

Abstract
The mechanism by which gastrin promotes pancreatic cancer cell metastasis is unclear. The process of directing polarized cancer cells toward the extracellular matrix is principally required for invasion and distant metastasis; however, whether gastrin can induce this process and its underlying mechanism remain to be elucidated. In this study, we found that gastrin-induced phosphorylation of paxillin at tyrosine 31/118 and RhoA activation as well as promoted the metastasis of PANC-1 cancer cells. Depletion of Gα12 and Gα13 inhibited the phosphorylation of paxillin and downstream activation of GTP-RhoA, blocked the formation and aggregation of focal adhesions and facilitated polarization of actin filaments induced by gastrin. Suppression of RhoA and ROCK also exhibited identical results. Selective inhibition of the CCKBR–Gα12/13–RhoA–ROCK signaling pathway blocked the reoriented localization of the Golgi apparatus at the leading edge of migrated cancer cells. YM022 and Y-27632 significantly suppressed hepatic metastasis of orthotic pancreatic tumors induced by gastrin in vivo. Collectively, we demonstrate that gastrin promotes Golgi reorientation and directional polarization of pancreatic cancer cells by activation of paxillin via the CCKBR–Gα12/13–RhoA–ROCK signal pathway.
Pancreatic cancer: Hormone gives cancer a helping hand
A hormone found in high levels in pancreatic cancer sufferers helps the disease spread by co-ordinating cellular migration. Pancreatic cancer is one of the most deadly forms of cancer, being highly aggressive and likely to metastasize. Honggang Yu at Renmin Hospital of Wuhan University and scientists across China have demonstrated that gastrin, a hormone expressed at higher levels in patients with pancreatic cancer, helps to co-ordinate directional cell migration and ensure the disease spreads effectively. By activating two key molecules via a specific signalling pathway, gastrin ensures the correct orientation of the Golgi apparatus, a cellular organelle tasked with packaging proteins for transportation. This in turn activates directional migration of the cancer cells. The results explain why gastrin is over-expressed in both tumors and blood in cancer patients, and may inform future therapies.
Introduction
Pancreatic cancer is one of the most common malignancies and is a leading cause of cancer-related death worldwide1. The incidence rate of pancreatic cancer continues to approximate the death rate, implying that most patients with pancreatic cancer die as a result of this cancer largely because it is highly aggressive and likely to metastasize2. A better understanding of the mechanisms underlying pancreatic cancer metastasis is essential for exploring novel strategies to enhance the current treatment efficacy and improve the prognosis of patients.
Directional cell migration is required for many important physiological processes, such as embryonic development, immune surveillance, and wound healing3,4. Additionally, directional cell migration plays a key role in pivotal steps that promote tumor metastasis, such as cellular migration and invasion into the surrounding stroma5,6. During directional cell migration, cancer cells acquire a highly polarized phenotype (with membrane protrusion and a retracting tail), form focal adhesions and reorient the Golgi apparatus to move proteins to specific intracellular locations7,8.
Cholecystokinin B receptor (CCKBR), a member of the family of G protein-coupled receptors (GPCR), couples with gastrin and cholecystokinin, which are principally expressed in the gastrointestinal tract9. CCKBR was first regarded as a regulator of gastric acid secretion and the calcium signaling pathway, and now CCKBR has been identified and characterized as a stimulator in multiple malignancies, including pancreatic cancer9-11. Compared with normal tissues, the expression level of CCKBR is significantly increased in pancreatic cancerous tissues12. The human pancreas produces gastrin during fetal development, and no gastrin is expressed in the healthy adult pancreas; however, gastrin is reexpressed in pancreatic cancerous tissues, where it enhances proliferation and migration through an autocrine mechanism11,13. However, the role of CCKBR in pancreatic cancer metastasis still remains to be clarified.
The Rho family of small GTPases, including RhoA, Rac1, Cdc42, and Rab43, exerts important functions in cancer progression by affecting multiple aspects, such as promoting cytoskeletal reorganization, intracellular trafficking, and Golgi orientation14,15. It has been reported that following gastrin binding, activated CCKBR undergoes a conformational change that exchanges GDP for GTP on the Gα subunits16,17. The GTP-bound Gα subunit then interacts with downstream signaling effectors, resulting in the activation of various second messenger molecules that are responsible for eliciting cellular responses16,17. On the other hand, paxillin is one of the most important proteins in focal adhesion formation and is essential for cellular adhesion, motility, and invasion18. In highly aggressive tumors, high levels of phosphorylated paxillin indicate a stronger ability to migrate and metastasize18. Previous work by others and ourselves showed that gastrin can induce rapid phosphorylation of paxillin19,20. Inspired by these findings, we hypothesize that gastrin/CCKBR may trigger the activation of RhoA and paxillin, induce directional cell migration, and in turn, promote metastasis of pancreatic cancer cells.
In this study, we showed that by co-ordinating paxillin activation and Golgi apparatus reorientation, gastrin plays a crucial role in the acquisition of a polarized phenotype and, accordingly, in directional cell migration of PANC-1 cells. Furthermore, during these events, activation of Gα12/13–RhoA–ROCK signaling is a pivotal mechanism. Thus, our findings elucidate a potential explanation for the tumor microenvironment in modulating the directional migration of pancreatic cancer cells at the molecular level.
Materials and methods
Antibodies and reagents
Antibodies were obtained from the following commercial sources: anti-paxillin monoclonal, anti-paxillin p-Tyr31 polyclonal, and anti-paxillin p-Tyr118 polyclonal (Invitrogen, CA, USA); anti-RhoA monoclonal, anti-FAK polyclonal, anti-β-actin polyclonal, and goat anti-mouse IgG, F(ab′)2-TRITC (Santa Cruz, CA, USA); goat anti-rabbit IgG (H + L), F(ab′)2 Fragment (Alexa Fluor® 555 Conjugate) antibody (Cell Signaling Technology, USA). Horseradish peroxidase-labeled antibodies were purchased from Thermo Pierce (Rockford, USA). Gastrin and Y-27632 were obtained from Sigma-Aldrich (St. Louis, USA). Rhosin was obtained from Calbiochem® (La Jolla, CA, Germany). Acti-stain™ 488 Fluorescent Phalloidin and Rho Activation Assay Biochem Kit were obtained from Cytoskeleton, Inc. (Japan). 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) was obtained from Roche Diagnostics (Japan).
Cell culture
The human pancreatic PANC-1 cancer cells, obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), were cultivated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, NY, USA) in a humidified incubator containing 5% CO2 at 37 °C.
Transfection experiments
The synthetic small interfering MISSION esiRNA targeting Gα12 and Gα13 subunits of the G protein-coupled receptor family was obtained from Sigma-Aldrich Biotechnology. In brief, PANC-1 cells were treated with 200 nM gastrin for 1 h after transfection with 50 ng/μl of targeting siRNA for 48 h. The depletion of specific proteins was analyzed by western blotting.
Protein extraction and western blotting
At the designated time points, cell protein extraction and protein concentration quantification were performed following the manufacturer’s instructions. In brief, equal amounts of protein were separated on polyacrylamide gels and transferred to nitrocellulose membranes. The blots were incubated with 1 μg of primary antibodies at 4 °C overnight. After incubation with horseradish peroxidase-linked secondary antibodies, the signals of target proteins were visualized by an ECL system (Millipore, CA).
Transwell migration and invasion assays
Cell migration and invasion were determined by Transwell® Inserts pre-coated with Matrigel (Corning, NY, USA). The tumor cells were incubated for 24 h and then the medium and cells remaining in the top chambers were removed. After fixing with methanol and staining with Hoechst 33342, the cells that penetrated the lower-membrane surface were counted by an inverted microscope. To assess invasion ability, filters were pre-coated with diluted Matrigel while the filters in the migration experiments were not pre-coated with Matrigel.
Endogenous RhoA activity assay
Active RhoA (GTP-bound) was captured using the Rho Activation Assay Biochem Kit (Cytoskeleton, Inc., Japan). PANC-1 cells that were grown in logarithmic phase in a 10-cm plate were starved in serum-free medium containing Y-27632/Rhosin for 24 h and were subsequently stimulated with gastrin. Cells were lysed in a buffer containing 100 mM NaCl, 20 mM Tris-HCL, 1% Triton X-100, 2 mM NaF, and protease inhibitors (Roche Diagnostics). Cell extracts were homogenized and incubated overnight with GST-rhotekin-RBD fusion protein (Cytoskeleton, Japan). Active GTP-RhoA in the lysates was evaluated by immunoblotting with a monoclonal RhoA antibody.
Immunofluorescence assay
For immunofluorescent staining, PANC-1 cells were seeded onto coverslips at 1 × 105 cells/ml and were maintained to adhere to the slides. Cells were pre-treated with gastrin or DMSO for the indicated time, were fixed in preheated 3.7% paraformaldehyde (PFA) and permeabilized with 0.4% Triton X-100 for 5 min before blocking with goat serum in PBS. Afterward, cells were incubated with primary antibodies at 4 °C overnight and were visualized by fluorescent-labeled secondary antibodies as indicated. Finally, cells were embedded with 1 μg/ml of DAPI, and fluorescent images were acquired using an upright Olympus fluorescence microscope.
Assay for Golgi orientation
PANC-1 cells were treated with siRNA before seeding on sterile coverslips and were left to grow to confluence before wounding. Monolayers were wounded by scratching with 20–200 μl pipette tips and were washed three times with PBS. The wounded monolayer was treated with gastrin for the indicated time. For Golgi apparatus reorientation assays, the first row of cells facing wounding was determined. The Golgi apparatus, nucleus, and F-actin were labeled by β-cop (red), DAPI (blue), and Acti-stain™ 488 Phalloidin reagent (green), respectively. Golgi orientation was evaluated for the first row of cells, and analysis was performed using Cell Scan software. As indicated in the schematic plot (Supplementary Figure 1), the dotted arrow denotes the vertical direction toward the scratched front, and the solid black arrow indicates the migrating direction of the cells. The vast majority of the Golgi apparatus (>70%) within a 60° angle facing the scratched line was considered as the positive orientation, while less than 70% Golgi apparatus in a 60° angle or dispersed in cytoplasm was considered as the negative orientation. To calculate angles between the Golgi migrated direction (>70% Golgi apparatus migrating direction) and the 0° vertical scratched line clockwise, 5° is classified as one unit. For quantitative analysis, at least 100 cells and Golgi stacks were assessed per experiment, and data analysis was performed using Origin 9.1 software.
Animal experiments and orthotopic implantation of pancreatic cancer cells
All in vivo studies were approved by the Committee on the Use of Live Animals in Teaching and Research at Wuhan University. PANC-1 cells were harvested and resuspended into a single-cell suspension with PBS containing 0.1% Matrigel (BD Biosciences). Then, suspensions containing 1 × 106 cells/50 μl were injected into the head or neck region of the pancreatic parenchyma of BALB/c nude mice (6 weeks, female). All mice were killed on day 42, and the pancreatic tumors in situ were dissected and weighed. Isolated tumors were cut in half and one part was lysed for western blotting while the other part was formalin-fixed for immunohistochemical assay. Livers of the mice were also prepared for H&E staining to determine the level of metastasis.
Statistical analysis
For all cell experiments, the mean values ± SD of at least triplicate experiments were calculated. Statistical significance between values of different experimental conditions was analyzed by Student’s t test. The value of P < 0.05 was considered to indicate statistical significance.
Results
Gastrin phosphorylates and activates paxillin in a concentration- and time-dependent manner in pancreatic cancer cells
The phosphorylation of Tyr31 and Tyr118 sites is crucial for the activation of paxillin18. In this study, we observed that gastrin can phosphorylate Tyr31 and Tyr118 of paxillin in the PANC-1 pancreatic cancer cell line in a concentration-dependent manner. As shown in Fig. 1a, at a low concentration (10−8 M), gastrin can significantly increase the phosphorylation of both Tyr31 and Tyr118. As the concentration of gastrin increased, the level of phosphorylation was increased accordingly and reached a peak level at 10−7 M and 10−6 M for Tyr31 and Tyr118, respectively. No significant change in the total level of paxillin was observed.
Fig. 1. Gastrin regulates paxillin activation and migration of pancreatic cancer cells.

a Gastrin dose dependently induces elevated tyrosine phosphorylation of paxillin in PANC-1 cells. b Gastrin time dependently induces elevated tyrosine phosphorylation of paxillin in PANC-1 cells. c 200 nM gastrin treatment for 60 min can activate paxillin in HGC cells. d 200 nM gastrin treatment for 60 min can activate paxillin in HT-29 cells. e, f Gastrin (200 nM) promotes the invasion (e) and migration (f) of PANC-1 cells, as indicated by Boyden chamber assays. g The wound healing assay indicates the elevated migration of PANC-1 cells following 48 h of gastrin stimulation. β-actin was used as a loading control. The values indicate the means ± SD from three independent experiments
The function of gastrin in paxillin phosphorylation is also time dependent. As shown in Fig. 1b, Tyr118 and Tyr31 began to show obvious phosphorylation after culturing with 200 nM gastrin for 60 min and 15 min, respectively, and the phosphorylation had positive relevance with the inducing time. Notably, the total expression level of paxillin was stable during these assays (Fig. 1b). Hence in the subsequent assays, we cultured the cells for 60 min in 200 nM gastrin to activate paxillin. These experimental conditions were also verified in the gastric cancer cells HGC and the colon cancer cells HT-29 (Fig. 1c, d).
Gastrin promotes the vertical migration, vertical invasion, and horizontal movements of PANC-1 cells
Previous studies have demonstrated that gastrin can promote the migration of many types of cancer cells10,11. Herein, we explored whether gastrin exerted similar effects on PANC-1 cells. We utilized Transwell chambers without or with the addition of Matrigel to evaluate vertical migration and invasion ability, respectively, and utilized the scratched wound healing assay in a culture dish covered with fibronectin to analyze horizontal movement. As shown in Fig. 1e–g, 200 nM gastrin significantly enhanced the motility of PANC-1 cells.
Gastrin activates GTP-RhoA in a time-dependent manner
The Rho GTPase family is associated with actin cytoskeleton rearrangement, cell polarity, and cellular directional movement, and RhoA is one of the most crucial members of this family14. It was reported that gastrin can induce the activation of RhoA in colonic cancer11; however, it is unclear whether RhoA has a similar function in pancreatic cancer. Herein, we showed that with 200 nM gastrin, the level of GTP-RhoA increased continuously with the culture time (Supplementary Figure 2). Our results indicate that gastrin can also promote the activation of RhoA in pancreatic cancer cells.
Gastrin phosphorylates paxillin, activates RhoA, modulates the formation of focal adhesions and cell polarization via its G protein-coupled receptor Gα12/13
To determine the underling mechanism by which gastrin activates paxillin and RhoA in pancreatic cancer, we began by investigating the gastrin receptor, CCKBR. The heterotrimeric G proteins Gα12 and Gα13 are widely expressed in human tissues17,21–23. We hypothesize that Gα12/13 may be the crucial mediator in the function of gastrin in pancreatic cancer. After validating the knockdown effects of Gα12 siRNA, we used it to inhibit the expression of Gα12 in PANC-1 cells. As shown in Fig. 2a, b, compared with the control group, Gα12 inhibition significantly reduced the activation of paxillin and RhoA, and the effects of gastrin on stimulating paxillin and RhoA were partially or totally rescued when Gα12 siRNA was transected. Immunofluorescence analysis further validated our western blot results. As shown in Fig. 2c, d, in the control (DMSO) group, paxillin, which is the marker protein of focal adhesions, was mainly located in the cytoplasm without clustering, and F-actin was also scattered and not bundling or polarized. After stimulation with gastrin, the numbers of focal adhesions were obviously increased with a larger area and more brightness, especially in the protruding lamellipodia, and F-actin was bundle-like in the same direction. After Gα12 siRNA was added, phenomena such as focal adhesions, F-actin assembly, and cellular protrusion were all significantly reduced. The scratch wound healing assay and cell invasion assay were also conducted to validate that Gα12 is crucial for gastrin’s function: after Gα12 inhibition, the effects of gastrin were remarkably alleviated (Fig. 2e, f). In addition, we observed similar results using Gα13 siRNA to treat the cell (Supplementary Figure 3A–F), and collectively, we demonstrated that CCKBR–Gα12/13 were pivotal to transmit the signal from gastrin in pancreatic cancer.
Fig. 2. Gastrin regulates the focal adhesion (FA) formation, F-actin polarization, and migration of PANC-1 cells through the Gα12 pathway.

PANC-1 cells were treated with control or Gα12 siRNA as indicated. a As indicated by immunoprecipitation experiments, RNAi-mediated Gα12 protein depletion significantly suppressed the phosphorylation of paxillin activated by gastrin in PANC-1 cells. b RNAi-mediated Gα12 protein depletion also restrained PANC-1 cells from the elevated GTP-RhoA level that was activated by gastrin. c, d Focal adhesion formation and F-actin polarization were evaluated by immunofluorescence (c) and were represented by the average number of focal adhesions that were labeled by paxillin per cell (d). White arrows show the focal adhesion formation and aggregation at the leading edge of the cells. e, f The scratched wound healing assay (e) and Boyden invasion assay (f) were conducted to show the migration and invasion of PANC-1 cells. The average number of focal adhesions from a minimum of 20 cells was quantified. The other values indicate the means ± SD from three independent experiments. *P < 0.05
Gastrin promotes directional pancreatic cancer cell migration via RhoA/ROCK activation
Others have established that RhoA and its downstream effector, ROCK, modulate cell polarization and cellular directional movement24–26. However, in pancreatic cancer, the roles of RhoA and ROCK in cell directional migration are not clear. After using the RhoA inhibitor Rhosin to block the RhoA–ROCK pathway (Fig. 3a), we noted that phosphorylation on Tyr31 and Tyr118 of paxillin was significantly reduced, although with gastrin induction (Fig. 3b). In immunofluorescence assays, we also observed that Rhosin significantly reduced the cellular polarization, focal adhesion formation, and F-actin assembly induced by gastrin (Fig. 3c, d) and promoted decreased wound healing and invasion ability of the cells (Fig. 3e, f). We also used an inhibitor of ROCK, Y27632, to treat the PANC-1 cells and obtain similar results (Fig. 4a–e). We also utilized another focal adhesion protein marker, FAK, and as expected, the results were in agreement (Supplementary Figures 4 and 5). Our results confirm that RhoA–ROCK is the main manipulator in activating paxillin and FAK and is crucial for cell directional migration induced by gastrin in pancreatic cancer.
Fig. 3. RhoA is necessary for gastrin-stimulated paxillin tyrosine phosphorylation, cell polarization, and FA formation.

PANC-1 cancer cells were pre-treated with Rhosin, an effective inhibitor of RhoA, for 24 h and were then incubated with gastrin for 1 h. a The gastrin-stimulated and endogenous activation of RhoA was significantly inhibited by Rhosin. b Rhosin pre-treatment suppressed the tyrosine phosphorylation of paxillin that was activated by gastrin. c The images indicate increased paxillin-labeled focal adhesion formation and aggregation at the cell leading-edge stimulated by gastrin, and Rhosin pre-treatment inhibited these changes. Boxed regions indicate the area of the merged image used for the zoom. White arrows show the focal adhesion formation and aggregation at the leading edge of cells. d The histogram indicates the mean number of focal adhesions labeled by paxillin per cell. The average number of focal adhesions from a minimum of 20 cells was calculated. e, f The scratched wound healing assay (e) and Boyden invasion assay (f) were performed to show the migration and invasion of PANC-1 cells. Values are represented as the means ± SD from three independent experiments. *P < 0.05
Fig. 4. Gastrin-regulated PANC-1 cell polarization, migration, and invasion through activation of ROCK.

PANC-1 cancer cells were pre-incubated with Y27632 reagent, an effective inhibitor of ROCK, for 4 h and were then stimulated with gastrin for 1 h. a Activated paxillin with high levels of phosphorylation at Tyr118 and Tyr31 that were induced by gastrin were significantly inhibited by Y27632 pre-treatment, while total paxillin was unchanged. b Immunofluorescence images indicate an increased polarization of F-actin and aggregation of focal adhesions at the leading edge in gastrin-treated PANC-1 cells. Y27632 pre-treatment obviously blocked the focal adhesion enrichment and cell polarization. Boxed regions represent the area of the merged image used for the zoom; c The mean number of large-sized focal adhesions labeled by paxillin per cell are indicated, and the average number of focal adhesions from a minimum of 20 cells was calculated. d, e The migration and invasion of PANC-1 cells pre-treated with Y27632 were investigated by scratched wound healing (d) and the Boyden invasion assay (e). Values indicate the means ± SD from three independent experiments. *P < 0.05
Gastrin induced Golgi apparatus reorientation via the CCKBR–Gα12/13–RhoA–ROCK pathway
Reorientation of the secretory machinery is involved in directional cell migration8. This process is in coordination with rearrangement of the cytoskeleton and focal adhesion formation8. However, the molecular mechanisms that dominate this reorientation are not well characterized. As mentioned above, we have shown that Gα12/13–RhoA–ROCK signaling is crucial for the morphological changes and high metastasis potential of gastrin-induced cells. We then tried to delineate the role of Gα12/13–RhoA–ROCK signaling in reorientation of the Golgi apparatus. We used a scratch wound healing model to induce the migration of PANC-1 cells and utilized a β-COP antibody to mark the location of the Golgi apparatus. As shown in the immunofluorescence and wind rose plot (Fig. 5), 200 nM gastrin treatment for 6 h significantly promoted the reorientation of the Golgi apparatus to the leading edge of the cells; however, in the DMSO group, the Golgi apparatus was disordered with no directivity. We then individually treated the cells with Gα12 siRNA, Gα13 siRNA, Rhosin, or Y27632, and we demonstrated that any missing link in Gα12/13–RhoA–ROCK signaling impaired the polarization of F-actin and impeded the reorientation of the Golgi in response to wounding. These results reveal a previously unidentified pathway that regulates the polarity of migrating cancer cells through Golgi reorientation.
Fig. 5. Gastrin promotes Golgi aggregation and cell polarization at the leading edge in PANC-1 cells through activation of the Gα12/13–RhoA–ROCK signaling pathway.

PANC-1 cells were allowed to grow to confluence for 24 h and were then pre-treated with Gα12 siRNA, Gα13 siRNA, Rosin, and Y27632 to inhibit expression or activity of Gα12, Gα13, RhoA, and ROCK, respectively. Cells that were scratched with tips were then stimulated with gastrin. Representative images from left to right indicate the Golgi marker protein β-cop (red), cytoskeletal F-actin (green), and cell nucleus (blue). Merged images and wind rose plots of Golgi orientation are shown. Gastrin promoted the reorganization and polarization of the Golgi apparatus at the leading edge of PANC-1 cells facing the detached line; however, a highly disorganized and fragmented Golgi apparatus was distributed in the cytoplasm. Gastrin also induced the polarization of cytoskeletal F-actin accompanied with a non-polarized morphology. Gα12 RNAi, Gα13 RNAi, Rosin, and Y27632 pre-treatment blocked the polarization of the Golgi and F-actin that was induced by gastrin. Wind rose plots indicate the Golgi orientation in cells, and only the gastrin treatment group restored proper cell polarity and Golgi orientation
Gastrin promotes progression of pancreatic cancer in nude mice with orthotopic transplantation
Next, we tested whether gastrin is capable of driving progression of pancreatic cancer cells in vivo. We transplanted PANC-1 cells under the pancreas envelope in nude mice. During the experiment, mice in different groups were administered different treatments (Fig. 6a). After 7 weeks, the mice were killed. Western blot analysis and immunohistochemistry staining showed that, compared with the control group (DMSO treatment), paxillin activation in the orthotopic tumor was significantly enhanced in the gastrin treatment group. YM022, a specific blocker of the CCKBR, and Y27632 reduced the phosphorylation level of paxillin induced by gastrin (Fig. 6b, c). Consistent with the results of western blot analysis, the weight of orthotopic tumors in the gastrin treatment group was the highest and YM022 and Y27632 inhibited the growth of the tumors (Fig. 6c, d).
Fig. 6. Gastrin promotes tumor progression of orthotopic tumors in nude mice.

PANC-1 cells (1 × 107 cells) were injected into the head/neck region of the nude mouse pancreas. a Mice were divided into six groups and treated according to schedule. b, c Immunoblotting (b) and immunohistochemistry (c) results are indicated for paxillin phosphorylation of orthotopic pancreatic cancer tissues. d–f YM022 and Y-27632 significantly inhibited the growth of the primary pancreatic tumor (d, e) and prevented liver metastasis (f). Data are represented as the mean ± SD, *P < 0.05
Pancreatic cancer spreads easily to distant organs, and the liver is its main target organ27; therefore, we checked the number of metastasis sites in the liver of the treated mice. As expected, gastrin significantly promoted the metastasis of pancreatic cancer cells, and YM022 and Y27632 inhibited this process (Fig. 6e). From these results, we further confirm that gastrin can promote pancreatic cancer progression via the CCKBR and its downstream signaling pathway.
Discussion
In this study, we demonstrated that gastrin promotes the reorientation of the Golgi apparatus and directional migration of pancreatic cancer cells by inducing the activation of paxillin and FAK via the CCKBR–Gα12/13–RhoA–ROCK signaling pathway (as illustrated in Fig. 7).
Fig. 7.
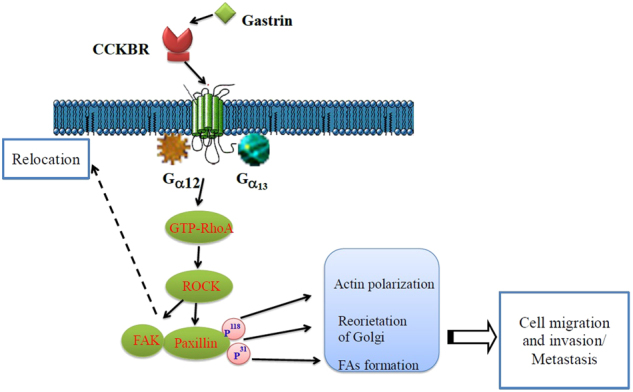
Schematic model depicting the role of gastrin-CCKBR–Gα12/13–RhoA–ROCK signaling in pancreatic cancer progression
The functions of gastrin and the CCKBR in cancer biology have been investigated and discussed for decades. It has been demonstrated that the downstream signaling pathway of the gastrin/CCKBR includes JNK, JAK2, STAT3, ERKs, and NF-kappaB28–30. In pancreatic cancer, gastrin induces the upregulation of beta1-integrin via the Src family of kinases and the PI3K pathway, resulting in enhanced invasion ability31. Further, αv integrin is upregulated by gastrin in pancreatic cancer cells and contributes to cell adhesion32. It has also been reported that gastrin regulates the oncogenic protein ABCG2 via NF-kappaB signaling in pancreatic cancer30. However, the roles of gastrin and the CCKBR in cellular directional migration had not been investigated. To our best knowledge, this study was the first to discover that gastrin can induce directional migration of cancer cells. Our research provides a supplementary explanation for how aberrant enrichment of gastrin in the tumor microenvironment and peripheral blood affects the morphological changes and invasion ability of malignant cells.
Reorientation of the Golgi apparatus is a key characteristic of the directional migration of cells7,8. Importantly, we observed that the location of the Golgi apparatus was responsive to gastrin. As we know, the Golgi apparatus is crucial for protein processing and membrane dynamics, so our results indicate that gastrin might promote cancer metastasis by facilitating polarized secretion and providing membrane to sites of directional migration. The molecular events that modulate the reorientation of the Golgi during cell migration have not been determined. Studies have shown that repositioning of the Golgi results from the reorganization of the microtubule33,34. Among the regulators of the cytoskeleton, the Rho family of small GTPases is principle24. Our results showed that in pancreatic cancer, Golgi reorientation, which is accompanied by F-actin reorganization, is induced by gastrin and is due to activation of RhoA–ROCK signaling. Our findings are consistent with previously established ideas.
During the formation of focal adhesion, paxillin is an adapter protein that functions in an important scaffolding role by recruiting structural and signaling molecules when it is phosphorylated on specific Tyr and Ser residues18,35,36. Among these residues, Tyr31 and Tyr118 are the most important18. Paxillin phosphorylation results in the generation of specific SH2 and SH3 interaction sites that make a link between integrin receptors and cytoskeletal elements, such as actin and vinculin37,38. Additionally, paxillin interacts with and facilitates the phosphorylation of FAK, which is important for binding to downstream signaling molecules, such as Src, PI3K, and SHC18,39. Mutation, amplification, overexpression, and aberrant activation of paxillin have been involved in many cancers. Paxillin has been associated with proliferation, chemoresistance, and epithelial–mesenchymal transition in cancer research40–42. Diverse stimuli can induce paxillin phosphorylation, such as Src and FAK18,43. Previously, we demonstrated that in human colon cancer cells, gastrin-induced tyrosine phosphorylation of paxillin in a time- and dose-dependent manner20, while its mechanism is obscure. Herein, we found that gastrin-activated paxillin through the Gα12/13–RhoA–ROCK signaling pathway, which provides a reasonable explanation.
The heterotrimeric G proteins Gα12 and Gα13 are widely expressed in human tissues, have been described as RhoA activators via RH-containing RhoGEFs, and can promote actin stress fiber formation and focal adhesion assembly22,23. Gα12/13 have consistently been demonstrated to play a key role in oncogenic transformation in multiple cancers, such as prostate cancer, breast cancer, and cervical cancer44-46, while its role in pancreatic cancer is largely unknown. However, another main finding of this current work is that Gα12/13 are responsible for gastrin’s function in pancreatic cancer. Intriguingly, as shown in Fig. 2a, b, compared with the control group, Gα12 knockdown resulted in reduction of the basic levels of phosphorylated paxillin and activated RhoA (Gα13 knockdown only reduced the basic levels of phosphorylated paxillin, Supplementary Figure 2A–B), and this indicated that Gα12/13 also modulated endogenous or alternative activation of paxillin and RhoA in pancreatic cancer. This result suggested that Gα12/13 may be potential therapy targets for pancreatic cancer.
Target therapy has been successful in a number of malignancies. Unfortunately, the results of pancreatic cancer treatments have been extremely disappointing to date. The GPCR family, with over 800 members, is the largest and most diverse protein family in the human proteome47. GPCRs play important roles in numerous cellular and physiological processes, and consequently, aberrant receptor expression or activity is demonstrated in many disorders and diseases47. Importantly, GPCRs are considered to be druggable proteins, and have been the most successful pharmaceutical target class in cancer therapy. For example, degarelix targets GnRH in prostate cancer48, sonidegib and vismodegib target SMO in basal cell carcinoma49,50, and mogamulizumab targets CCR4 in lymphoma51. Previous studies have demonstrated that gastrin and its receptor, CCKBR, are involved in the tumorigenesis and progression of pancreatic cancer. CCKBR, a GPCR member, has been regarded as a promising target in the therapy of pancreatic adenocarcinoma10. The CCKBR antagonists JB95008 and Z-360 have been tested in clinical trial, but the results have been unsatisfactory52,53. Our research elucidated a key signaling pathway in pancreatic cancer progression mediated by gastrin and/or CCKBR. Hopefully, our findings can provide clues for novel CCKBR targeting-drug development and can improve the prognosis of pancreatic cancer patients.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was supported by grants from the Fundamental Research Funds for the Chinese Central Universities (no. 2012302020214), National Natural Science Foundation of China (nos. 81602116, 81672387, and 81703030) and Hubei Provincial Natural Science Foundation (no. 2016CFB195).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contribute equally: Ganggang Mu, Qianshan Ding, Hongyan Li.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s12276-018-0081-6.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Torre LA, et al. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Gaianigo, N., Melisi, D. & Carbone C. EMT and treatment resistance in pancreatic cancer. Cancers 9, E122 (2017). [DOI] [PMC free article] [PubMed]
- 3.Mizoguchi T, Ikeda S, Watanabe S, Sugawara M, Itoh M. Mib1 contributes to persistent directional cell migration by regulating the Ctnnd1-Rac1 pathway. Proc. Natl Acad. Sci. USA. 2017;114:e9280–e9289. doi: 10.1073/pnas.1712560114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grande-García A, del Pozo MA. Caveolin-1 in cell polarization and directional migration. Eur. J. Cell Biol. 2008;87:641–647. doi: 10.1016/j.ejcb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Yuan X, et al. Directional migration in esophageal squamous cell carcinoma (ESCC) is epigenetically regulated by SET nuclear oncogene, a member of the inhibitor of histone acetyltransferase complex. Neoplasia. 2017;19:868–884. doi: 10.1016/j.neo.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lupo B, et al. Tankyrase inhibition impairs directional migration and invasion of lung cancer cells by affecting microtubule dynamics and polarity signals. BMC Biol. 2016;14:5. doi: 10.1186/s12915-016-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardakheh FK, Self A, Marshall CJ. RHO binding to FAM65A regulates Golgi reorientation during cell migration. J. Cell Sci. 2016;129:4466–4479. doi: 10.1242/jcs.198614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing M, et al. GOLPH3 drives cell migration by promoting Golgi reorientation and directional trafficking to the leading edge. Mol. Biol. Cell. 2016;27:3828–3840. doi: 10.1091/mbc.e16-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am. J. Physiol. 1998;274:G607–G613. doi: 10.1152/ajpgi.1998.274.4.g607. [DOI] [PubMed] [Google Scholar]
- 10.Boyce M, Lloyd KA, Pritchard DM. Potential clinical indications for a CCK2 receptor antagonist. Curr. Opin. Pharmacol. 2016;31:68–75. doi: 10.1016/j.coph.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Masià-Balagué M, et al. Gastrin-stimulated Gα13 activation of Rgnef protein (ArhGEF28) in DLD-1 colon carcinoma cells. J. Biol. Chem. 2015;290:15197–15209. doi: 10.1074/jbc.M114.628164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith JP, Hamory MW, Verderame MF, Zagon IS. Quantitative analysis of gastrin mRNA and peptide in normal and cancerous human pancreas. Int. J. Mol. Med. 1998;2:309–315. doi: 10.3892/ijmm.2.3.309. [DOI] [PubMed] [Google Scholar]
- 13.Smith JP, Shih A, Wu Y, McLaughlin PJ, Zagon IS. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am. J. Physiol. 1996;270:R1078–R1084. doi: 10.1152/ajpcell.1996.270.3.C939. [DOI] [PubMed] [Google Scholar]
- 14.Li C, et al. The GTPase Rab43 controls the anterograde ER-Golgi trafficking and sorting of GPCRs. Cell Rep. 2017;21:1089–1101. doi: 10.1016/j.celrep.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donnelly SK, Bravo-Cordero JJ, Hodgson L. Rho GTPase isoforms in cell motility: don’t fret, we have FRET. Cell Adh. Migr. 2014;8:526–534. doi: 10.4161/cam.29712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stepan V, et al. Role of small GTP binding proteins in the growth-promoting and antiapoptotic actions of gastrin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G715–G725. doi: 10.1152/ajpgi.00169.2003. [DOI] [PubMed] [Google Scholar]
- 17.Le Page SL, Bi Y, Williams JA. CCK-A receptor activates RhoA through G alpha 12/13 in NIH3T3 cells. Am. J. Physiol. Cell Physiol. 2003;285:C1197–C1206. doi: 10.1152/ajpcell.00083.2003. [DOI] [PubMed] [Google Scholar]
- 18.López-Colomé AM, Lee-Rivera I, Benavides-Hidalgo R, López E. Paxillin: a crossroad in pathological cell migration. J. Hematol. Oncol. 2017;10:50. doi: 10.1186/s13045-017-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leyton J, Garcia-Marin LJ, Tapia JA, Jensen RT, Moody TW. Bombesin and gastrin releasing peptide increase tyrosine phosphorylation of focal adhesion kinase and paxillin in non-small cell lung cancer cells. Cancer Lett. 2001;162:87–95. doi: 10.1016/S0304-3835(00)00639-X. [DOI] [PubMed] [Google Scholar]
- 20.Yu HG, Schrader H, Otte JM, Schmidt WE, Schmitz F. Rapid tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130Cas by gastrin in human colon cancer cells. Biochem. Pharmacol. 2004;67:135–146. doi: 10.1016/j.bcp.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki N, Hajicek N, Kozasa T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals. 2009;17:55–70. doi: 10.1159/000186690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Z, et al. CYP2J2 metabolites, epoxyeicosatrienoic acids, attenuate Ang II-induced cardiac fibrotic response by targeting Gα12/13. J. Lipid Res. 2017;58:1338–1353. doi: 10.1194/jlr.M074229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie S, Mason FM, Martin AC. Loss of Gα12/13 exacerbates apical area dependence of actomyosin contractility. Mol. Biol. Cell. 2016;27:3526–3536. doi: 10.1091/mbc.e16-05-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marjoram RJ, Lessey EC, Burridge K. Regulation of RhoA activity by adhesion molecules and mechanotransduction. Curr. Mol. Med. 2014;14:199–208. doi: 10.2174/1566524014666140128104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y, et al. The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. J. Immunol. 2009;182:3837–3845. doi: 10.4049/jimmunol.0803838. [DOI] [PubMed] [Google Scholar]
- 26.Cao X, et al. A phosphorylation switch controls the spatiotemporal activation of Rho GTPases in directional cell migration. Nat. Commun. 2015;6:7721. doi: 10.1038/ncomms8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito T, et al. The diagnostic advantage of EOB-MR imaging over CT in the detection of liver metastasis in patients with potentially resectable pancreatic cancer. Pancreatology. 2017;17:451–456. doi: 10.1016/j.pan.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Beales IL, Ogunwobi O. Glycine-extended gastrin inhibits apoptosis in colon cancer cells via separate activation of Akt and JNK pathways. Mol. Cell Endocrinol. 2006;247:140–149. doi: 10.1016/j.mce.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, et al. Gastrin acting on the cholecystokinin2 receptor induces cyclooxygenase-2 expression through JAK2/STAT3/PI3K/Akt pathway in human gastric cancer cells. Cancer Lett. 2013;332:11–18. doi: 10.1016/j.canlet.2012.12.030. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, et al. Gastrin regulates ABCG2 to promote the migration, invasion and side populations in pancreatic cancer cells via activation of NF-κB signaling. Exp. Cell Res. 2016;346:74–84. doi: 10.1016/j.yexcr.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Cayrol C, et al. Cholecystokinin-2 receptor modulates cell adhesion through beta 1-integrin in human pancreatic cancer cells. Oncogene. 2006;25:4421–4428. doi: 10.1038/sj.onc.1209484. [DOI] [PubMed] [Google Scholar]
- 32.Cayrol C, et al. v integrin: a new gastrin target in human pancreatic cancer cells. World J. Gastroenterol. 2011;17:4488–4495. doi: 10.3748/wjg.v17.i40.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang P, et al. The protein encoded by the CCDC170 breast cancer gene functions to organize the Golgi-microtubule network. EBio Med. 2017;22:28–43. doi: 10.1016/j.ebiom.2017.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wesolowski J, et al. Chlamydia hijacks ARF GTPases to coordinate microtubule posttranslational modifications and Golgi complex positioning. MBio. 2017;8:e02280–16. doi: 10.1128/mBio.02280-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fusté NP, et al. Cytoplasmic cyclin D1 regulates cell invasion and metastasis through the phosphorylation of paxillin. Nat. Commun. 2016;7:11581. doi: 10.1038/ncomms11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin R, et al. Phosphorylation and turnover of paxillin in focal contacts is controlled by force and defines the dynamic state of the adhesion site. Cytoskeleton. 2015;72:101–112. doi: 10.1002/cm.21209. [DOI] [PubMed] [Google Scholar]
- 37.Turner CE, Miller JT. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125Fak-binding region. J. Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- 38.Deakin NO, Ballestrem C, Turner CE. Paxillin and Hic-5 interaction with vinculin is differentially regulated by Rac1 and RhoA. PLoS ONE. 2012;7:e37990. doi: 10.1371/journal.pone.0037990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wade R, Vande Pol S. Minimal features of paxillin that are required for the tyrosine phosphorylation of focal adhesion kinase. Biochem. J. 2006;393:565–573. doi: 10.1042/BJ20051241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang LL, et al. Phosphatase and tensin homolog (PTEN) represses colon cancer progression through inhibiting paxillin transcription via PI3K/AKT/NF-κB pathway. J. Biol. Chem. 2015;290:15018–15029. doi: 10.1074/jbc.M115.641407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanteti R, Batra SK, Lennon FE, Salgia R. FAK and paxillin, two potential targets in pancreatic cancer. Oncotarget. 2016;7:31586–31601. doi: 10.18632/oncotarget.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu DW, Huang CC, Chang SW, Chen TH, Lee H. Bcl-2 stabilization by paxillin confers 5-fluorouracil resistance in colorectal cancer. Cell Death Differ. 2015;22:779–789. doi: 10.1038/cdd.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kratimenos P, et al. FAK-Src-paxillin system expression and disease outcome in human neuroblastoma. Pediatr. Hematol. Oncol. 2017;34:221–230. doi: 10.1080/08880018.2017.1360969. [DOI] [PubMed] [Google Scholar]
- 44.Kelly P, et al. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 2006;281:26483–26490. doi: 10.1074/jbc.M604376200. [DOI] [PubMed] [Google Scholar]
- 45.Juneja J, Cushman I, Casey PJ. G12 signaling through c-Jun NH2-terminal kinase promotes breast cancer cell invasion. PLoS ONE. 2011;6:e26085. doi: 10.1371/journal.pone.0026085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan B, Cui J, Wang W, Deng K. Gα12/13signaling promotes cervical cancer invasion through the RhoA/ROCK-JNK signaling axis. Biochem. Biophys. Res. Commun. 2016;473:1240–1246. doi: 10.1016/j.bbrc.2016.04.048. [DOI] [PubMed] [Google Scholar]
- 47.Nieto Gutierrez A, McDonald PH. GPCRs: emerging anti-cancer drug targets. Cell Signal. 2018;41:65–74. doi: 10.1016/j.cellsig.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Sayyid RK, et al. A phase II, randomized, open-label study of neoadjuvant degarelix versus LHRH agonist in prostate cancer patients prior to radical prostatectomy. Clin. Cancer Res. 2017;23:1974–1980. doi: 10.1158/1078-0432.CCR-16-1790. [DOI] [PubMed] [Google Scholar]
- 49.Casey D, et al. FDA approval summary: sonidegib for locally advanced basal cell carcinoma. Clin. Cancer Res. 2017;23:2377–2381. doi: 10.1158/1078-0432.CCR-16-2051. [DOI] [PubMed] [Google Scholar]
- 50.Basset-Séguin N, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer. 2017;86:334–348. doi: 10.1016/j.ejca.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 51.Makita S, Tobinai K. Mogamulizumab for the treatment of T-cell lymphoma. Expert Opin. Biol. Ther. 2017;17:1145–1153. doi: 10.1080/14712598.2017.1347634. [DOI] [PubMed] [Google Scholar]
- 52.Chau I, et al. Gastrazole (JB95008), a novel CCK2/gastrin receptor antagonist, in the treatment of advanced pancreatic cancer: results from two randomised controlled trials. Br. J. Cancer. 2006;94:1107–1115. doi: 10.1038/sj.bjc.6603058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ueno M, et al. A randomized phase II study of gemcitabine plus Z-360, a CCK2 receptor-selective antagonist, in patients with metastatic pancreatic cancer as compared with gemcitabine plus placebo. Cancer Chemother. Pharmacol. 2017;80:307–315. doi: 10.1007/s00280-017-3351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.