

Abstract
Traditional radiosynthetic optimization faces the challenges of high radiation exposure, cost and inability to perform serial reactions due to tracer decay. To accelerate tracer development, we have developed a strategy to simulate radioactive 18F-syntheses by using tracer-level (nanomolar) non-radioactive 19F-reagents and LC-MS/MS analysis. The methodology was validated with fallypride synthesis under tracer-level 19F-conditions, which showed reproducible and comparable results with radiosynthesis, and proved the feasibility of this process. Using this approach, the synthesis of [18F]MDL100907 was optimized under 19F-conditions with greatly improved yield. The best conditions were successfully transferred to radiosynthesis. A radiochemical yield of 19–22% was achieved with the radiochemical purity >99% and the molar activity 38.8 – 53.6 GBq/μmol (n = 3). The tracer-level 19F-approach provides a high-throughput and cost-effective process to optimize radiosynthesis with reduced radiation exposure. This new method allows medicinal and synthetic chemists to optimize radiolabeling conditions without the need to use radioactivity.
Keywords: 19F, 18F, optimization, tracer-level, fallypride, MDL100907
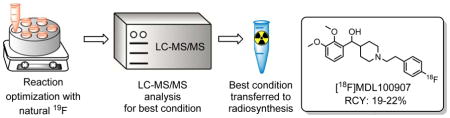
Graphical Abstract
A strategy to simulate radioactive 18F-syntheses with non-radioactive 19F-reagents was developed. Reaction optimization was performed with tracer-level (nanomolar) 19F-reagents and analyzed by high-throughput screening with LC-MS/MS. The best conditions were then transferred to 18F-radiosynthesis. This approach provides a high-throughput and cost-effective process to optimize radiosyntheses with reduced radiation exposure.
1. Introduction
Molecular imaging is an active research area that allows the visualization of molecular probes in biological processes of a living subject.1, 2 In the past few decades, molecular imaging techniques have made significant achievements in both methodological and instrumental development.3 In particular, positron emission tomography (PET) has evolved into a powerful scientific and clinical tool for non-invasively monitoring molecular pathways in vivo.4, 5 PET utilizes radiopharmaceuticals labeled with positron-emitting radionuclides. Among the commonly used PET radionuclides, fluorine-18 (18F) is the most popular PET isotope, often referred to as the radionuclide of choice in PET imaging. In the last decade, the number of bioactive fluorine containing drugs has significantly increased, providing an expanded pool of fluorinated bioactive candidates for PET tracer development.6,7 To produce novel tracers and to optimize the syntheses of known tracers with low efficiency, there is an urgent need to develop a practical and robust way to explore radiosynthetic conditions.
We propose and demonstrate an approach to optimize radiochemical reactions by tracer-level (nanomolar) [19F]KF and quantitation using liquid chromatography/tandem mass spectrometry (LC-MS/MS). Optimizing radiochemical reactions with natural isotope has been a known approach for decades, which helped radiochemists to establish radiolabeling conditions.8–11 But to our knowledge no quantitation method at no-carrier-added (NCA) level has been reported, primarily due to the lack of available analytical methods to quantify tracer-level of product. We believe that tandem MS spectrometry could be applied to tracer-level reactions for product quantification. Tandem quadrupole instruments such as the triple quadrupole combine high sensitivity and high selectivity, allowing quantitation of trace levels of analytes in very complex sample matrices. MS technique is routinely used in research and clinical settings to quantitate drug levels for pharmacokinetic studies of various biological matrixes.12–14 One of the key advantages to LC-MS/MS is the high-throughput ability to evaluate numerous samples quickly. It allows medicinal and synthetic chemists to quickly assay multiple tracer-level reactions, so that the reaction optimization can be concluded in a short time before transferring to radiochemists for tracer production. It is especially useful when transferring synthetic methods to radiochemistry. Although purchase of a triple quadruple mass spectrometer is a significant investment, LC-MS/MS based assays are rountinely used in drug discovery and development.15 There is increasing availability of LC-MS/MS for other applications such as proteomics and metabolomics studies and clinical analysis.16–18 Labs who are routinely developing new radiochemistry will find it indispensable for the rapid analysis of parallel reaction optimization.19,20 Another advantage of the proposed approach is that no exposure to radioactivity is necessary as compared to the radioactive 18F-approach, as all reaction screenings are conducted with natural 19F-reagents. Once the conditions with the highest yield had been identified by LC-MS/MS, 18F-labeled radiosynthesis was performed for validation purposes. This feature not only greatly reduced exposure for the radiochemists, but also lowered the cost of purchasing radioactivity which offsets the initial investment in the LC-MS/MS instrument. In addition, the proposed approach can be potentially applied to all imaging isotopes, offering a broad scope of potential applications.
2. Results and Discussion
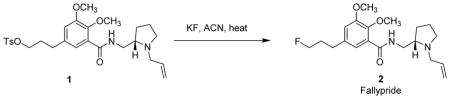
To evaluate the correlation of natural 19F to radioactive 18F-reaction yields, we have selected the tracer-level synthesis of fallypride as a proof-of-concept study. [18F]Fallypride is a PET probe used to quantify dopamine D2/D3 receptors in the brain and has been extensively employed in many clinical PET centers.21,22 The radiosynthesis of [18F]fallypride has been optimized by several groups and well-established with up to 70% radiochemical yield (RCY).23,24 In this study, we performed a series of fallypride syntheses with tracer-level 19F, and found the yields were comparable with radioactive 18F-syntheses thereby validating the 19F-protocol. Once the protocol was validated, the tracer-level 19F-approach was applied to the optimization of MDL100907 synthesis. [18F]MDL100907 is a promising 5-HT2a receptor ligand which exhibits excellent selectivity, affinity, and favorable brain metabolism.25,26 However, the radiosynthesis of [18F]MDL100907 is limited by extremely low yield and long synthesis time, as the electron-rich aryl group hinders the nucleophilic substitution of F−. Ren and coworkers reported an improved radiolabeling approach using Ni-mediated fluorination. However, the overall yield is about 3%. A more robust synthesis of this promising tracer is needed.26,27 By optimizing the synthesis of [18F]MDL100907 under tracer-level 19F-conditions, we demonstrate the great potential of the 19F-process for PET tracer development.
For the LC-MS/MS analysis, we chose to use two distinct MS/MS transitions for each analyte (fallypride and MDL100907) to increase the selectivity for the desired analyte. The transition giving the highest response was used for quantitation. The second transition was used to confirm the identity of the analyte.
Fallypride served as the model compound to validate the feasibility of the 19F-approach. To evaluate the possibility whether other detection methods such as UV and electrochemical detectors are sufficient for tracer-level detection, we performed the comparison between UV and MS/MS detection. Calibration curves were prepared for both detection methods at various fallypride concentration (Figure 1), and limit of quantification (LOQ) was determined. For UV detection, the LOQ is 188 ng/mL which is in the same level as NCA reactions. In theory it is possible to quantify 19F-fallypride synthesis with UV detector since it provides sufficient sensitivity for nanomolar concentrations. The analysis of a representative 19F-synthesis was attempted on UV detector. However, it lacks selectivity for the nanomolar -level product in crude reactions, which is fully obscured by enormous amount of impurities at much higher concentrations (Figure 1b). Similar outcome was predicted for electrochemical detector, that the selectivity is insufficient for quantification purpose, but we did not have access to one to test our hypothesis. It may be possible to quantitate other tracers with much less impurities, or in cases that baseline separation was established for the compound of interest. However, it is case specific and not applicable to all tracers. Under MS/MS condition, the LOQ for fallypride is 0.98 ng/mL, 200-fold higher than UV method (Figure 1c). More importantly, quantification was successfully achieved on the same crude reaction sample. In multiple reaction monitoring (MRM) mode, the trace fallypride signal was selectively recorded among other impurity peaks in much higher concentration (Figure 1d). This mass selection feature allows the detection and quantification of tracer-level products in complex mixtures. MS/MS provides a more reliable and versatile approach for NCA level detection.
Figure 1.

Comparison between UV and MS/MS detection. a) Calibration curve for UV detection, LOQ = 188 ng/mL; b) UV chromatogram of a representative 19F-synthesis (solid) vs. fallypride standard at 1000 ng/mL (dot); c) Calibration curve for MS/MS detection, LOQ = 0.98 ng/mL; d) MS/MS chromatogram of a representative 19F-synthesis: total ion chromatogram (TIC, dot), UV 254 nm (solid) and fallypride signal in MRM mode (×100 amplified, dash)
Next, an experiment was carried out to survey the level of background fluoride contributed from the reaction vial (Figure 2). In this regard, fallypride synthesis was performed with the same process as other tracer-level 19F-procedure, but no fluorine source (KF) was added. The reaction mixture was then analyzed by LC-MS/MS. When the glass reaction vials were used without any washing, it was found the fallypride concentration reached as high as 253 ± 34 ng/mL (n = 3), which represented ~0.7 nmol of fallypride formation attributed from background F−. However, when another group of glass vials were pretreated by washing with K2CO3 and rinsing with water, fallypride concentration was highly reduced by up to 70–80% (85 ± 6 ng/mL). This experiment indicated that some glassware does contain high amount of F− which may participate in the fluorination reaction and potentially affect the SA. A base wash of the glassware can reduce the residual F−. In all later experiments the background F− has been subtracted for yield calculation.
Figure 2.

Fallypride concentration in unwashed and washed reaction vials with no KF added. When the glass vial was washed with K2CO3 and rinsing with water, fallypride concentration was significantly reduced by up to 70–80%.
The correlation between the amount of KF and yield was also studied (Figure 3). Five parallel syntheses of fallypride were performed with various amount of KF (0.0375, 0.075, 0.15, 0.50, 1.0 μg). However, the yield of the 5 syntheses remained in the same level (39–44%, n =3). The result suggested that at this mass range, the amount of KF is not a major contributing factor to yield. For the non-radioactive optimization work in this project, the amount of fluorine (0.15 μg KF) was selected to match with the corresponding radioactive batches, considering a typical molar activity (Am) of 0.18–0.3 GBq (5–8 mCi)/nmol for the [18F]F− produced at our institute and 0.27–0.54 GBq (10–20 mCi) of [18F]F− used in each validation reaction.
Figure 3.

The correlation between amount of KF and yield. Similar yield was found with different amount of KF in each reaction (0.0375 – 1 μg). Reaction condition: K2CO3 (3 mg), K222 (7–8 mg), ACN, 100 °C, 30 min.
It has been reported by Moon et al. that the amount of base plays a crucial role in improving the yield of fallypride.24 Excess base may lead to unwanted thermal degradation of the precursor and potentially lower the product yield and purity. The improved synthesis of [18F]fallypride under low-base conditions gave up to 70% RCY. In our study, these conditions were evaluated as the model reaction to validate tracer-level 19F-approach (Table 1). Initially, the high-base condition was tested for the 19F-reaction (entry 2). A 41% yield was obtained which was considerably lower than all other low-base conditions. This finding is consistent with literature report. For the low-base conditions (entries 3 and 4), 63% and 55% yield was obtained respectively, which are comparable with reported 18F-radiosynthesis. Since the reaction time of 30 min is long for a typical 18F-radiosynthesis (entry 3), we have evaluated the shortened time of 10 min (entry 5). Under these conditions, a similar yield of 64% was achieved. Though the yield is similar to that of entry 2, it will be more practical in 18F-tracer production due to shorter synthesis time. In addition, this condition was replicated multiple times (n = 4), with highly reproducible results. The use of the milder base KHCO3 also generated similar yield to the K2CO3 and tetrabutylammonium hydrogen carbonate (TBAB) approaches (entries 6 and 7).
Table 1.
Preparation of fallypride using tracer-level [19F]KF and the comparison with 18F-radiosynthesis
| ||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | T (°C) | Time (min) | Yield (%) | 18F RCY (%)a |
| 1b | K2CO3 (0.8 mg) | ACN | 100 | 30 | n/a | n/a |
| 2 | K2CO3 (3.0 mg) | ACN | 100 | 30 | 41 | 60 |
| 3 | K2CO3 (0.8 mg) | ACN | 100 | 30 | 63 | 74 |
| 4 | TBAB (4.0 mg) | ACN | 100 | 10 | 55 | 68 ± 1.6c |
| 5 | K2CO3 (0.8 mg) | ACN | 100 | 10 | 64 ± 4d | 77 |
| 6 | KHCO3 (0.8 mg) | ACN | 100 | 10 | 55 | 82 |
| 7 | KHCO3 (1.6 mg) | ACN | 100 | 10 | 63 | 79 |
RCY was determined by HPLC analysis of the crude product.
No KF was used to test background reaction. Glass reaction vial was washed with K2CO3. Fallypride concentration of 85 ± 6 ng/mL was detected.
RCY obtained from Ref. 17
Reaction was run in multiple times (n = 4) and found good reproducibility.
To evaluate the correlation between natural 19F and radioactive 18F-reactions, the radiosynthesis of [18F]fallypride was performed under each reaction condition of Table 1, and general agreement in the RCY between 19F and 18F-reactions was found. For example, under high-base conditions (entry 2), a 60% yield was obtained, which is substantially lower than those of low-base conditions (entries 3, 5–7). Under the conditions which gave the highest yield (entries 3, 5–7), the isolated RCY of [18F]fallypride reached 74–82% (n = 5), the radiochemical purity was >99%, and the Am was 47.3 – 108.4 GBq/μmol (1.28–2.93 Ci/μmol) at the end of synthesis (Figure S1). This study has confirmed the feasibility of tracer-level 19F-approach, in that the 19F-reactions displayed comparable results with radiosyntheses, and the LC-MS/MS is able to provide reproducible data to analyze product in nanogram scale.
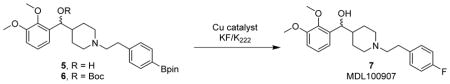
With the protocol validated, we further evaluated the synthesis of MDL100907 using the tracer-level 19F-approach. Since the fluorine atom is on an electron-rich aromatic system, the radiofluorination via conventional nucleophilic approach is not practical. Recently, copper-mediated aromatic nucleophilic radiofluorination reported by Gouverneur et. al. has provided a robust approach to achieve fluorination on both electron-rich and electron-deficient arenes.28 Starting from the aryl boronic ester, copper-mediated radiofluorination has successfully produced a wide range of products, including 6-[18F]fluoro-L-DOPA and [18F]DAA1106. This method was improved by several groups so that it is more suitable for radiochemical processes and automated synthesizers.29–31 The application of the copper-catalyzed method may provide a superior synthetic approach of [18F]MDL100907 as compared to previously reported synthesis.
The Cu-catalyzed synthesis of MDL100907 requires the aryl boronic ester 6 as the precursor. A two-step synthesis for the pinacol boronate was developed (Scheme 1). Briefly, commercially available compounds, (2,3-dimethoxyphenyl)(piperidin-4-yl)methanol (3) and (4-(2-bromoethyl)phenyl)-boronic acid (4) were coupled under basic conditions. Without work-up or purification, the newly generated boronic acid was treated with pinacol to yield 5. Compound 6 was prepared by protecting the benzylic hydroxyl group with di-t-butyl dicarbonate.
Scheme 1.

Synthesis of the boronic ester 6, the precursor for MDL100907
Reagents and conditions: a) NaHCO3, DMF (23 mL), 85 °C, 90 min; b) pinacol, 85 °C, 20 min; c) Boc2O, DMAP, pyridine (5 mL), 60 °C, 30 min.
Tracer-level 19F-reactions were carried out to optimize the Cu mediated synthesis (Table 2). Several factors were evaluated in this study, such as different precursors (unprotected/protected), the relative amounts of base and precursor, selection of catalyst, solvent, and temperature, etc. Firstly, the unprotected alcohol 5 was tested as precursor using various catalysts and amount of base (entries 1–4). However, no product was detected by LC-MS/MS analysis. This result is in agreement with the literature, that unprotected hydroxyl or amino functionalities will interfere with the desired C-C coupling.28 Next, our effort was focused on the Boc-protected pinacol boronate 6 as the precursor (entries 5–14). Under high-base and Cu(OTf)2(py)4 condition, the reaction also did not occur which confirmed low reactivity of high-base protocol (entry 5).29 To overcome this problem, the rest of experiments were performed under low-base conditions, in which a small amount of K2CO3 (0.060 mg, 0.43 μmol) was used. Under the low-base condition (entry 6), the reactivity was dramatically increased, a yield of 18.1% was observed by LC-MS/MS. Other approaches such as increased amount of precursor, various temperature and solvent did not increase the yield (entries 7–10). Increasing catalyst loading has an adverse effect, resulting in a decreased yield of 11.3% (entry 11). When the precursor loading was reduced, the yield was also significantly dropped to 8.3% (entry 12). Other conditions such as using Cu(OTf)2 as catalyst, or exchanging the solvent for a ACN/DMF mixture were also evaluated, and led to no product formation (entries 13–14).
Table 2.
Optimization of MDL100907 synthesis using tracer-level [19F]KFa
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Precursor (mg) | K2CO3 (mg) | K222 (mg) | Catalystb | Solvent (mL) | T (˚C) | % conv. | |
| R = H | 1 | 5 | 2.8 | 14 | Cu(OTf)2(py)4 | DMF 0.3 | 110 | n/a |
| 2 | 5 | 2.8 | 14 | Cu(OTf)2 | DMF 0.3 | 110 | n/a | |
| 3 | 5 | 0.060 | 0.28 | Cu(OTf)2 | DMF 0.3 | 110 | n/a | |
| 4 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.3 | 110 | n/a | |
|
| ||||||||
| R = Bocc | 5 | 5 | 2.8 | 14 | Cu(OTf)2(py)4 | DMF 0.3 | 110 | n/a |
| 6 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.3 | 110 | 18.1 | |
| 7 | 10 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.3 | 110 | 17.0 | |
| 8 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.3 | 120 | 16.9 | |
| 9 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.3 | 100 | 15.5 | |
| 10 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.2 | 110 | 14.8 | |
| 11 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4d | DMF 0.3 | 110 | 11.3 | |
| 12 | 2.5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF 0.2 | 110 | 8.3 | |
| 13 | 5 | 0.060 | 0.28 | Cu(OTf)2 | DMF 0.3 | 110 | n/a | |
| 14 | 5 | 0.060 | 0.28 | Cu(OTf)2(py)4 | DMF:ACN 1:1 | 110 | n/a | |
In all experiments, 0.15 μg of KF was used as the fluorine source.
Catalyst: precursor = 1:3 (molar ratio)
Deprotection of Boc group was carried out with TFA.
Catalyst: precursor = 1: 2
With the optimized conditions in hand (Table 2, entry 6), the tracer-level 19F-approach was transferred to 18F-radiosynthesis (Scheme 2). The radiosynthesis was performed by the 2-step protocol. Applying the optimized conditions, [18F]MDL100907 was obtained in 19–22% isolated RCY after HPLC purification (n = 3). The radiochemical purity was >99%, and the Am was 38.8 – 53.6 GBq/μmol (1.05–1.450 Ci/μmol) at the end of synthesis (Figure S2). The synthesis time was approximately 70 min. The identity of [18F]7 was confirmed by analytical HPLC. The residual Cu content in the final formulation was analyzed. It was found to be ≤ 1 ppm, well below the concentration limit of copper for parenteral drug products under both US Pharmacopeia and ICH guidelines.32 Compared with the reported yield of 3%,26 this optimized radiosynthesis offers significantly higher RCY. More importantly, the entirely optimization process was performed under natural 19F-conditions.
Scheme 2.

Radiosynthesis of [18F]MDL100907 ([18F]7).
The 19F-process provides high-throughput capability, allowing many conditions to be tested and analyzed within a 24-hour period. In our lab, a batch of 10 reactions has been performed during the day, and the LC-MS/MS analysis was completed overnight through the auto-sampling process. A generic LC method was used for both fallypride and MDL100907 (Figure S3), which can be easily adopted to other tracers without further modification. The efficient process of optimizing radiochemical reaction can be used to significantly improve existing methods of radiosynthesis. In addition, for both tracers evaluated in this study, the 19F-approach provided consistent data, which was reproduced with similar yield in radiosynthesis. The 18F-reaction is only necessary to validate the optimized condition from the 19F-approach, which will greatly reduce the radiation exposure for the chemists. A major benefit of this approach is that it provides techniques that can be used by medicinal or synthetic chemists to discover new methods of preparing PET probes with no need to handle radioactivity.
3. Experimental
3.1. General
Unless otherwise noted, all chemicals and solvents were purchased from Sigma-Aldrich (Milwaukee, WI, USA) or Fisher Scientific (Hanover Park, IL, USA) and used without further purification. Fallypride and tosyl-fallypride were purchased from ABX GmbH (Radeberg, Germany). (2,3-dimethoxyphenyl)(piperidin-4-yl)methanol was purchased from Aurum Pharmatech LLC (Howell, NJ, USA). (4-(2-bromoethyl)phenyl)-boronic acid was purchased from Combi-Blocks, Inc (San Diego, CA, USA). Non-carrier added [18F]fluoride was obtained from the National Institutes of Health cyclotron facility (Bethesda, MD, USA). Glass reaction vials (catalog: 352016) were obtained from Biotage (Charlotte, NC, USA). Ultra-pure water was produced with the Milli-Q® Integral aater purification system (Billerica, MA, USA). The residual copper concentration in final dose was analyzed by Robertson Microlit Laboratories (Ledgewood, NJ, USA).
Flash chromatography was performed on a Biotage Isolera™ flash system (Charlotte, NC, USA). NMR was recorded on a Bruker 400 MHz spectrometer (Billerica, MA, USA). LC-MS/MS analysis was performed on Agilent 6460C triple quadrupole mass spectrometry with an ESI source. The LC inlet was Agilent 1200 series chromatographic system equipped with 1260 binary pump, 1290 thermostatted column compartment and 1260 high performance autosampler. Instrument control and data processing were performed using Aglient’s MassHunter Software. Semi-preparative and analytical high-pressure liquid chromatography (HPLC) were conducted on the Agilent 1260 HPLC system equipped with multi-wavelength UV detector and radiation detector. All RCY were determined by HPLC analysis of the crude product, unless stated otherwise.
3.2. Chemical synthesis
(R)-(2,3-dimethoxyphenyl)(1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenethyl)piperidin-4-yl)methanol (5)
4-(2-Bromoethyl)phenylboronic acid 4 (0.91 g, 3.98 mmol) was added to a solution of (2,3-dimethoxyphenyl)(piperidine-4-yl)methanol 3 (1.00 g, 3.98 mmol), NaHCO3 (0.50 g, 5.97 mmol) and molecular sieves (5 – 6 g) in DMF (23 mL). The reaction solution was stirred at 85 °C for 90 min. The reaction was monitored via LC-MS. Pinacol (0.470 g, 3.98 mmol) was added to the solution and stirred at 85 °C for 20 min. The solution was filtered through celite and the solvent was removed under reduced pressure. The resulting oil was purified using a Biotage ACI system on a 25g KP-Sil SNAP cartridge (450 ml 25% EtOAc in hexanes then 300 mL 1% triethylamine in EtOAc) yielding a light yellow oil (900 mg, 47%). 1H NMR (400MHz, CDCl3): δ 7.71 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 7.9 Hz, 2H), 7.04 (t, J = 7.9 Hz, 1H), 6.89 (dd, J = 7.9, 1.5 Hz, 1H), 6.84 (dd, J = 8.1, 1.5 Hz, 1H), 4.62 (d, J = 8.1 Hz, 1H), 3.86 (s, 6H), 3.07 (d, J = 11.4 Hz, 1H), 2.92 (d, J = 11.1 Hz, 1H), 2.83 – 2.77 (m, 2H), 2.57 – 2.51 (m, 2H), 2.11 – 2.04 (m, 1H), 2.01 – 1.85 (m, 2H), 1.73 – 1.61 (m, 1H), 1.33 (s, 16H). 13C NMR (101 MHz, CDCl3) δ 152.47, 146.51, 143.93, 136.39, 134.90, 128.14, 123.97, 119.70, 111.41, 99.99, 83.66, 75.02, 74.54, 60.89, 60.58, 55.71, 53.65, 42.84, 33.84, 28.79, 28.73, 24.86. HRMS (ESI): Calcd for C28H40BNO5 (M+H)+ 482.3072, found 482.3079.
(R)-tert-butyl ((2,3-dimethoxyphenyl)(1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenethyl) piperidin-4-yl)methyl) carbonate (6)
Di-tert-butyl dicarbonate (160 mg, 0.734 mmol) was added to a solution of compound 5 (334 mg, 0.668 mmol) and 4-(dimethylamino)pyridine (24.4 mg, 0.2 mmol) in pyridine (5 mL). The resulting mixture was stirred at 60 °C for 30 min. The volatile was removed under reduced pressure. The remaining oil was purified on a Biotage flash system on a 25 KP-Sil SNAP cartridge (10 – 25% EtOAc in hexanes) yielding product as a yellow foam (85 mg, 38%) and recovered unreacted starting material (150 mg). 1H NMR (400MHz, CDCl3): δ 7.71 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.03 (t, J =8.0 Hz, 1H), 6.92 (dd, J = 7.9, 1.6 Hz, 1H), 6.83 (dd, J = 8.1, 1.5 Hz, 1H), 5.73 (d, J = 7.9 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.02 (d, J = 10.6 Hz, 1H), 2.91 (d, J = 11.4 Hz, 1H), 2.83 – 2.72 (m, 2H), 2.57 – 2.47 (m, 2H), 1.99 – 1.83 (m, 3H), 1.77 (m, 1H), 1.47 (d, J = 6.5 Hz, 4H), 1.42 (s, 9H), 1.32 (d, J = 8.0 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ 153.10, 152.39, 146.46, 143.90, 134.90, 133.32, 128.14, 123.99, 118.77, 111.45, 83.65, 81.75, 76.89, 75.02, 60.60, 60.52, 55.63, 53.51, 53.35, 41.31, 33.81, 28.29, 27.82, 24.86. HRMS (ESI): Calcd for C33H48BNO7 (M+H)+ 582.3596, found 582.3599.
3.3. Instrumental configuration
Chromatographic condition
Chromatographic separations were carried out with gradient elution on an Agilent RRHT Zorbax SB-C18 column (2.1×50mm, 1.8 μm) or Waters Acquity UPLC BEH-C18 (2.1x50 mm, 1.7 μm). The mobile phase was composed of water (A) and acetonitrile (B) each containing 0.1 % formic acid. The analytes were eluted from the column by a linear gradient which started at 5% B, held at initial conditions for 0.30 min, then increased from 5 to 95% B within 2.95 min and held at 95% B for 1.0 min then returned to the initial conditions. The total run time was 6.65 min. The flow rate was set at 0.6 mL/min. The column oven was kept at 40 °C throughout the analysis. The injection volume was 1 μL and the autosampler rack temperature was 20 °C.
UV quantitation of fallypride
A calibration curve in the range of 200 – 8,000 ng/mL (ppb) was prepared for fallypride. The curve was fitted by least square linear regression using the peak areas. The acceptable criterion for the calibration curve was a correlation coefficient (r2) of 0.98 or better, and that each back-calculated standard concentration must be within 20% deviation from the nominal value. The S/N ratio of the lowest calibration level was greater than 10. The limit of detection (LOD) was estimated as 3 × SD for the lowest calibrant. LOQ was calculated as 3 × LOD. A LOD value of 62.8 ng/mL was calculated, and LOQ was 188 ng/mL.
Mass spectrometric conditions
Mass spectrometric data were acquired in positive ion mode with the following ESI-MS parameters: gas temperature 350 °C; gas flow 13 L/min; nebulizer 60 psi; capillary voltage 3000 V. Nitrogen was used as desolvation gas and collision gas, dwell time were set at 75 ms (MDL100907/Verapamil) and 80 ms (fallypride/verapamil) for each transition. Cell accelerator voltage was set to 2 for fallypride/verapamil and 6 for MDL100907/verapamil. Quantification was done using MRM mode. The product ion giving the highest response was used for quantitation (quan). The qualifier ion (qual) was the next highest responding ion. The ratio between the quan ion and the qual ion was used, along with retention time, to confirm the identity of the analyte in the sample. The precursor ions and MS/MS parameters for each compound are displayed in Table 3. Yields of natural 19F-reactions were determined by LC-MS/MS analysis. Briefly, a calibration curve in the range of 2 – 12,000 ng/mL (ppb) was prepared for the standard compounds (fallypride and MDL100907) containing an internal standard (verapamil). The curves were fitted by a weighted (1/x or 1/x2) least squares linear regression method by measuring the peak area ratio of the analyte to the internal standard. The acceptable criterion for a calibration curve was a correlation coefficient (r2) of 0.98 or better, and that each back-calculated standard concentration must be within 20% deviation from the nominal value. S/N of the lowest calibration level was greater than 10. All calibrators and samples were run in triplicate. LOD was estimated as 3 × SD for the lowest calibrant. LOQ was calculated as 3 × LOD. A LOD value of 0.32 ng/mL was calculated, and LOQ was 0.98 ng/mL.
Table 3.
Mass Spec conditions for Fallypride, MDL100907 and Verapamil
| Fallypride | MDL100907 | Verapamil | |
|---|---|---|---|
| MW | 364.46 | 373.47 | 454.61 |
| MRM transition (quan) | 365.5 → 225.0 | 374.2 → 123.1 | 455.3 → 165.1 |
| Collision Energy (quan) | 88 | 36 | 20 |
| MRM transition (qual) | 365.2 → 124.1 | 374.2 → 103.1 | 455.3 → 150.0 |
| Collision Energy (qual) | 24 | 60 | 44 |
| Fragmentor | 154 | 134 | 162 |
3.4. General method for fallypride optimization under 19F-condition
For K2CO3/KHCO3 method: to a 2-mL microwave vial was added KF (0.15 μg, 2.59 nmol) in 15 μL water, K2CO3 or KHCO3 (selected amount), K222 (7–8 mg), and 1 mL of acetonitrile. For tetrabutylammonium hydrogen carbonate (TBAB) method: KF (0.15 μg, 2.59 nmol) in 15 μL of water, TBAB (0.18 mL, 0.075 M), and 1 mL of acetonitrile was added to the reaction vial. The solution was azeotropically dried under vacuum and nitrogen flow at 100 ˚C. To the dried mixture, acetonitrile (1.0 mL) was added. The mixture was further azeotropically dried under nitrogen and vacuum at 100 °C. After the drying sequence was completed, the tosyl-precursor in acetonitrile (1.0 mL) was added. The reaction vial was stirred at elevated temperature for selected time and cooled to 50 °C. The yield was analyzed via LC-MS/MS quantification. The MS sample was prepared by diluting 10 μL of the reaction solution with 80 μL of acetonitrile and 10 μL of verapamil (internal standard, 166 ng/mL).
3.5. Radiosynthesis of [18F]fallypride
A sample of 0.42 GBq [18F]/fluoride (11.3 mCi) in water (50–100 μL) was added to a reaction vessel containing selected amount of base and Kryptofix 2.2.2 (7 – 8 mg) in acetonitrile (1.0 mL). The mixture was azeotropically dried under nitrogen and vacuum at 100 °C. To the dried mixture, acetonitrile (1.0 mL) was added. The mixture was further azeotropically dried under nitrogen and vacuum at 100 °C. After the drying sequence was completed, the tosyl-precursor (2 mg) in acetonitrile (1.0 mL) was added. The reaction vial was stirred at elevated temperature for selected time and cooled to 50 °C. The reaction mixture was quenched with 1 mL H2O (with 0.1% triethylamine) and transferred to the HPLC for purification. HPLC conditions: Agilent XDB C18, 250 × 10 mm, 5 μ; mobile phase: 50% acetonitrile and 50% water (with 0.1% triethylamine) at flow rate 4 mL/min. The labeled product was eluted between 15–16 min, which was collected in a collection flask. The chemical identity of the product was checked by analytical HPLC. HPLC conditions: Agilent XDB C18, 150 × 4.6 mm, 5 μ; mobile phase: 60% acetonitrile and 40% H2O (with 0.1% triethylamine) at flow rate 1 mL/min. The total synthesis time was approximately 70–75 min.
3.6. General method for MDL100907 optimization under 19F-condition
To a 2-mL microwave vial was added KF (0.15 μg, 2.59 nmol) in 15 μL water, K2CO3 or KHCO3 (as in table) and K222 in acetonitrile (1.0 mL). The solution was azeotropically dried under vacuum and nitrogen flow at 110 °C. The azeotropic drying process was repeated by adding acetonitrile (1 mL × 3). In another vial, the boronic ester precursor was mixed with copper (II) catalyst in DMF and vortexed for 5 s. The mixture was added to the reaction vial containing the dried F−, and the resulting mixture was heated at elevated temperature for a selected time. TFA (200 μL) was added to the reaction vial, which was heated at 110 ˚C for 20 min for hydrolysis. NaOH (20%, 0.5 mL) was added to the reaction vial to quench the reaction. The yield was analyzed via LC/MS-MS quantification. The MS sample was prepared by diluting 10 μL of the neutralized solution with 80 μL of acetonitrile and 10 μL of verapamil (internal standard, 100 ng/mL).
3.7. Radiosynthesis of [18F]MDL100907 ([18F]7)
A sample of 0.54 GBq [18F]/fluoride (14.5 mCi) in water (50–100 μL) was added to a reaction vessel containing K2CO3 (0.060 mg) and K222 (0.28 mg) in water (0.2 mL) and acetonitrile (1.0 mL). The solution was azeotropically dried under vacuum and nitrogen flow at 110 ˚C. The azeotropic drying process was repeated by adding acetonitrile (1 mL × 3). In another vial, the boronic ester precursor, 6 (5 mg) was mixed with Cu(OTf)2(py)4 (2.3 mg) in DMF (0.3 mL) and vortexed for 5 s. The mixture was added to the reaction vial containing the dried activity, and the resulting mixture was heated on a microwave reactor at 110 °C for 20 min. TFA (200 μL) was added to the reaction vial, which was heated at 110 °C for 5 min for hydrolysis. NaOH (20%, 0.75 mL) was added to the reaction vial to adjust the pH to basic, the product was purified by semi-preparative HPLC. Conditions: Phenomenex Luna C18 (2) column, 250 × 10 mm, 5 μ; mobile phase: 50% acetonitrile and 50% water (with 0.1% triethylamine) at flow rate 4 mL/min. The labeled product was eluted between 21–22 min. The chemical identity of the product was checked by analytical HPLC. HPLC conditions: Phenomenex Luna C18 (2) column, 100 × 4.6 mm, 5 μ; mobile phase: 50% acetonitrile and 50% H2O (with 0.1% triethylamine) at flow rate 1 mL/min. The labeled product was eluted between 6–7 min. The total synthesis time was approximately 70 min.
4. Conclusions
A reliable 19F-approach to simulate 18F-radiosynthesis under similar molar activity level has been developed. Using this strategy, the radiosynthesis of [18F]MDL100907 has been improved with 19–22% RCY. With high-throughput ability and decreased radiation exposure, the tracer-level 19F-approach will find great potential in 18F-tracer development process. Besides fluorine-18, this technique can be applied on all imaging radionuclide, which will accelerate future tracer development process.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH. Intramural research funds for the Imaging Probe Development Center were administered by the National Heart, Lung, and Blood Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We thank Dr. Carolyn Woodroofe for her assistance in proofreading.
Abbreviations
- 18F
fluorine-18
- 19F
fluorine-19
- LC-MS/MS
liquid chromatography/tandem mass spectrometry
- PET
positron emission tomography
- Am
molar activity
- LOD
limit of detection
- LOQ
limit of quantification
- MRM
multiple reaction monitoring
- NCA
no-carrier-added
- RCY
radiochemical yield
- S/N
signal-to-noise
- TBAB
tetrabutylammonium hydrogen carbonate
- TIC
total ion chromatogram
References
- 1.Herschman HR. Molecular imaging: looking at problems, seeing solutions. Science. 2003;302:605–608. doi: 10.1126/science.1090585. [DOI] [PubMed] [Google Scholar]
- 2.Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr Opin Chem Biol. 2010;14:71–79. doi: 10.1016/j.cbpa.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 3.Hagooly A, Rossin R, Welch MJ. Small molecule receptors as imaging targets. Handb Exp Pharmacol. 2008:93–129. doi: 10.1007/978-3-540-77496-9_5. [DOI] [PubMed] [Google Scholar]
- 4.Elsinga PH, Dierckx RA. Small molecule PET-radiopharmaceuticals. Curr Pharm Des. 2014;20:2268–2274. doi: 10.2174/13816128113196660661. [DOI] [PubMed] [Google Scholar]
- 5.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of C-11, F-18, O-15, and N-13 Radiolabels for Positron Emission Tomography. Angew Chem Int Ed. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 6.Bégué J-P, Bonnet-Delpon D. Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products. J Fluorine Chem. 2006;127:992–1012. [Google Scholar]
- 7.Kirk KL. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J Fluorine Chem. 2006;127:1013–1029. [Google Scholar]
- 8.Levy S, Livni E, Elmaleh D, Curatolo W. Direct displacement with anhydrous fluoride of the C-2 trifluoromethanesulphonate of methyl 4,6-O-benzylidene-3-O-methyl-2-O-trifluoromethylsulphonyl-β-D-mannopyranoside. J Chem Soc Chem Commun. 1982;0:972–973. [Google Scholar]
- 9.Brown LJ, Ma N, Bouvet DR, Champion S, Gibson AM, Hu Y, et al. Synthesis of the positron-emitting radiotracer [18F]-2-fluoro-2-deoxy-d-glucose from resin-bound perfluoroalkylsulfonates. Org Biomol Chem. 2009;7:564–575. doi: 10.1039/b816032e. [DOI] [PubMed] [Google Scholar]
- 10.Bhalla R, Levason W, Luthra SK, McRobbie G, Sanderson G, Reid G. Radiofluorination of a pre-formed gallium(III) aza-macrocyclic complex: towards next-generation positron emission tomography (PET) imaging agents. Chem Eur J. 2015;21:4688–4694. doi: 10.1002/chem.201405812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Liu W, Hooker JM, Groves JT. Targeted fluorination with the fluoride ion by manganese-catalyzed decarboxylation. Angew Chem Int Ed. 2015;54:5241–5245. doi: 10.1002/anie.201500399. [DOI] [PubMed] [Google Scholar]
- 12.Björkhem I. Selective ion monitoring in clinical chemistry. CRC Crit Rev Clin Lab Sci. 1979;1:53–105. doi: 10.3109/10408367909105854. [DOI] [PubMed] [Google Scholar]
- 13.Axen U, Gréen K, Hörlin D, Samuelsson B. Mass spectrometric determination of picomole amounts of prostaglandins E2 and F2α using synthetic deuterium labeled carriers. Biochem Biophys Res Commun. 1971;45:519–525. doi: 10.1016/0006-291x(71)90850-3. [DOI] [PubMed] [Google Scholar]
- 14.Joshi EM, Need A, Schaus J, Chen Z, Benesh D, Mitch C, et al. Efficiency gains in tracer identification for nuclear imaging: can in vivo LC-MS/MS evaluation of small molecules screen for successful PET tracers? ACS Chem Neurosci. 2014;5:1154–1163. doi: 10.1021/cn500073j. [DOI] [PubMed] [Google Scholar]
- 15.Di L, Kerns EH. Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization. 2. Elsevier Inc; 2016. [Google Scholar]
- 16.Xiao JF, Zhou B, Ressom HW. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends Analyt Chem. 2012;32:1–14. doi: 10.1016/j.trac.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calderón-Santiago M, Priego-Capote F, Luque de Castro MD. Enhanced Detection and Identification in Metabolomics by Use of LC–MS/MS Untargeted Analysis in Combination with Gas-Phase Fractionation. Anal Chem. 2014;86:7558–7565. doi: 10.1021/ac501353n. [DOI] [PubMed] [Google Scholar]
- 18.Grebe SK, Singh RJ. LC-MS/MS in the Clinical Laboratory – Where to From Here? Clin Biochem Rev. 2011;32:5–31. [PMC free article] [PubMed] [Google Scholar]
- 19.Diagne AB, Li S, Perkowski GA, Mrksich M, Thomson RJ. SAMDI Mass Spectrometry-Enabled High-Throughput Optimization of a Traceless Petasis Reaction. ACS Comb Sci. 2015;17:658–662. doi: 10.1021/acscombsci.5b00131. [DOI] [PubMed] [Google Scholar]
- 20.Bellomo A, Celebi-Olcum N, Bu X, Rivera N, Ruck RT, Welch CJ, et al. Rapid Catalyst Identification for the Synthesis of the Pyrimidinone Core of HIV Integrase Inhibitors. Angew Chem Int Ed. 2012;51:6912–6915. doi: 10.1002/anie.201201720. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee J, Yang ZY, Das MK, Brown T. Fluorinated benzamide neuroleptics--III. Development of (S)-N-[(1-allyl-2-pyrrolidinyl)methyl]-5-(3-[18F]fluoropropyl)-2, 3-dimethoxybenzamide as an improved dopamine D-2 receptor tracer. Nucl Med Biol. 1995;22:283–296. doi: 10.1016/0969-8051(94)00117-3. [DOI] [PubMed] [Google Scholar]
- 22.Elsinga PH, Hatano K, Ishiwata K. PET tracers for imaging of the dopaminergic system. Curr Med Chem. 2006;13:2139–2153. doi: 10.2174/092986706777935258. [DOI] [PubMed] [Google Scholar]
- 23.Christian BT, Vandehey NT, Fox AS, Murali D, Oakes TR, Converse AK, et al. The distribution of D2/D3 receptor binding in the adolescent rhesus monkey using small animal PET imaging. Neuroimage. 2009;44:1334–1344. doi: 10.1016/j.neuroimage.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon BS, Park JH, Lee HJ, Kim JS, Kil HS, Lee BS, et al. Highly efficient production of [18F]fallypride using small amounts of base concentration. Appl Radiat Isot. 2010;68:2279–2284. doi: 10.1016/j.apradiso.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Mathis CA, Mahmood K, Huang Y, Simpson NR, Gerdes JM, Price JC. Synthesis and preliminary in vivo evaluation of [C-11]MDL 100907: A potent and selective radioligand for the 5-HT2A receptor system. Med Chem Res. 1996;6:1–10. [Google Scholar]
- 26.Ren H, Wey HY, Strebl M, Neelamegam R, Ritter T, Hooker JM. Synthesis and Imaging Validation of [F-18]MDL100907 Enabled by Ni-Mediated Fluorination. ACS Chem Neurosci. 2014;5:611–615. doi: 10.1021/cn500078e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mühlhausen U, Ermert J, Herth MM, Coenen HH. Synthesis, radiofluorination and first evaluation of (±)-[18F]MDL100907 as serotonin 5-HT2A receptor antagonist for PET. J Labelled Compd Radiopharm. 2009;52:6–12. [Google Scholar]
- 28.Tredwell M, Preshlock SM, Taylor NJ, Gruber S, Huiban M, Passchier J, et al. A general copper-mediated nucleophilic 18F fluorination of arenes. Angew Chem Int Ed. 2014;53:7751–7755. doi: 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]
- 29.Zlatopolskiy BD, Zischler J, Krapf P, Zarrad F, Urusova EA, Kordys E, et al. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem Eur J. 2015;21:5972–5979. doi: 10.1002/chem.201405586. [DOI] [PubMed] [Google Scholar]
- 30.Mossine AV, Brooks AF, Makaravage KJ, Miller JM, Ichiishi N, Sanford MS, et al. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org Lett. 2015;17:5780–5783. doi: 10.1021/acs.orglett.5b02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preshlock S, Calderwood S, Verhoog S, Tredwell M, Huiban M, Hienzsch A, et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem Commun. 2016;52:8361–8364. doi: 10.1039/c6cc03295h. [DOI] [PubMed] [Google Scholar]
- 32.Copper is an elemental impurity with a residual concentration limit of 30 ppm. Guideline for Elemental Impurities Q3D. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2014; United States Pharmacopeia, National Formulary; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.