabolite, likely acetate, which can be further metabolized to cytosolic AcCoA20,21 (Fig. 6). Unfortunately, [1,2-13C]glucose and several other commonly used glucose tracers cannot provide a reliable estimate of the TKTL1 flux. To address this limitation, alternative glucose-tracing strategies have been developed to better resolve the three glucose metabolism pathways, glycolysis, PPP, and TKTL173. One of the best tracer strategies was based on mixtures of 50% [4,5,6-13C]glucose and 50% of either [1-13C]glucose, [2-13C]glucose, or [3-13C]glucose (Fig. 6b). With these tracers, it is possible to determine precise fluxes of all three metabolic pathways, as recently demonstrated in Chinese hamster ovary cells73. Other optimal glucose tracers have also been proposed for analysis of specific metabolic pathways; for example, [3,4-13C]glucose was determined to be a particularly good tracer for quantifying the anaplerotic flux of glucose into the TCA cycle57,77,78.
Quantifying fluxes in lower metabolism
For analysis of fluxes in lower part of central carbon metabolism, i.e., downstream of pyruvate, fully labeled [U-13C]glutamine is often used. Glutamine is a the second most highly consumed carbon substrate by many cancer cells (after glucose)79; as a result, [U-13C]glutamine produces high labeling in metabolites, especially in TCA cycle intermediates, and rich labeling patterns for flux estimation using 13C-MFA (Fig. 7). Another advantage of using 13C-glutamine as a tracer is that labeling dynamics of 13C-glutamine are not affected by the buffering effect of extracellular lactate. Since 13C-glutamine labels mainly metabolites downstream of pyruvate, isotopic steady state is reached for the labeled TCA cycle metabolites within a few hours after [U-13C]glutamine addition, even when external lactate concentration is high56.
Fig. 7. [U-13C]Glutamine tracer experiments produce rich labeling patterns in TCA cycle metabolites that allow precise quantification of metabolic fluxes in lower part of central metabolism, i.e., downstream of pyruvate, using 13C-MFA.
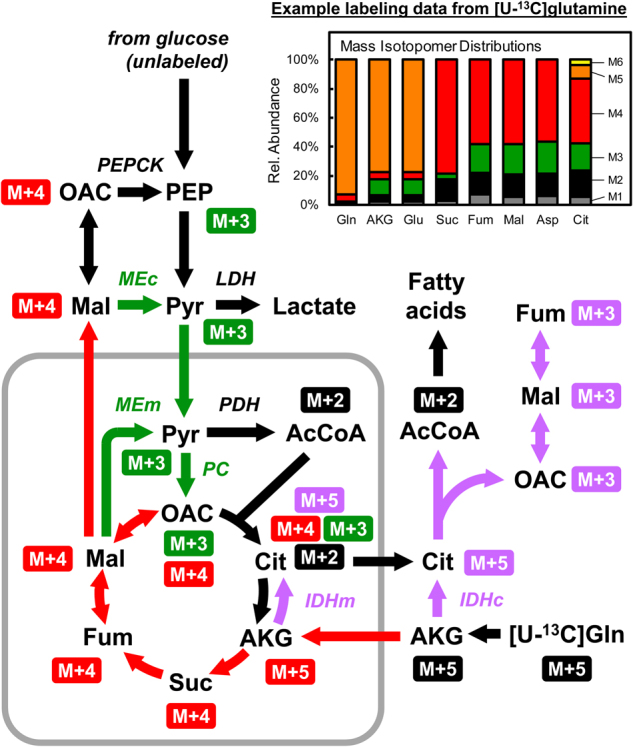
The diagram shows schematically the flow of 13C-labeling from [U-13C]glutamine into relevant metabolic pathways in cancer cells. The insert shows an example of labeling data obtained from a [U-13C]glutamine tracer experiment. Colors of arrows indicate different metabolic pathways: reductive carboxylation of glutamine (purple); glutaminolysis (red); conversion of malate to oxaloacetate via malic enzyme and pyruvate carboxylase (green)
In the past decade, [U-13C]glutamine tracing has played an important role in elucidating the contribution of glutamine to lipogenesis via reductive carboxylation pathway13,42, i.e., via the conversion of glutamine to AKG, then to citrate (i.e., in the reverse direction of TCA cycle, catalyzed by isocitrate dehydrogenases), and finally to AcCoA after cleavage by ATP citrate lyase. To highlight additional flux information that can be obtained from [U-13C]glutamine tracer experiments, Fig. 7 shows schematically the flow isotopic labeling from [U-13C]glutamine into relevant metabolic pathways. The insert in Fig. 7 shows an example of labeling data set obtained from a [U-13C]glutamine tracer experiment. Metabolism of [U-13C]glutamine via reductive carboxylation (purple arrows in Fig. 6) results in the production of M + 5-labeled citrate42; after cleavage of citrate by ATP citrate lyase, M + 2-labeled AcCoA and M + 3-labeled oxaloacetate are produced (while labeling of oxaloacetate cannot be measured directly, it can be inferred from the labeling of aspartate). In contrast, metabolism of [U-13C]glutamine via the glutaminolysis pathway along the normal oxidative direction of the TCA cycle (red arrows in Fig. 6) results in the production of M + 4-labeled succinate, fumarate, malate, and oxaloacetate. M + 4 malate can also produce M + 3-labeled oxaloacetate, after conversion to pyruvate via malic enzyme, followed by carboxylation of pyruvate to oxaloacetate by pyruvate carboxylase (green arrows in Fig. 6). Taken together, [U-13C]glutamine tracer experiments produce rich labeling patterns in TCA cycle metabolites that permit precise quantification of metabolic fluxes in these pathways using 13C-MFA. In addition to [U-13C]glutamine, [5-13C]glutamine and [1-13C]glutamine have also been used for 13C-MFA13,36,77. However, in general, these singly labeled glutamine tracers are not as informative as [U-13C]glutamine for comprehensive analysis of cellular metabolism.
Concluding remarks
The isotopic tracing strategies and 13C-MFA methods reviewed here present powerful tools for elucidating metabolic flux rewiring in cancer cells. Technically, other stable isotopes such as 2H, 18O, and 15N can also be used to study metabolic phenotypes, and for certain applications these alternative isotope tracers may be preferred80,81. From a modeling perspective, the application of multiple isotopes will not cause any problems for MFA. In fact, one of the motivations for developing the EMU framework was to permit and encourage the application of multiple isotopes for flux analysis39. Several pioneering studies have already made use of this45,82. However, there are several drawbacks and limitations that should be considered when contemplating the use of alternative stable isotopes. For example, 18O tracers are generally much more expensive than 13C tracers and at present the number commercially available 18O tracers is limited. While 15N can be used to investigate metabolic pathways where the metabolic intermediates contain N atoms, such as amino-acid pathways, they cannot be used to study central carbon metabolism. Finally, interpretation of 2H labeling data is complicated by the presence of significant deuterium kinetic isotope effects. In contrast to 13C tracers, where it has been demonstrated that the kinetic isotope effects are negligible83, the kinetic isotope effects for 2H are substantial84. Thus, determining fluxes from 2H labeling data is strongly influenced by specific assumptions made regarding the magnitude of kinetic isotope effects for various enzymatic reactions. Still, 2H tracers can be valuable in resolving specific aspects of metabolism such as NADPH metabolism in different cellular compartments, which cannot be elucidated with 13C tracers85,86.
Currently, one of the biggest challenges for 13C-MFA in mammalian cells is to resolve compartment-specific fluxes87. While certain compartment-specific metabolic fluxes can be determined precisely with 13C-MFA, e.g., mitochondrial vs cytosolic malic enzyme fluxes, other fluxes are much more difficult to resolve, e.g., mitochondrial vs cytosolic isocitrate dehydrogenase fluxes. In theory, resolving compartment-specific fluxes would be easier if compartment-specific labeling data could be collected88. However, with current protocols for quenching metabolism and extracting intracellular labeling, all intracellular metabolite pools are sampled. As a result, the measured labeling data must be modeled as mixtures from multiple cellular pools36,61,89. To resolve compartmentalized metabolism, alternative approaches such as organelle isolation may be valuable in the future90–92.
When interpreting 13C-MFA results, it is also important to keep in mind that the accuracy of 13C-MFA calculations depends strongly on the validity of several modeling assumptions that collectively form the basis for the underlying isotopomer models. These inherent assumptions include the following: (1) metabolic steady-state assumption—it is assumed that metabolic fluxes are constant during the labeling experiment; (2) isotopic steady-state assumption—it is assumed that isotopic labeling does not change in time; (3) no kinetic isotope effect for 13C tracers—it is assumed that enzymes cannot discriminate between unlabeled (12C) and labeled (13C) atoms83,93; (4) no metabolite channeling—it is assumed that substrate tunneling via multi-enzyme complexes can be ignored; (5) homogeneous metabolite pools—it is assumed that metabolites within a particular compartment are perfectly mixed; (6) homogeneous cell population—it is assumed that all cells in a culture have the same metabolic phenotype; and (7) no turnover of macromolecules—it is assumed that cellular macromolecules such as proteins, lipids, RNA, and DNA are not broken down and produced at the same time. If one or more of these assumptions are shown to be incorrect for a given biological system, then the 13C-MFA methodology must be adjusted to account for these effects. For example, the isotopic 13C-NMFA was developed for analysis of systems where labeling data are not constant in time41,94, and dynamic MFA methodologies (DMFA and 13C-DMFA) were developed for analysis of systems where fluxes are not constant in time46,95–97. More recently, the co-culture 13C-MFA methodology was developed for analysis of non-homogeneous cell cultures89. Turnover of macromolecules such as glycogen, lipids, and RNA has also been observed in many biological systems98–100, and these effects can be captured in 13C-MFA by adding appropriate dilution fluxes99.
Lastly, we want to emphasize the importance of full transparency in reporting 13C-MFA results by providing full access to data, models, methods, results, and statistics. As described in this review, 13C-MFA results are highly dependent on assumptions and models used for data analysis. As cancer research progresses and new insights are obtained into the unique metabolic features of cancer cells, we may discover additional reactions or pathways that have not been considered before. Reanalyzing past data using updated metabolic models could provide a powerful approach for testing new hypotheses. A recent review paper has proposed minimum data standards to facilitate dissemination of methods, data, and results from 13C-MFA studies44.
Acknowledgements
This work was supported by NSF MCB-1616332 grant.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol. Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong W, Keibler MA, Stephanopoulos G. Review of metabolic pathways activated in cancer cells as determined through isotopic labeling and network analysis. Metab. Eng. 2017;43:113–124. doi: 10.1016/j.ymben.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badur MG, Metallo CM. Reverse engineering the cancer metabolic network using flux analysis to understand drivers of human disease. Metab. Eng. 2017;45:95–108. doi: 10.1016/j.ymben.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metallo CM, Deberardinis RJ. Engineering approaches to study cancer metabolism. Metab. Eng. 2017;43:93. doi: 10.1016/j.ymben.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Hiller K, Metallo CM. Profiling metabolic networks to study cancer metabolism. Curr. Opin. Biotechnol. 2013;24:60–68. doi: 10.1016/j.copbio.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between metabolism and cancer biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 10.Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vander Heiden MG, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeBerardinis RJ. Serine metabolism: some tumors take the road less traveled. Cell Metab. 2011;14:285–286. doi: 10.1016/j.cmet.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacold ME, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016;12:452–458. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 20.Coy JF, Dressler D, Wilde J, Schubert P. Mutations in the transketolase-like gene TKTL1: clinical implications for neurodegenerative diseases, diabetes and cancer. Clin. Lab. 2005;51:257–273. [PubMed] [Google Scholar]
- 21.Diaz-Moralli S, et al. A key role for transketolase-like 1 in tumor metabolic reprogramming. Oncotarget. 2016;7:51875–51897. doi: 10.18632/oncotarget.10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schug ZT, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mashimo T, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comerford SA, et al. Acetate dependence of tumors. Cell. 2014;159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014;2:23. doi: 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson SM, et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 2016;23:517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Achreja A, et al. Exo-MFA—a 13C metabolic flux analysis framework to dissect tumor microenvironment-secreted exosome contributions towards cancer cell metabolism. Metab. Eng. 2017;43(Pt B):156–172. doi: 10.1016/j.ymben.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hensley CT, et al. Metabolic heterogeneity in human lung tumors. Cell. 2016;164:681–694. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nilsson A, Nielsen J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017;43:103–112. doi: 10.1016/j.ymben.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Thiele I, et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swarup A, Lu J, DeWoody KC, Antoniewicz MR. Metabolic network reconstruction, growth characterization and 13C-metabolic flux analysis of the extremophile Thermus thermophilus HB8. Metab. Eng. 2014;24:173–180. doi: 10.1016/j.ymben.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Long CP, Antoniewicz MR. Metabolic flux analysis of Escherichia coli knockouts: lessons from the Keio collection and future outlook. Curr. Opin. Biotechnol. 2014;28:127–133. doi: 10.1016/j.copbio.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haverkorn van Rijsewijk BR, Nanchen A, Nallet S, Kleijn RJ, Sauer U. Large-scale 13C-flux analysis reveals distinct transcriptional control of respiratory and fermentative metabolism in Escherichia coli. Mol. Syst. Biol. 2011;7:477. doi: 10.1038/msb.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Young JD. Metabolic flux rewiring in mammalian cell cultures. Curr. Opin. Biotechnol. 2013;24:1108–1115. doi: 10.1016/j.copbio.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang L, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–258. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Templeton N, et al. Application of (13)C flux analysis to identify high-productivity CHO metabolic phenotypes. Metab. Eng. 2017;43(Pt B):218–225. doi: 10.1016/j.ymben.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Maier K, et al. Quantification of statin effects on hepatic cholesterol synthesis by transient (13)C-flux analysis. Metab. Eng. 2009;11:292–309. doi: 10.1016/j.ymben.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng. 2007;9:68–86. doi: 10.1016/j.ymben.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab. Eng. 2006;8:324–337. doi: 10.1016/j.ymben.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Young JD, Walther JL, Antoniewicz MR, Yoo H, Stephanopoulos G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008;99:686–699. doi: 10.1002/bit.21632. [DOI] [PubMed] [Google Scholar]
- 42.Yoo H, Antoniewicz MR, Stephanopoulos G, Kelleher JK. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem. 2008;283:20621–20627. doi: 10.1074/jbc.M706494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young JD. INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics. 2014;30:1333–1335. doi: 10.1093/bioinformatics/btu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crown SB, Antoniewicz MR. Publishing 13C metabolic flux analysis studies: a review and future perspectives. Metab. Eng. 2013;20:42–48. doi: 10.1016/j.ymben.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crown SB, Antoniewicz MR. Parallel labeling experiments and metabolic flux analysis: Past, present and future methodologies. Metab. Eng. 2013;16:21–32. doi: 10.1016/j.ymben.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antoniewicz MR. Methods and advances in metabolic flux analysis: a mini-review. J. Ind. Microbiol. Biotechnol. 2015;42:317–325. doi: 10.1007/s10295-015-1585-x. [DOI] [PubMed] [Google Scholar]
- 47.Ahn WS, Antoniewicz MR. Towards dynamic metabolic flux analysis in CHO cell cultures. Biotechnol. J. 2012;7:61–74. doi: 10.1002/biot.201100052. [DOI] [PubMed] [Google Scholar]
- 48.Antoniewicz MR, Stephanopoulos G, Kelleher JK. Evaluation of regression models in metabolic physiology: predicting fluxes from isotopic data without knowledge of the pathway. Metabolomics. 2006;2:41–52. doi: 10.1007/s11306-006-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crown SB, Long CP, Antoniewicz MR. Optimal tracers for parallel labeling experiments and 13C metabolic flux analysis: a new precision and synergy scoring system. Metab. Eng. 2016;38:10–18. doi: 10.1016/j.ymben.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antoniewicz MR. 13C metabolic flux analysis: optimal design of isotopic labeling experiments. Curr. Opin. Biotechnol. 2013;24:1116–1121. doi: 10.1016/j.copbio.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Buescher JM, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt K, Carlsen M, Nielsen J, Villadsen J. Modeling isotopomer distributions in biochemical networks using isotopomer mapping matrices. Biotechnol. Bioeng. 1997;55:831–840. doi: 10.1002/(SICI)1097-0290(19970920)55:6<831::AID-BIT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 53.Zupke C, Stephanopoulos G. Intracellular flux analysis in hybridomas using mass balances and in vitro (13)C nmr. Biotechnol. Bioeng. 1995;45:292–303. doi: 10.1002/bit.260450403. [DOI] [PubMed] [Google Scholar]
- 54.Wiechert W, Mollney M, Isermann N, Wurzel M, de Graaf AA. Bidirectional reaction steps in metabolic networks: III. Explicit solution and analysis of isotopomer labeling systems. Biotechnol. Bioeng. 1999;66:69–85. doi: 10.1002/(SICI)1097-0290(1999)66:2<69::AID-BIT1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 55.Ahn WS, Antoniewicz MR. Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab. Eng. 2011;13:598–609. doi: 10.1016/j.ymben.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 56.Ahn WS, Antoniewicz MR. Parallel labeling experiments with [1,2-(13)C]glucose and [U-(13)C]glutamine provide new insights into CHO cell metabolism. Metab. Eng. 2013;15:34–47. doi: 10.1016/j.ymben.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Crown SB, Ahn WS, Antoniewicz MR. Rational design of (1)(3)C-labeling experiments for metabolic flux analysis in mammalian cells. BMC Syst. Biol. 2012;6:43. doi: 10.1186/1752-0509-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Metallo CM, Walther JL, Stephanopoulos G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J. Biotechnol. 2009;144:167–174. doi: 10.1016/j.jbiotec.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Antoniewicz MR. Parallel labeling experiments for pathway elucidation and 13C metabolic flux analysis. Curr. Opin. Biotechnol. 2015;36:91–97. doi: 10.1016/j.copbio.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 60.Crown SB, Kelleher JK, Rouf R, Muoio DM, Antoniewicz MR. Comprehensive metabolic modeling of multiple 13C-isotopomer data sets to study metabolism in perfused working hearts. Am. J. Physiol. Heart Circ. Physiol. 2016;311:H881–H891. doi: 10.1152/ajpheart.00428.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeWaal D, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018;9:446. doi: 10.1038/s41467-017-02733-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crown SB, Marze N, Antoniewicz MR. Catabolism of branched chain amino acids contributes significantly to synthesis of odd-chain and even-chain fatty acids in 3T3-L1 adipocytes. PLoS ONE. 2015;10:e0145850. doi: 10.1371/journal.pone.0145850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crown SB, Long CP, Antoniewicz MR. Integrated 13C-metabolic flux analysis of 14 parallel labeling experiments in Escherichia coli. Metab. Eng. 2015;28:151–158. doi: 10.1016/j.ymben.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Long CP, Gonzalez JE, Sandoval NR, Antoniewicz MR. Characterization of physiological responses to 22 gene knockouts in Escherichia coli central carbon metabolism. Metab. Eng. 2016;37:102–113. doi: 10.1016/j.ymben.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Long CP, Antoniewicz MR. Quantifying biomass composition by gas chromatography/mass spectrometry. Anal. Chem. 2014;86:9423–9427. doi: 10.1021/ac502734e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McConnell BO, Antoniewicz MR. Measuring the composition and stable-isotope labeling of algal biomass carbohydrates via gas chromatography/mass spectrometry. Anal. Chem. 2016;88:4624–4628. doi: 10.1021/acs.analchem.6b00779. [DOI] [PubMed] [Google Scholar]
- 67.Long CP, Au J, Sandoval NR, Gebreselassie NA, Antoniewicz MR. Enzyme I facilitates reverse flux from pyruvate to phosphoenolpyruvate in Escherichia coli. Nat. Commun. 2017;8:14316. doi: 10.1038/ncomms14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakahigashi K, et al. Systematic phenome analysis of Escherichia coli multiple-knockout mutants reveals hidden reactions in central carbon metabolism. Mol. Syst. Biol. 2009;5:306. doi: 10.1038/msb.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cordova LT, Cipolla RM, Swarup A, Long CP, Antoniewicz MR. 13)C metabolic flux analysis of three divergent extremely thermophilic bacteria: Geobacillus sp. LC300, Thermus thermophilus HB8, and Rhodothermus marinus DSM 4252. Metab. Eng. 2017;44:182–190. doi: 10.1016/j.ymben.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clasquin MF, et al. Riboneogenesis in yeast. Cell. 2011;145:969–980. doi: 10.1016/j.cell.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crown SB, et al. Resolving the TCA cycle and pentose-phosphate pathway of Clostridium acetobutylicum ATCC 824: isotopomer analysis, in vitro activities and expression analysis. Biotechnol. J. 2011;6:300–305. doi: 10.1002/biot.201000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng X, et al. Characterization of the central metabolic pathways in Thermoanaerobacter sp. strain X514 via isotopomer-assisted metabolite analysis. Appl. Environ. Microbiol. 2009;75:5001–5008. doi: 10.1128/AEM.00715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahn WS, Crown SB, Antoniewicz MR. Evidence for transketolase-like TKTL1 flux in CHO cells based on parallel labeling experiments and (13)C-metabolic flux analysis. Metab. Eng. 2016;37:72–78. doi: 10.1016/j.ymben.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kharroubi AT, Masterson TM, Aldaghlas TA, Kennedy KA, Kelleher JK. Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. Am. J. Physiol. 1992;263(4 Pt 1):E667–E675. doi: 10.1152/ajpendo.1992.263.4.E667. [DOI] [PubMed] [Google Scholar]
- 75.Green CR, et al. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016;12:15–21. doi: 10.1038/nchembio.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fendt SM, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang L, et al. Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab. Eng. 2017;43(Pt B):198–207. doi: 10.1016/j.ymben.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Crown SB, Antoniewicz MR. Selection of tracers for 13C-metabolic flux analysis using elementary metabolite units (EMU) basis vector methodology. Metab. Eng. 2012;14:150–161. doi: 10.1016/j.ymben.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeBerardinis RJ, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl Acad. Sci. USA. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Previs SF, et al. New methodologies for studying lipid synthesis and turnover: looking backwards to enable moving forwards. Biochim. Biophys. Acta. 2014;1842:402–413. doi: 10.1016/j.bbadis.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 81.Landau BR, et al. Contributions of gluconeogenesis to glucose production in the fasted state. J. Clin. Invest. 1996;98:378–385. doi: 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hasenour CM, et al. Mass spectrometry-based microassay of (2)H and (13)C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am. J. Physiol. Endocrinol. Metab. 2015;309:E191–E203. doi: 10.1152/ajpendo.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sandberg TE, et al. Evolution of E. coli on [U-13C]glucose reveals a negligible isotopic influence on metabolism and physiology. PLoS ONE. 2016;11:e0151130. doi: 10.1371/journal.pone.0151130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu L, et al. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat. Chem. Biol. 2016;12:345–352. doi: 10.1038/nchembio.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis CA, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fan J, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wahrheit J, Nicolae A, Heinzle E. Eukaryotic metabolism: measuring compartment fluxes. Biotechnol. J. 2011;6:1071–1085. doi: 10.1002/biot.201100032. [DOI] [PubMed] [Google Scholar]
- 88.Niklas J, Schneider K, Heinzle E. Metabolic flux analysis in eukaryotes. Curr. Opin. Biotechnol. 2010;21:63–69. doi: 10.1016/j.copbio.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 89.Gebreselassie NA, Antoniewicz MR. 13)C-metabolic flux analysis of co-cultures: a novel approach. Metab. Eng. 2015;31:132–139. doi: 10.1016/j.ymben.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen WW, Freinkman E, Wang T, Birsoy K, Sabatini DM. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell. 2016;166:1324–1337 e11. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nicolae A, Wahrheit J, Nonnenmacher Y, Weyler C, Heinzle E. Identification of active elementary flux modes in mitochondria using selectively permeabilized CHO cells. Metab. Eng. 2015;32:95–105. doi: 10.1016/j.ymben.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 92.Nonnenmacher Y, et al. Analysis of mitochondrial metabolism in situ: combining stable isotope labeling with selective permeabilization. Metab. Eng. 2017;43:147–155. doi: 10.1016/j.ymben.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 93.Wasylenko TM, Stephanopoulos G. Kinetic isotope effects significantly influence intracellular metabolite (13) C labeling patterns and flux determination. Biotechnol. J. 2013;8:1080–1089. doi: 10.1002/biot.201200276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wiechert W, Noh K. From stationary to instationary metabolic flux analysis. Adv. Biochem. Eng. Biotechnol. 2005;92:145–172. doi: 10.1007/b98921. [DOI] [PubMed] [Google Scholar]
- 95.Antoniewicz MR. Dynamic metabolic flux analysis--tools for probing transient states of metabolic networks. Curr. Opin. Biotechnol. 2013;24:973–978. doi: 10.1016/j.copbio.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 96.Leighty RW, Antoniewicz MR. Dynamic metabolic flux analysis (DMFA): a framework for determining fluxes at metabolic non-steady state. Metab. Eng. 2011;13:745–755. doi: 10.1016/j.ymben.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 97.Antoniewicz MR, et al. Metabolic flux analysis in a nonstationary system: fed-batch fermentation of a high yielding strain of E. coli producing 1,3-propanediol. Metab. Eng. 2007;9:277–292. doi: 10.1016/j.ymben.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Long CP, Au J, Gonzalez JE, Antoniewicz MR. 13C metabolic flux analysis of microbial and mammalian systems is enhanced with GC-MS measurements of glycogen and RNA labeling. Metab. Eng. 2016;38:65–72. doi: 10.1016/j.ymben.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gonzalez JE, Long CP, Antoniewicz MR. Comprehensive analysis of glucose and xylose metabolism in Escherichia coli under aerobic and anaerobic conditions by 13C metabolic flux analysis. Metab. Eng. 2017;39:9–18. doi: 10.1016/j.ymben.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guzman S, et al. 13)C metabolic flux analysis shows that resistin impairs the metabolic response to insulin in L6E9 myotubes. BMC Syst. Biol. 2014;8:109. doi: 10.1186/s12918-014-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]