

We report familial cardiac conduction disease (FCCD) in a 3-generation pedigree, including some family members with congenital junctional ectopic tachycardia (JET) (Fig. 1A). The proband presented during a febrile illness at 3.5-years of age with variable atrio-ventricular (AV) block (1st, 2nd, and 3rd degrees) together with left anterior fascicular block (LAFB) and right bundle branch block (RBBB) (Fig. 1B). Viral studies and echocardiogram were normal and he received a pacemaker; he subsequently developed clinical junctional tachycardia. Family history revealed a maternal grandmother with RBBB diagnosed at 5 years of age. The electrocardiogram (ECG) from the proband’s mother revealed sinus rhythm (SR), 1st degree AV block and a nonspecific intraventricular conduction delay. Clinical genetic testing of SCN5A did not reveal any disease-associated mutations. Subsequently she gave birth to identical twin girls. Both girls had rapid JET in the immediate post-natal period that was initially difficult to control. There was rate dependent RBBB and LAFB in both children prior to use of antiarrhythmic drugs. The arrhythmia has resolved in one twin but persists in the other as a catecholamine dependent accelerated junctional rhythm/congenital JET that is modified by propranolol therapy (Fig. 1C). The resting ECGs now both show LAFB and intermittent incomplete RBBB. The mother has subsequently developed symptomatic palpitations and shows electrocardiographic features consistent with typical atrioventricular nodal re-entrant tachycardia (AVNRT) on symptom-rhythm correlation observed on loop recorder. Thus, the conduction defects in this family were of varying severity and the relationship to congenital JET in the three children is important as it is well recognized that congenital JET may progress to heart block [1,2]. At this stage the conduction abnormalities are minor in the twins who presented with the JET and have not progressed.
Fig. 1.
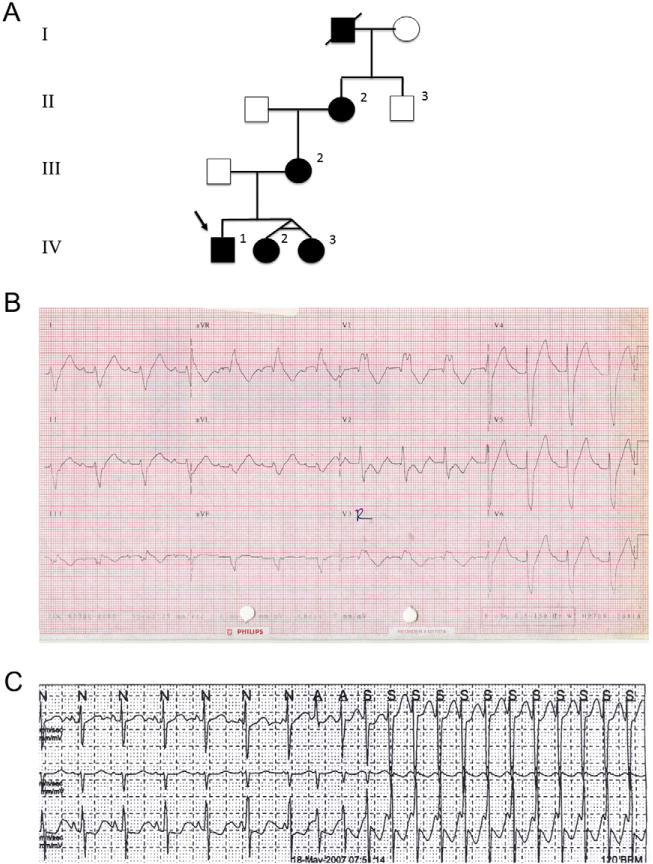
A) Pedigree of the family with novel cardiac conduction system disease. Affected individuals are represented by black symbols. B) Electrocardiogram (ECG) from proband (IV-1) at presentation during a period of stable rhythm. The ECG shows sinus rhythm with first degree AV block, left anterior fascicular block (LAFB) and RBBB with QRS duration of 200 ms. C) Strip from Holter tape showing onset of tachycardia with AV dissociation and accelerating rate consistent with congenital junctional ectopic tachycardia (JET) (IV-2).
To determine the genetic basis of this novel FCCD and congenital JET, with the approval from the Children’s Hospital of Eastern Ontario Ethics Board and informed consent from the family, we performed whole exome sequencing (WES) on peripheral blood DNA samples from the affected boy, one of the affected twin girls, and the unaffected father using methods previously described [3]. Coding variants and those present in intron–exon boundaries were filtered to a frequency of less than 1% for all 3 family members. Given the dominant inheritance from the maternal lineage, we then looked for heterozygous variants shared by the two patients but not the father, which resulted in 34 rare candidate variants (Supplementary Table 1). Of the 34 rare variants, one was present in a gene recently associated with cardiac arrhythmia in humans: TNNI3K (NM_015978.2:c.1615A>G; p.Thr539Ala). The previous report of a p.Gly526Asp mutation in TNNI3K associated with variable atrial arrhythmias, conduction disease and dilated cardiomyopathy [4] suggests that this gene is causative in the family reported here. We confirmed that the variant was inherited from the affected mother (Fig. 2A).
Fig. 2.
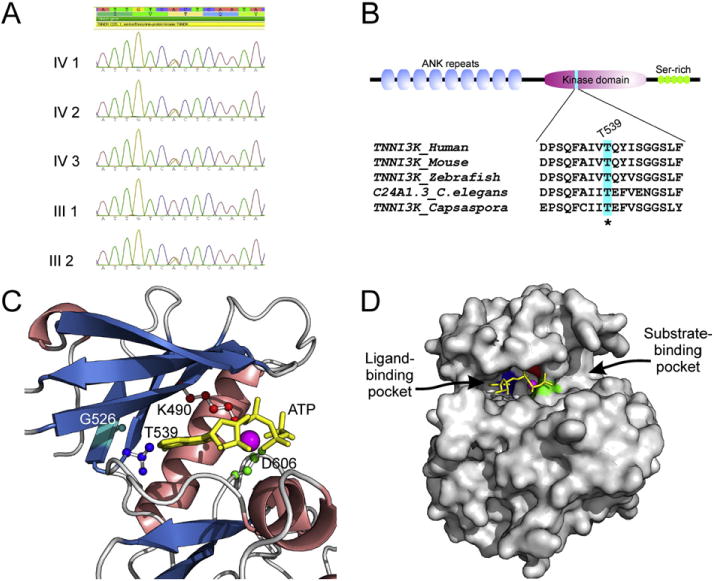
A) Sanger sequencing validation of TNNI3K (NM_015978.2:c.1615A>G; p.Thr539Ala) heterozygous variant identified by whole exome sequencing. B) Domain architecture of human TNNI3K and conservation of Thr539 position in animal lineage. C) Visualization of active site residues of human TNNI3K kinase domain together with ATP and Mg2+. Homology model was constructed using the Modeller9v11 program on the basis of four PDB templates, 3PPZ, 3KMW, 2EVA and 3BLQ, and evaluated by the ModFOLD program (score 0.81; any score >0.4 is a generally confident model). The α helices are shown in salmon, β sheets in blue, and loops in gray; catalytic residues Lys (K490) shown in red, Asp (D606) in green, Gly (G526) in light blue and gatekeeper residue Thr (T539) in dark blue; ATP in yellow and Mg2+ in purple. D) Surface view of ATP-binding pocket of human TNNI3K kinase domain in which the ATP and Mg2+ were hold by three key residues K490 in red, D606 in green and T539 in dark blue.
TNNI3K is a cardiac-specific gene, encoding a cardiac troponin I-interacting MAP kinase. In mice, Tnni3k is shown to play an important role in the regulation of cardiac differentiation and cardiac contractility [5]. Overexpression of Tnni3k accelerated disease progression in mouse models of cardiomyopathy [6]. Moreover, increased level of Tnni3k resulted in prolonged PR interval duration, indicating its critical role in cardiac conduction [7]. The TNNI3K protein is comprised of nine N-terminal ankyrin repeats, followed by a serine/threonine kinase domain and a C-terminal serine-rich region (Fig. 2B). Thr539 is located in the kinase domain, and sequence analysis combined with phylogenetic analysis shows that conservation of Thr539 can be traced back to the basal member of the animal lineage namely Capsaspora (Fig. 2B). This conservation over large evolutionary time is suggestive of a functional constraint on this residue. We generated the homology model for the TNNI3K kinase domain and found that Thr539 is in the ATP-binding pocket (Fig. 2C). In contrast to the active site residues Lys490 and Asp606, which contact the triphosphate moiety and Mg2+ on one side of the pocket, Thr539 interacts with the adenine ring of the ATP on the side opposite to them (Fig. 2C and D). Importantly, this position has been shown to be a key determinant controlling the kinase activity and the accessibility of the ligand-binding pocket to selective kinase inhibitors (a “gatekeeper” residue) [8–10]. As suggested by previous experimental studies on other kinases [8,9], any substitution of the gatekeeper Thr to a hydrophobic amino acid (including Thr539Ala substitution) increases activity of the kinase domain. On the other hand, a mutation of the equivalent gatekeeper residue in the Tec kinase Itk to glycine or alanine inactivates the kinase [10]. Thus, alteration of the gatekeeper Thr in TNNI3K could lead to altered phosphorylation of its substrates, such as cardiac troponin I, and may result in cardiac dysfunction. In addition, the TNNI3K p.Gly526Asp mutation recently associated with cardiac arrhythmia and dilated cardiomyopathy [4] is located at the β strand structurally adjacent to the one containing Thr539 (Fig. 2C) and is extremely conserved across kinase domains, suggesting an important structural role for the residue. It is possible that the p.Gly526Asp mutation might affect the positioning of Thr539 in the active site pocket via disruption of interactions with the adjacent β strand.
In summary, using WES we identified a heterozygous p.Thr539Ala mutation in TNNI3K, a cardiac-specific kinase, as the likely cause for a novel FCCD including congenital JET. Comparative sequence analysis and in silico modeling of TNNI3K kinase domain indicates the p.Thr539Ala substitution could result in an alternation of its kinase activity, but further experiments are required to investigate this hypothesis and how this alteration of TNNI3K kinase activity results in changes of specialized conduction tissues and arrhythmogenesis. This is the second family reported to date with variants in this gene associated with FCCD and arrhythmias. Additional families will need to be identified to understand the spectrum of disease associated with disruption of TNNI3K.
Supplementary Material
Footnotes
Funding: This work was performed under the Care4Rare Canada Consortium funded by Genome Canada (OGI-064), the Canadian Institutes of Health Research (GPH-129346), the Ontario Genomics Institute (OGI-064), Ontario Research Fund (REG1-4404), Genome Quebec (OGI-064), and Children’s Hospital of Eastern Ontario Foundation (to K.M.B.). Additional funding was provided by the Intramural Research Program of the National Library of Medicine, NIH (to L.A.).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ijcard.2015.03.130.
Conflict of interest
The authors report no relationships that could be construed as a con-flict of interest.
References
- 1.Henneveld H, Hutter P, Bink-Boelkens M, et al. Junctional ectopic tachycardia evolving into complete heart block. Heart. 1998;80:627–628. doi: 10.1136/hrt.80.6.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villain E, Vetter VL, Garcia JM, et al. Evolving concepts in the management of congenital junctional ectopic tachycardia. A multicenter study. Circulation. 1990;81:1544–1549. doi: 10.1161/01.cir.81.5.1544. [DOI] [PubMed] [Google Scholar]
- 3.Srour M, Putorti ML, Schwartzentruber J, et al. Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve. 2014;50:775–779. doi: 10.1002/mus.24224. [DOI] [PubMed] [Google Scholar]
- 4.Theis JL, Zimmermann MT, Larsen BT, et al. TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy. Hum Mol Genet. 2014;23:5793–5804. doi: 10.1093/hmg/ddu297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai ZF, Chen YZ. Evidence, hypotheses and significance of MAP kinase TNNI3K interacting with its partners. World J Hypertens. 2012;2:22–28. [Google Scholar]
- 6.Wheeler FC, Tang H, Marks OA, et al. Tnni3k modifies disease progression in murine models of cardiomyopathy. PLoS Genet. 2009;5:e1000647. doi: 10.1371/journal.pgen.1000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodder EM, Scicluna BP, Milano A, et al. Dissection of a quantitative trait locus for PR interval duration identifies Tnni3k as a novel modulator of cardiac conduction. PLoS Genet. 2012;8:e1003113. doi: 10.1371/journal.pgen.1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azam M, Seeliger MA, Gray NS, et al. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyers PA, Craxton M, Morrice N, et al. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol. 1998;5:321–328. doi: 10.1016/s1074-5521(98)90170-3. [DOI] [PubMed] [Google Scholar]
- 10.Joseph RE, Andreotti AH. Controlling the activity of the Tec kinase Itk by mutation of the phenylalanine gatekeeper residue. Biochemistry. 2011;50:221–229. doi: 10.1021/bi101379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.