

Abstract
Introduction
In looking for novel non-amyloid-based etiologies for Alzheimer’s disease we explore the hypothesis that age-related myelin loss is an attractive explanation for age-associated cognitive decline and dementia.
Methods
We performed a meta-analysis of data in the NACC database accompanied by quantitative histopathology of myelin and oligodendrocytes in frontal cortices of 24 clinically characterized individuals. Pathological findings were further validated in an AD mouse model and in culture.
Results
Myelin lesions increased with cognitive impairment in an amyloid-independent fashion with signs of degeneration appearing prior to neuronal loss. Myelinating oligodendrocytes in the gray matter showed greater vulnerability than those in white matter, and the degenerative changes correlated with evidence of DNA damage. Similar results were found in myelinating oligodendrocyte cultures where DNA damage caused aberrant oligodendrocyte cell cycle reentry and death.
Discussion
We present the first comprehensive analysis of the cell biology of early myelin loss in sporadic Alzheimer’s disease.
1. Introduction
Sporadic Alzheimer’s disease (AD) is a dementing illness that is associated with the appearance of β-amyloid peptide deposits, phosphorylated tau protein aggregates, synaptic loss and neuroinflammation [1]. To offer an explanation of this complex condition, it has been proposed that the amyloid abnormalities drive a cascade of events that leads to the other pathological features [2]. Indeed, CSF amyloid changes are found a full 15–20 years before the onset of dementia, appearing as early as the mid-fifties [3]. Yet while the amyloid cascade hypothesis has become the dominant AD model, the recent failures of clinical trials based on this model have stimulated a search for alternative hypothesis for this multifactorial disease in additional to the amyloid cascade [4, 5]. Observations, primarily from magnetic resonance imaging (MRI), have suggested that loss of myelin represents one such alternative.
The natural history of myelination across the human lifespan closely tracks cognitive capacity [6, 7]. Cortical myelin, in particular in frontal lobes, does not fully mature in humans until their late-thirties [8, 9]. Afterwards myelination regresses and this naturally occurring age-related loss is accelerated in AD [10]. While myelin is associated mostly with white matter, myelination of axons begins and ends in gray matter. This is significant as gray matter (intracortical) myelin is the brain feature that is most highly correlated with decline in cognitive performance in healthy humans as they age [6, 11–13]. Indeed, it is thought that the gray matter portions of myelinated axons are the main determinants of axon conduction velocity [14]. Myelin loss, like tangles of phosphorylated tau, is not likely to be AD-specific as it occurs in a wide spectrum of amyloid-free neurological diseases. For example in multiple sclerosis (MS), patients with severe cortical demyelination have dementia symptoms that highly resemble those of AD, despite the virtual absence of amyloid plaques and tangles [15, 16]. Myelin loss is therefore a strong correlate of cognitive decline in the aging brain, and as such deserves our attention as a contributor to the etiology of AD [17].
A second attractive candidate as an age-associated contributor to the Alzheimer’s disease process is the loss of genomic integrity. DNA damage may be one of the most powerful drivers of aging, particularly in brain [18–20]. Human mutations in DNA repair genes (e.g., Cockayne syndrome) virtually all cause premature aging. Like neurons, oligodendrocytes (OLs) accumulate DNA damage with age and in DNA repair mutants, myelin abnormalities appear as part of the premature aging syndrome [21]. We recently proposed that the accumulation of unrepaired DNA drives age-related losses of OLs and myelin and that this process potentially contributes to the loss of cognition in AD and other dementias [17]. One of the ways in which DNA damage exerts its influence is through the process of cell cycle regulation. Our laboratory and others have shown that DNA damage in postmitotic neurons can drive aberrant cell cycle re-entry and that in AD this is followed by neuronal degeneration [22–25]. A similar involvement of ectopic neuronal cell cycling in neurodegeneration is found conditions such as ataxia-telangiectasia (A–T) [26, 27], and others [28]. As mature OLs are also permanently postmitotic [29], we set out to ask whether DNA damage in these cells might have the same effects.
Earlier studies suggested that myelin degradation in sporadic AD or its mouse models is the direct consequence of amyloid pathology [10, 30, 31], but these findings are difficult to reconcile with the timing of myelin loss in normal aging cerebral cortex, which is detected as early as the mid-forties [6, 8, 32]. Our central hypothesis is that myelin degradation is one of the earliest structural changes in the sporadic AD brain and is driven by age-associated DNA damage, independent of amyloid plaque formation. Here, our findings from MRI meta-analysis, histopathological observations of human autopsy material, longitudinal studies of mouse AD models as well as oligodendrocyte cell culture not only support this hypothesis, but also offer an alternative explanation and pathway to sporadic AD in addition to and beyond the amyloid cascade.
2. Method and materials
2.1 NACC-MRI datasets
A cross-sectional dataset of a longitudinal study cohort was obtained from the National Alzheimer’s Coordinating Center (NACC). This is a database with standardized research data from 39 Alzheimer’s Centers in the United States [33] (http://www.alz.washington.edu/). A summary dataset of MRI scans of 507 subjects from one of the centers was analysed. The analysis protocol is summarized in supplementary methods and the demographic details of the subjects are listed in Table S1.
2.2 Human brain tissue
Postmortem brain tissue was obtained from the National Institutes of Health NeuroBioBank at The University of Maryland and the Human Brain and Spinal Fluid Resource Center, CA with approvals from Tissue Access Committee of NeuroBioBank and The Hong Kong University of Science and Technology (HKUST) human research committee. Subjects with amyloid plaque-free dementia (DEM), Alzheimer’s disease (AD) and age-matched normal controls (NC) were requested; any cases with Parkinsonian disorder or depression were excluded. A total of 24 subjects meeting the search criteria was obtained. All tissues were from left frontal cortex that had been frozen without prior fixation and stored at −80°C. The demographics and neuropathological characteristics of the subjects are listed in Table S2. TissueTek medium-embedded frontal cortices were cryosectioned at 16 μm and stored at −80°C until use.
2.3 Animals
A colony of R1.40 transgenic mice (B6.129-Tg(APPSw)40Btla/Mmjax) was established from mice originally purchased from The Jackson Laboratory. The mice carry a 650 kb insert from a yeast artificial chromosome clone that contains the full genomic copy of the human APP gene with the familial AD Swedish mutation (K670M/N671L) [34]. Our R1.40 colony is maintained by backcrossing hemizygous animals to wild type mice of the C57BL/6J genetic background. Hemizygote mice were used for the reported studies. All animal experiments were conducted with license from the Government of Hong Kong SAR, and were approved by the Animal Ethics Committee of HKUST. Mice were maintained in the Animal and Plant Care Facility (APCF) at HKUST and all procedures comply with the guidelines of the institution and the Department of Health, Government of Hong Kong.
2.4 Histopathology and immunohistochemistry of tissue sections
Mice were deeply anesthetized by Avertin (1.25% tribromoethanol, Sigma, 375 mg/kg, I.P.) and sacrificed using transcardial saline perfusion delivered with a peristaltic pump. The right brain was isolated by dissection and post-fixed in paraformaldehyde (4%) for 24 hours. The tissue was then cryoprotected in sucrose (30% w/v) for 48 hours before embedding for cryosectioning. Sagittal 10 μm sections were cut beginning at the midline and kept at −80° C until use. For human tissue, the frozen sections described above were fixed for 30 mins in 4% paraformaldehyde at room temperature.
Visualization of fine intracortical myelin fibers in frozen sections was achieved by staining with Blackgold II compound according to the manufacturer’s instructions; cell nuclei were counterstained with cresyl violet (0.2% w/v – HistoChem Inc.).
For immunohistochemistry (IHC), all tissues were washed with PBS, then incubated in blocking solution consisting of PBS with normal donkey serum (10%) plus Triton X-100 (0.3%) for 1 h at room temperature. Tissues were next incubated with specific primary antibodies overnight at 4° C. After washing in PBS, the specimens were incubated with the appropriate secondary antibodies conjugated with Alexa Fluor 488, 555 or 647 fluorescent dye (Life Technologies), for 1 h at room temperature. Nuclei were counterstained with DAPI. All sections were mounted under glass coverslips using Hydromount (National Diagnostics). The primary antibodies used for immunohistochemistry are listed in Table S3. All tissue sections were examined and imaged on a fluorescent microscope (BX53 with DP80 camera, Olympus) equipped with 20× (UPlanSApo, 0.75 N.A.) and 40× objectives (UPlanSApo, 0.95 N.A., all Olympus) using an X-Cite® 120Q light source (Excelitas Tech Corp). The procedures for quantification of the images are detailed in supplementary methods.
2.5 Oligodendrocyte cell culture
Cultures of mouse OPC were obtained by following protocols described previously [35, 36] with minor modifications (see Supplementary Methods) After seven days in vitro, the expanded OPCs were collected and plated on coated 12 mm glass coverslips (35,000 cells each) for differentiation. To differentiate OPC into mature OL (mOL), the cells were cultured for a further 14 days in OPC growth media without PDGF or CNTF and a reduced FBS concentration (0.1%), but with the addition of triiodothyronine (T3 – 34 ng/mL).
2.6 Immunocytochemistry of cultured cells
Oligodendrocytes were cultured on coverslips in 24-well tissue culture plates. After rinsing with PBS, cells were fixed in paraformaldehyde (4%) for 20 min. The cells were permeabilized and blocked by incubation with PBS containing normal donkey serum (5%) plus 0.3% triton X-100 for 30 min. The cells were incubated with primary antibodies for 2 h at room temperature. The coverslip was then stained with the appropriate secondary antibody, mounted and examined as described above. Coverslips incubated without primary antibody served as negative controls. For high resolution imaging, specimens were visualized on a Leica TCS SP8 confocal laser scanning platform equipped with Leica HyD hybrid detector and visualized through a HC PL APO CS2 63x/1.40 N.A Oil lens. In each experiment, sections were imaged at a preset laser power with steps of 0.3–0.5 μm per optical slice. The maximum z-projection of the optical stacks are presented.
2.7 Western blots
The left neocortex of each animal was collected for protein extraction after saline perfusion and disection. Tissue was homogenized in RIPA buffer supplemented with protease and phosphatase inhibitors (Roche Diagnostic). The protein concentration of the cortical lysate was measured and equal amounts (20 – 50 μg per lane) were loaded onto 10 – 15% SDS-polyacrylamide gels for electrophoresis. Proteins were transferred onto PVDF membranes (Bio-rad) for antibody staining. Membranes were incubated with primary antibodies after blocking with non-fat milk (Table S3), and probed with the appropriate horseradish peroxidase-linked secondary immunoglobulin (Cell signalling). GAPDH served as a loading control. The immunoblots were visualized by chemiluminescent substrates (SuperSignal™ West Pico or West Femto Substrates, Thermo Scientific) and signals were detected using medical blue sensitive X-ray films. The films were digitalized and the protein intensities were quantified using Image J software.
2.8 Gene expression analysis
Messenger RNA extracted from the human frontal cortex or mouse neocortex was analysed using SYBR-green based real-time PCR reactions. For human frontal cortex, the total RNA from the cortical gray matter (GM) and subcortical white matter (WM) were isolated by microdissection under light microscope. The procedures of extraction, reverse transcription and PCR reactions are summarized in Supplementary Methods. The primers used in this study are listed in Supplementary Table S4.
2.9 Transmission electron microscopy (TEM)
TEM preparation was performed as described earlier [37] and the detail procedure is described in Supplementary Methods. All specimen were visualized by JEM 100CXII (JEOL) at 100kV. G-ratio analysis was performed as described previously [38].
2.10 Statistical analysis
All data are presented as means ± SEM from experiments repeated at least three times. Comparisons between pairs were made by unpaired t-test. Comparisons between multiple groups were made using one-way analysis of variance with Newman-Keuls post hoc tests. Correlations between observations were examined by Pearson’s test. All statistics were computed using GraphPad Prism software version 5.0 (GraphPad Software Inc.). Statistical significance was taken as P < 0.05.
3. Results
3.1 Myelin lesions increase with age and cognitive decline
We queried a cross-sectional MRI-dataset from NACC (the NIH National Alzheimer’s Coordinating Center). As expected, our analysis found that the volume of total gray matter and of hippocampus were significantly correlated with cognitive decline as graded by MMSE scores (Fig. 1A(i–ii)). When stratified into normal control (NC), mild cognitive impairment (MCI) and dementia (DEM) groups (Table S1), the gray matter (GM) and hippocampal volume also declined with age (Fig. 1A(v–vi)). Unlike previous reports, total white matter (WM) volume was reduced with normal aging, but was not significantly correlated with MMSE score (Fig 1A(iii, vii)). Possible reasons for this are discussed below. We did, however, find a significant association between WM hyperintensities (WMH) and cognitive decline (Fig 1A(iv)) and aging (Fig 1A(viii)) in all groups. As WMH include frank myelin lesions as well as vascular anomalies [39–42], the correlation between myelin injury, cognitive decline and aging is affirmed.
Fig. 1. Myelin injury and oligodendrocyte degeneration are associated with cognitive decline, but independent of amyloid plaques.
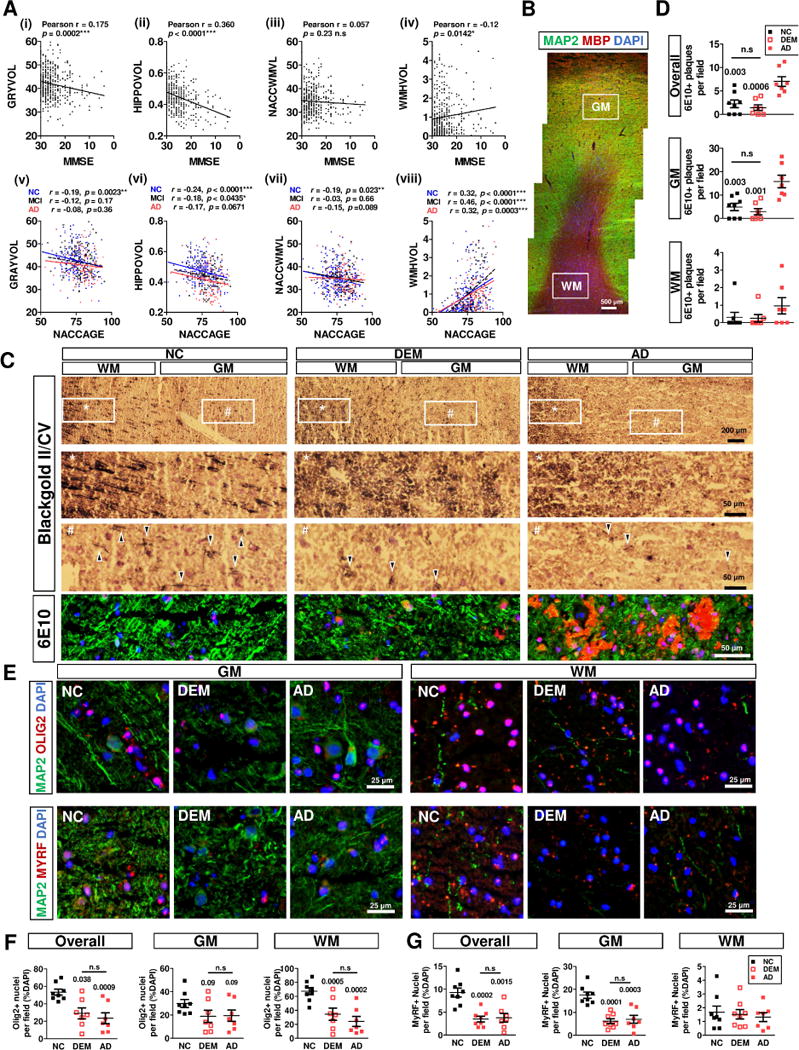
A The summarized MRI data of GM (GRAYVOL), Hippocampus (HIPPOVOL), Total WM (NACCWMVL) and WM hyperintensity (WMHVOL) were correlated with cognition graded by MMSE (i–iv) and age (v–viii) by Pearson test respectively. B Frontal cortex specimen with distinct cortical gray matter (GM, MAP2, Green) and subcortical white matter (WM, MBP, Red). C upper panel The myelin in the subcortical WM (*) were comparable among groups, but the extent of intracortical myelin fibers (Blackgold II/Cresyl violet, arrowheads) were similar reduced in DEM and AD cortices (#).C lower panel Amyloid plaques density (6E10, red) is significantly higher in the AD group, compared with DEM and age-matched NC subjects (g, h, i).D The density of the amyloid plaques in the fontal cortex were quantified and agrees with neuropathology reports (Table S2). E upper panel The OL lineage (Olig2, red) and lower panel the mature OL subpopulations (MyRF, red) were identified in both GM and WM from all subjects. F Quantification revealed a significant reductions of Olig2+ population in both GM and WM in in DEM and AD group; G the number of MyRF+ mature OL was only reduced in the GM in DEM and AD.
3.2 Characterization of the subjects
A second set of subjects was obtained from the NIH NeuroBioBank. The demography and neuropathological diagnosis of these subjects can be found in Table S2A. While individuals in the AD group each showed marked neuronal loss accompanied by severe amyloid and tau pathology, the individuals in the DEM group had minimal neuronal loss, tau pathology or amyloid-deposition. The DEM subjects represent a heterogeneous group of individuals diagnosed with dementias of different etiologies (e.g. mini-stroke in the brain stem or cerebral infarct – Table S2B). Their mean age was five years younger than the AD group and it is possible that some of them were progressing towards AD conversion.
3.3 Loss of intracortical OL is associated with cognitive status
We examined the myelin and cells of the OL lineage in human prefrontal cortex – Brodmann Areas 8 and 9 (Fig. 1B). Black Gold staining revealed few differences among the three groups in the appearance of the myelin in the subcortical WM (regions labeled * in Fig. 1C). By contrast, the density of myelinated fibers in GM (# in Fig 1C) was consistently reduced in both DEM and AD groups. To validate the NeuroBioBank information, we confirmed the absence of amyloid deposition in the NC and DEM groups by immunostaining (Fig 1D). Using Olig2 as a marker for all cells in the OL lineage [43] and MyRF as a marker for mature oligodendrocytes that are actively myelinating [29] we found that in gray matter of DEM subjects there was a reduction in the density of both Olig2+ and MyRF+ cells (Fig. 1E). The overall reduction of cells of the entire oligodendrocyte lineage (Olig2+) was due mainly to a highly significant loss of Olig2+ cells in the WM (P < .001), with only a downward trend in GM (P = .09 – Fig. 1F). Unexpectedly, the reduction of mature, actively myelinating oligodendrocytes (MyRF+) in both DEM and AD groups was due almost entirely to their depletion in the GM (P < .0003 – Fig. 1G).
3.4 Gray matter oligodendrocytes show enhanced DNA damage and repair
DNA damage is an oft-cited correlate of aging and degenerative illness. To test whether the myelin loss we observed was related to DNA damage, we used γH2A.X as a marker. In all groups we found that γH2A.X expression in the GM OLs was significantly higher than WM cells. In addition, in the DEM group, we found a significant increase in Olig2+ cells with nuclear γH2A.X labeling (p = 0.003 vs NC). We saw no such increase in the AD group (Fig. 2A, S1). To pursue this observation, we examined the phosphorylation status of the ATM (A-T mutated) kinase. DNA double strand breaks (DSB) lead to ATM phosphorylation on serine 1981 and its subsequent activation, Activated ATM kinase then serves as a part of a cell’s response to DNA damage [44]. We found a significant increase in the density of pATMser1981/Olig2-double positive cells in both DEM and AD groups (Fig 2B, S1).
Fig. 2. DNA damage and cell cycle protein re-expression in OLs of frontal cortex.
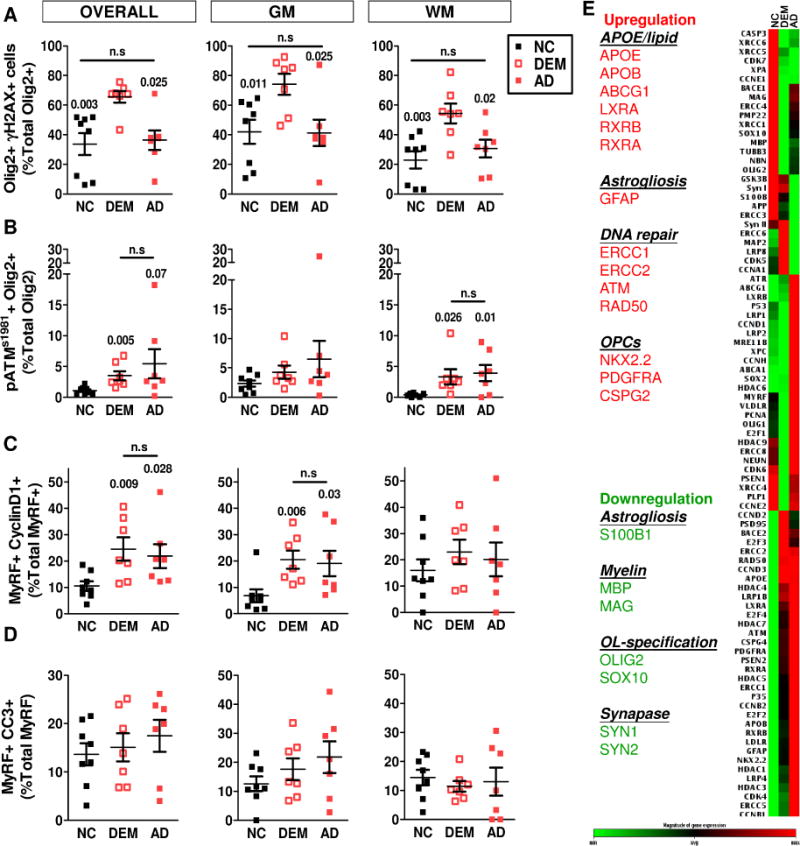
A The DSBs DNA damage in the OL lineage of human frontal cortex were analysed by γH2AX. Quantifications revealed a significantly increase of DSB-DNA damage in the DEM subjects. B DNA repair enzyme, ATM was found activated (pATMser1981) in the OL (Olig2+) lineage, and a significant upregulation of pATMser1981 in the OL lineage in both GM and WM, in AD (All unpaired t-test, vs NC). To test whether DNA damage was associated with aberrant cell cycle reentry and cell death in the mature OLs, human frontal cortex were analysed by Cyclin D1 and cleaved caspase-3 (CC3). C A significant increase of postmitotic mOL re-expressing Cyclin D1 was found in the GM of the DEM and AD subjects (All unpaired t-test, vs NC). D The cell cycle events (CCE) in intracortical mOL was accompanied by a mild, but not significant, increase of apoptosis in the GM of the DEM and AD subjects. E The gene expression profile of the whole cortex presented in a custergram normalized against clinical phenotype (NC, DEM and AD). Consistent pattern of change between DEM and AD listed on the left. Green, downregulation; red, upregulation.
3.5 DNA damage and aberrant re-expression of cell cycle proteins
As neuronal DNA damage can lead to ectopic cell cycle re-entry [22, 23, 25, 27, 45–48], we next asked whether the DNA damage burden in the mature MyRF+ OLs was associated with a similar phenomenon. In NC samples, using cyclin D1 as a cell cycle marker, only about 10% of the mature OLs were cell cycle-positive. By contrast, in DEM and AD groups, this percentage more than doubled (Fig. 2C, S2). Further, these increased cell cycle events (CCE) were restricted almost exclusively to the GM. In neurons, ectopic cell cycling leads to cell death. To determine whether mOL similarly undergo degeneration, we double stained the brain sections for both MyRF and cleaved (activated) caspase-3 (CC3), and found that the number of CC3/MyRF double-positive OL trended higher in the GM of the AD group (Fig. 2D, S2).
To validate these histological findings, we performed an analysis of gene expression of tissue microdissected from the samples (Fig. 2E). In both the DEM and AD groups, we detected an upregulation of DNA repair genes (ATM, RAD50), as well as a down-regulation of myelin structural genes (MBP, MAG) and OL-specification genes (SOX10, OLIG2). Cell cycle proteins, including cyclin D and members of the E2F family, were also up-regulated in the two dementia groups. Genes related to OL early progenitor cells (CSPG4, PDGFRA, NKX2.2) were upregulated as well.
We next tested for correlations among the datasets. We found that histological measures of OL DNA damage (Olig2+ γH2A.X+ immunostaining) were correlated with a decline in both MyRF+ (P < .02) and Olig2+ (P = .06) cell density (Fig. 3A, B) but an increase in the density of MyRF/cyclinD1 double-positive cells (Fig. 3C). Cell cycle re-entry was highly correlated with the loss of MyRF+ cells (Fig. 3D), though not with their visible apoptosis (CC3+ – Fig. 3E). Finally, ATM activation (p-ATM S1981) correlated with dramatic drops in the density of both MyRF+ and Olig2+ cells (Fig. 3F–H). None of these parameters was significantly correlated with amyloid plaque density (Fig. S3)
Fig. 3. DNA damage, CCE and OL degeneration are highly correlated in the frontal cortex.
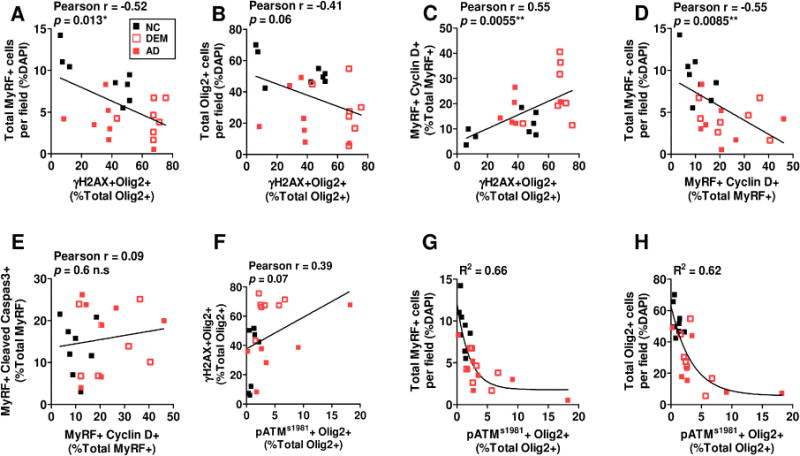
The histological data of OL degeneration, OL-specific DNA damage, CCE and ATM activation were examined for correlations using Pearson test. A–C DSB-DNA damage, OL degeneration and CCE were strongly correlated; D, E The CCE of mOL was significantly associated with their decline in number, but not their apoptosis. F–H A robust correlation between ATM activation, DSB-DNA damage and OL losses were observed. All Pearson test. Data categorized by clinical phenotype (dark square = NC, open square = DEM, red square = AD)
3.6 Activation of DNA damage, cell cycle and cell death in gray matter
We found significant histological differences between the GM and WM OLs in all subjects, without regard to cognitive status. Although the density of OLs was always greater in WM, the actively myelinating, MyRF+, cells were far more prevalent in GM (Fig. 4A, B). Further, the levels of DNA damage in the OL population were significantly higher in the GM, and coupled with ATM activation and apoptosis (Fig. 4C–E).
Fig. 4. Intracortical OL are more vulnerable to DNA damage and ectopic cell cycle associated degeneration than OL in the WM.
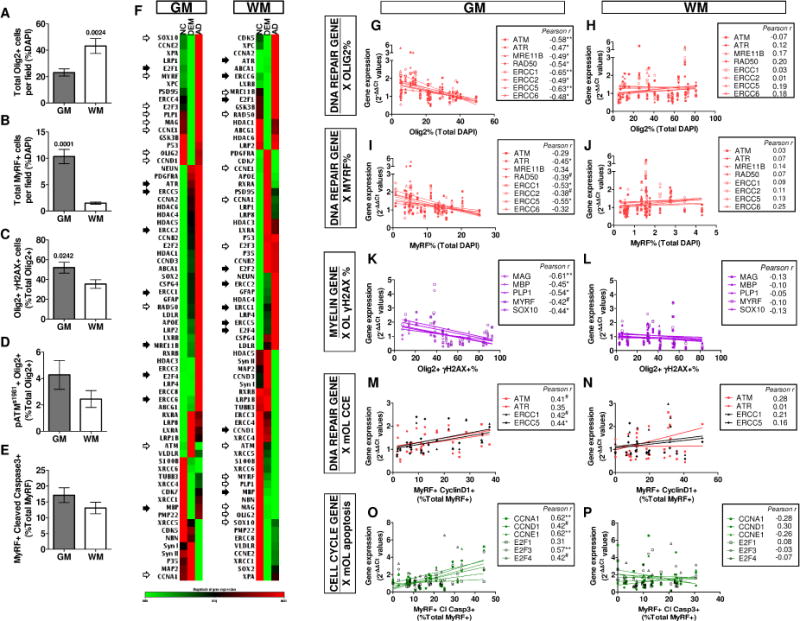
The regional differences in OL population between the intracortical region (GM) and subcortical WM were compared (n=24). A, B The OL (Olig2+) population was significantly higher in the subcortical WM, in contrast, the myelinating mOL (MyRF+) population was significantly higher in the GM. C The intracortical OL population suffer from a significantly higher degree of DSB and oxidative DNA damage. D, E Higher, but not significant, ATM activity and apoptosis were detectable the intracortical OL population in the GM (All unpaired t-test). F Gene expression profile clustergram of the intracortical GM and subcortical WM normalized against clinical phenotype (NC, DEM and AD) were compared. Open arrows: consistent trend of gene expression found between GM and WM in all groups. Closed arrows, opposing trend of gene expression found between GM and WM in all groups. Without arrows, genes with no consistent trend between GM and WM. G–P The differential histological data in GM and WM was plotted against the regional gene expression level for each subject. G–J The number of OL lineage and mOL population were negatively correlated with DNA repair gene expression. K–L The percentage of OL with DSB-DNA damage was negatively correlated with multiple myelin gene expression. M–N The percentage of mOL with CCE was directly proportional to DNA repair gene expression. O–P The expression of cell cycle related genes was positively correlated to the percentage of apoptotic mOL. All correlations were only significant in WM (All Pearson test, #P < .08 * P < .05, ** P < .01, *** P < .001)
To pursue this regional difference, we extracted mRNA from GM and WM for each individual and assayed for gene expression. While many changes were similar in GM and WM (open arrows, Fig. 4F), the expression associated with DNA damage repair, cell cycle initiation and OL lineage-specific genes are substantially different (closed arrows, Fig. 4F). Importantly, the gene expression of the neuronal markers SYN1, SYN2, MAP2 and TUBB3 were only down-regulated in the AD group, but not in the DEM group (Fig. 4F). We next plotted the gene expression data against the corresponding histological data in GM and WM for each subject. Correlations with Pearson coefficients ≥ 0.3 are shown (Fig. 4G–P). Among the genes analyzed, strong and significant correlations with OL cell number were exclusively observed in the GM (Fig. 4G, I, K, M), but not in the WM (Fig. 4H, J, L, N). Specifically, the expression of DNA repair genes increased as the density of gray matter Olig2+ and MyRF+ oligodendrocytes declined (Fig. 4G, I). Using OLs with DNA damage as the independent variable (X-axis), we found that with increasing DNA damage myelin gene expression was strongly down-regulated, but this relationship too was only found in GM (Fig. 4K, L). Focusing on cell cycle re-entry in the MyRF+ population, we found that in the GM, but not the WM, expression of DNA repair genes such as ATM, ERCC1 and ERCC5, increased (Fig 4M, N). Finally, consistent with our hypothesis, we found that the expression of cell cycle genes was highly correlated with an increase in apoptotic (CC3+/MyRF+) cells (Fig. 4O, P). Once again, this relationship was found in the gray matter, but not the white matter.
3.7 Loss of oligodendrocytes precedes neurodegenerative changes in an AD mouse model
The above findings in human brain suggest (1) that myelin loss is associated with OL degeneration in many forms of dementia; (2) that mature OLs in GM are more vulnerable than their neighbors in WM, and (3) that OL degeneration is associated with DNA damage and cell cycle dysregulation. While we would like to know the sequence of cellular events that produced these findings, observations from a relatively small group of subjects can offer only proof of correlation, not evidence for a path of causality. One problem with this or any human neuropathological study is that each sample is a snapshot of one moment in a unique individual’s lifetime. Mouse models of human disease are more informative in this regard as longitudinal studies are possible. Therefore, we examined the age-related changes in the OL population in R1.40 mice – an APP transgene-based model of AD. These animals develop neuronal cell cycle events and neuroinflammation by 6 months of age, even though the first amyloid plaques do not appear until 13–14 months [46, 49]; cognitive impairment is first seen only at 19 months [50]. Assuming a typical mouse life-span of 2.5 years, this temporal progression closely tracks that seen in human subjects with familial AD. We found that R1.40 animals showed myelin loss in the neocortex, but not in corpus callosum or cerebellum, by 12 months of age (Fig. 5A). Western blots of tissue lysates confirmed the loss of MBP, but not Olig2 or NG2 beginning at 6 months and becoming statistically significant by 12 months (Fig. 5B).
Fig. 5. Loss of myelin and OL precede neuronal and amyloid pathology in R1.40 mice.
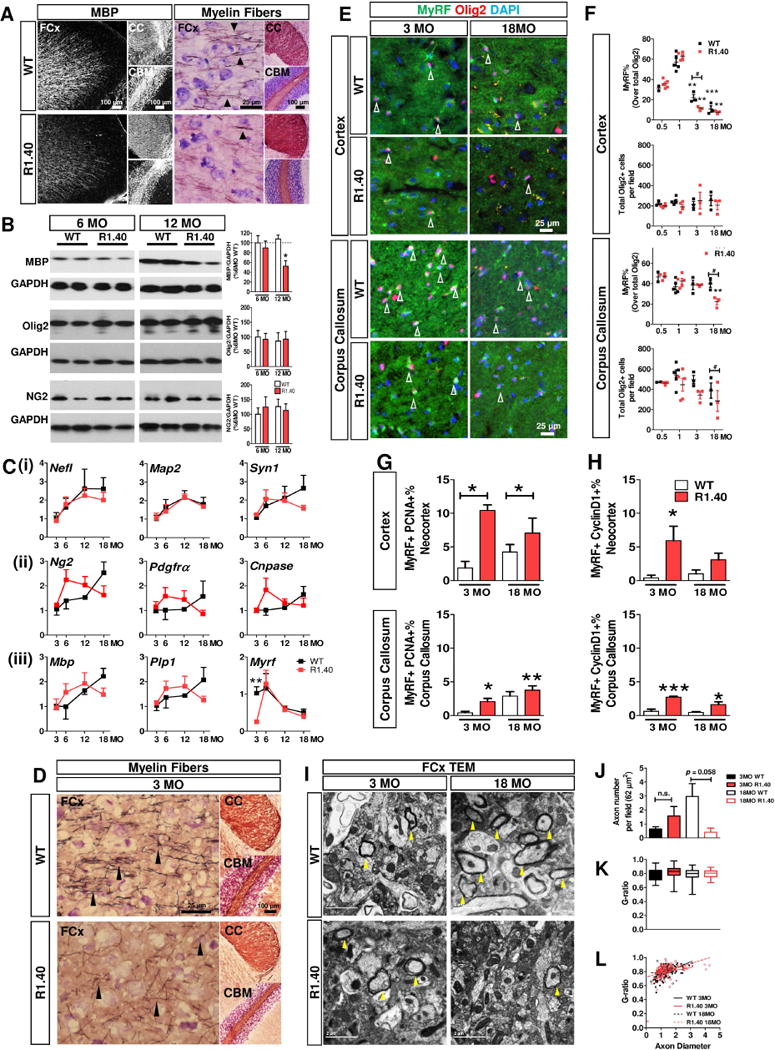
A Immunohistochemistry of MBP revealed reduction of myelination within the frontal cortex (FCx), but not in corpus callosum (CC) and cerebellum (CBM) in the R1.40 mice at 12 months of age (MO). The number of intracortical myelin fibers (arrowheads) were also reduced (Blackgold II/cresyl violet) B Immunoblot showing only MBP, but not Olig2 nor NG2 were significantly reduced in the neocortex of R1.40 mice (One way ANOVA with Newman-Keuls posthoc, *p < 0.05) C Age-dependent gene expression of (i) neuronal/axonal markers, (ii) OPC markers and (iii) myelin markers of the neocortex isolated from the WT (black) and R1.40 mice (red) (time course P < 0.05 WT vs R1.40, One way ANOVA with Newman-Keuls posthoc) (unpaired t-test R1.40 vs WT at 3MO, **p = 0.0034). A significant reduction of Myrf was found at 3MO. D Early loss of myelination in the frontal cortex (FCx), but not corpus callosum (CC) or cerebellum (CBM) in R1.40 mice at 3 MO E–F Reduction of cortical mOL (MyRF+/Olig2+) began by 3MO in the R1.40 mice, but no change was found in Olig2+ population (Olig2+) (vs Control at 0.5MO. No changes in OLs at the CC until 18 MO. G–H A subpopulation of MyRF+ re-express PCNA and Cyclin D1 in R1.40 mice, indicative of CCE in the Cx and CC in age- and genotype-dependent fashion. I TEM micrograph showing myelinated fibers in the frontal cortex of WT and R140 mice at 3 and 18 MO. J–L TEM analysis revealed a significant reduction of the myelinated axons in the R1.40 mice at 18 months, but not at 3 months. Remarkably, no differences in g-ratio was observed among all groups (unpaired t-test, * P < .05, ** P < .01, *** P < .001).
Longitudinal gene expression analysis of the neocortex enlarged upon these findings. We found minimal differences between R1.40 and wild type animals in axonal (Nefl), dendritic (Map2) or synaptic markers (Syn1) (Fig. 5C(i)). By contrast, for genes associated with OL progenitors or function, expression levels increased faster in R1.40 than wild type at younger ages (3 months), and then declined more rapidly (Fig. 5C(ii–iii)). One gene that stood out from this general trend was Myrf, which was significantly down-regulated in R1.40 mice at 3 months before rapidly increasing to wild type levels by 6 months of age.
This low expression of Myrf was consistent with histological findings. We used Black Gold myelin staining and found fewer intracortical myelin fibers in GM of frontal cortex, but not in WM, of 3 month old R1.40 mice (Fig. 5D). Further, we found an age- and genotype-dependent reduction in the density of MyRF+ OLs with no corresponding change in Olig2+ cell density (Fig. 5E). This reduction was especially prevalent in the GM of cortex where the density of MyRF+ cells was reduced by 50% at 3 months of age, but not at earlier ages or with aging. In the WM, a significant reduction of MyRF+ and Olig2+ cells was found in the R1.40 corpus callosum only at 18 months but not at younger ages (Fig. 5F, S4).
As in human, the loss of mature OLs in both neocortex and corpus callosum of R1.40 mice was accompanied by a significant re-expression of cell cycle proteins including cyclin D1 and PCNA (Fig. 5G, H; Fig. S5). Furthermore, aberrant cell cycling was accompanied by higher levels of DNA damage (γH2A.X) in gray matter OLs (Fig. S5). To test whether these changes in the oligodendrocytes were preceded by, and thus were possibly the consequence of, axonal or neuronal dystrophy, we examined the cortex of R1.40 mice in the electron microscope (Fig. 5I). This analysis revealed no significant reduction in axon number or myelin thickness (G-ratio) in R1.40 mice at 3 months of age (Fig. 5J–K). There was a strong trend towards decreased axon density in the older R1.40 animals, but this decrease did not reach statistical significance. These findings are consistent with the reported absence of neuronal/axonal damage in R1.40 mice before 6 months of age [46, 49] and strongly hint that the myelin changes precede those found in neurons.
3.8 DNA damage triggers cell cycle re-entry in mature oligodendrocytes
To test whether DNA damage was responsible for the aberrant cell cycle-re-entry in mature, OLs, we first queried the single-cell RNA-sequencing database of cultured OLs at different differentiation stages [51]. As OLs differentiate from an early progenitor cell state (Cspg4/Pdgfra) to a myelinating OL phenotype (Myrf, Plp1, Mag and Mbp), the expression of cell cycle genes such as cyclin D1 (Ccnd1) and proliferating cell nuclear antigen (Pcna) declines. Indeed, the level of Ccnd1 was reduced to negligible levels in the Myrf-expressing population. In addition, expression of the DNA repair gene, Atm, and its upstream partners in the MRN complex (Mre11a, Rad50, Nbs1) also declined in the more differentiated cell samples.
To prove that DNA damage can directly drive ectopic cell cycle re-entry and that abnormal cell cycle progression is lethal in these postmitotic cells, we treated primary MBP-expressing OL cultures with either etoposide (10 μM) to induce DNA damage or a combination of OL-specific mitogens (PDGF-AA and bFGF, 2 ng/mL each). Both treatments increased the formation of nuclear foci of γH2A.X and 53BP1 (a second marker of DNA double strand breaks) and a significant increase of cyclin D1 expression (Fig. 6B-D). Further, etoposide induced the re-expression of cyclin D1 and reduced the number of MBP-expressing mOL in a concentration-dependent fashion (Fig. 6E). To test whether the microenvironment in the AD brain will similarly induce ectopic cell cycle events in mOLs, bacterial lipopolysaccharides (LPS; 0.1 μg/mL) and the cytotoxic Aβ25–35 peptide (10 μM), were applied to mimic inflammation and amyloidosis. Only etoposide, but not LPS or soluble mixture of Aβ25–35 induced cell cycle reentry and reduced the number the MBP-expressing OL in vitro (Fig. 6F).
Fig. 6. DNA damage directly induced aberrant re-expression of cell cycle protein in postmitotic, mature oligodendrocyte.
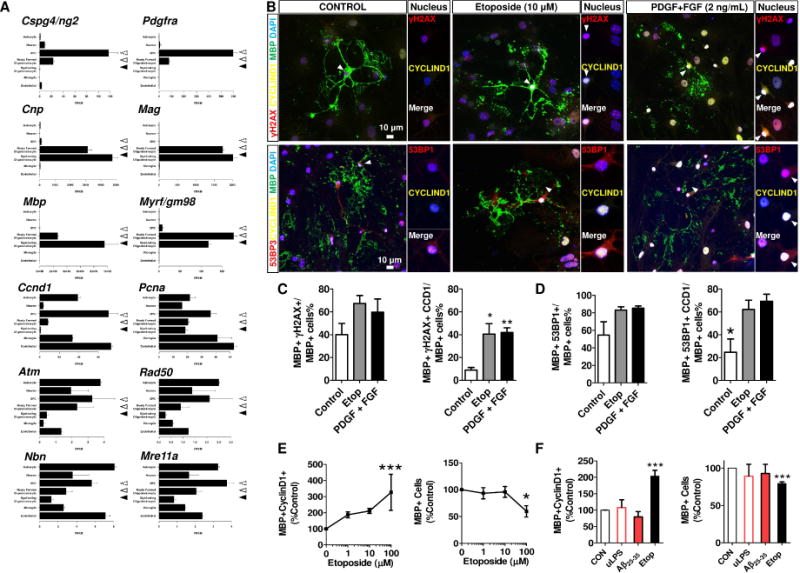
A The RNA-seq data was adopted from the database stabled by Barres Lab in the public domain – http://web.stanford.edu/group/barres_lab/brainseqMariko/brainseq2.html (Zhang et al 2014). The reproduction of these data were made with written permission from the corresponding author. The cell-stage specificity of the OL population was validated by the expression of OPC genes (Cspg4, and Pdgfra) in the progenitor cells (dashed arrowheads), andhe expression of myelin and mature OL genes (Cnp, Mag, Mbp and Myrf) in the newly formed oligodendrocytes (open arrowhead) and myelinating oligodendrocyte (filled arrowhead). Of note, the expression of cell cycle markers, Ccnd1 (cyclin D1) and Pcna were attenuated along the differentiation axis of OL, and the expression of ATM family were the lowest in myelinating oligodendrocytes B The direct application of DSB-inducing agent, etoposides (10 μM, 24h) or OL-specific mitogens (PDGF-AA+bFGF, 2 ng/mL), similarly induced DSB foci formation labelled by γH2AX, (upper panel, red) and 53BP1 (lower panel, red) in the MBP expressing mOL (green). Of note, the aberrant re-expression of Cyclin D1 (arrow, yellow) remarkably increased in these mature mOL with DSBs. C, D Quantifications of cyclin D1 in the mOL population with DSBs. E Etoposide induced aberrant Cyclin D1 expression and reduced mature OL population in a dose-dependent fashion (0 – 100 μM, 24 h). F Only application of etoposide (10 μM, 24h), but not Lipopolysaccharide-induced inflammation (uLPS, 0.1 μg/mL, 24 h) nor soluble Aβ25-35 peptide mixture (10 μM, 24 h) induced abnormal expression of cyclin D1 in mature OL (unpaired t-test, n=3).
4. Discussion
Alzheimer’s disease is a uniquely human condition, as is the tremendous expansion of cortical myelination; both are distinctive traits not found in most other primates [52, 53]. The loss of fine myelin fibers in the gray matter (also known as intracortical myelin) is specifically associated with declining cognitive performance during normal aging [6] and it was this association that prompted us to investigate myelin loss in the context of dementia, Alzheimer’s and the amyloid cascade hypothesis [54–56]. Our analysis of the NACC-MRI dataset and 24 additional samples of human frontal cortices obtained from the NeuroBioBank show that myelin/OL degeneration correlates with dementia status. The frequent concurrence of our AD and DEM samples illustrates that the myelin-related changes that we report are correlated with all types of dementia, not only individuals with an elevated amyloid plaque burden. In this regard, we provided human, animal and cell culture evidence in support of the hypothesis that DNA damage is a major pathogenic force driving these age-related myelin changes. We conclude from our data that it is DNA damage in mature OLs, both in vivo and in vitro, that leads to their unscheduled re-entry into a cell cycle process and ultimately to their death.
4.1 Intracortical oligodendrocytes are vulnerable to aging and Alzheimer’s dementia
Neuroimaging studies revealed WMH mainly in the periventricular and subcortical regions. Apart from one report [57], most earlier studies showed the presence of myelin degradation surrounding WMH lesions on top of the vascular changes [39, 40, 42, 58]. The strong associations between cognitive impairment and WMH volume found in our MRI data meta-analysis are consistent with previous findings during the preclinical and clinical state of AD [54, 59]. Here, we add cellular level resolution to these studies by showing that in patients with dementia, oligodendrocyte degeneration is more severe in the intracortical gray matter than in subcortical white matter. The same pattern is found in the R1.40 mouse model of AD. In both species the picture that emerges is that GM myelin is far more dynamic than that in the adjacent subcortical WM and that, as a consequence, gray matter OLs are more susceptible to cellular damage including the loss of DNA integrity (Fig. 4, 5). This fits well with the reduction in the expression of DNA repair enzyme genes found during OL differentiation in the Zhang dataset [51] (Fig. 6A and S6). The oligodendrocyte may well be uniquely sensitive to factors that lead to reductions in DNA repair proteins. This is because many of these proteins, such as members of the ERCC family, serve dual functions in both DNA repair and OL-specific transcription [60]. Finally, as myelin maintenance and remodeling are pre-requisites for learning [61–63], the unique dynamism of GM myelin, especially in human frontal cortex, may underlie species differences in cognitive abilities and their loss in the uniquely human Alzheimer’s disease [53].
4.2 DNA damage induced cell cycle re-entry in postmitotic OL
The finding of a cell cycle/cell death correlation in OLs represents an intriguing extension of an older hypothesis linking the two processes in postmitotic neurons [25, 27, 64–66]. Several groups have found that DNA damage can serve as one trigger for this illicit cell cycle behavior [22, 23, 47, 48]. As in neurons, mature OLs also re-expresses cell cycle proteins before their degeneration during the course of dementia, or after etoposide-induced DSB in vitro. MyRF is a master transcription factor expressed by postmitotic and mature OL to drive myelin gene expression [67] (Fig. 6). Once they have progressed to this stage of maturation, OLs have irreversibly exited cell cycle and cannot revert to a progenitor state, even when Myrf is conditionally ablated [29]. Early studies reported that mature OLs undergo apoptosis when forced to enter the cell cycle by exposure to OL-specific mitogens in vitro and in vivo [68, 69], and such apoptosis is exacerbated when cell cycle is blocked by an antimitotic agent [68]. We have reproduced these early studies and extended them by showing that the mitogen-induced cell cycle in mOLs results in DNA damage (Fig. 6). In the aging brain, vascular changes, metabolic changes, silent traumatic injury, inflammation or oxidative stress and a decline in DNA repair capacity could all contribute to DNA damage in the OLs [17, 21].Therefore, our findings reinforce the hypothesis that unscheduled cell cycle initiation in mOL will cause cell death and demyelination with DNA damage serving as both cause and consequence.
4.3 Myelin pathology is an alternative paradigm to explain AD pathogenesis
Intracortical myelin and WM volume start to decline by the mid-forties in humans – at least a decade earlier than any amyloid pathology [6, 70]. This makes it unlikely that myelin degradation is a downstream consequence of axonal and neuronal dystrophy or sub-clinical accumulation of Aβ aggregates. Our findings of significant myelin/OL abnormalities in the DEM group, where neocortical neuronal loss and amyloid-pathology are minimal (Table S2), support a model in which myelin degeneration is both a risk factor and potentially a causative element that defines a phase prior to the earliest stage of AD-related amyloid abnormalities. The finding of OL deficiencies in R1.40 mice at ages before nerve cell body or axon changes are found also supports this view. To investigate age-dependent histological changes of OLs prior to plaque formations, the use of animal model is imperative, even while it comes with caveats. Wild type mice do not normally exhibit any AD pathology with aging. The findings in R1.40 mice, while one of the least aggressive AD models, formally apply only for familial forms of the human disease. Compared with earlier reports using double and triple transgenic AD mouse models [31, 71], the R1.40 model offers a long, human-like, temporal progression allowing for a more detailed study of early myelin degeneration. The myelin/OL degeneration found months before signs of amyloid plaque formation or neurodegeneration in R1.40 model is consistent with our central hypothesis. The early loss of mOL and their cell cycle re-entry at 3 months were unlikely the effect of extensive amyloidosis, which would also injure neurons. The detailed mechanism(s) by which human APPSwe expression drives myelin pathology preceding plaque formation remains to be identified. One possible explanation is that the mutant APPswe places excessive oxidative stress on the highly vulnerable OLs, which then is followed by DNA damage [72, 73].
Based on the strong correlation between cognitive capacity and myelin integrity – in normal aging and in dementia – myelin loss has been proposed previously as one of the earliest degenerative changes in AD [30]. Our results in human and mice support this idea and extend it. Our data suggest that genomic damage in the OL lineage is a major underlying age-associated cause of cognitive decline and dementia. As such they offer an alternative disease model that is independent from, but not mutually exclusive with, the amyloid cascade hypothesis [74, 75]. Our data showing the failure of soluble Aβ to trigger a cell cycle in cultured OLs strongly supports its independence and argues that myelin degradation could be an early pathogenic driver of Alzheimer’s dementia. One intriguing way in which myelin degeneration might trigger AD neuronal loss involves APP itself. As a stress response protein, APP accumulates at axonal lesions after demyelination [76, 77]. This makes it at least plausible that in combination with other genetic risk factors, such as APOE Ԑ4 or a pathogenic TREM2 variant, APP accumulation might contribute to plaque formation. In addition to this APP-based mechanism, the loss of saltatory conduction after myelin loss places enormous energy demands on the neurons. Coupled with the loss of trophic support from the OL axonal metabolic homeostasis would be lost putting its structure and function in jeopardy [78–80], possibly exacerbating neuronal loss [81]. A final mechanism worth considering involves the fact that age-associated myelin debris is constantly cleared by phagocytic microglia [82]. Pathological amount of myelin breakdowns may not only interfere with other phagocytic function of microglia including amyloid clearance, but also trigger neuroinflammation.
One especially attractive feature of the myelin hypothesis, is that myelin integrity is independently linked with the strongest identified genetic risk factor for sporadic AD – apolipoprotein E (APOE). Myelin is compromised in APOE Ԑ4 carriers, resulting in negative effects on executive functions [55, 83]. The APOE Ԑ4 genotype is also associated with an enhanced innate immune response, metabolic stress and cerebrovascular dysfunction, all of which are likely contribute to increased stress on the aging brain. As differentiating OLs are poorly defended against oxidative stress due to their low expression of glutathione-related antioxidants [72, 84, 85] (Fig S6), they may be uniquely vulnerable to oxidative and DNA damage. These effects of the APOE Ԑ4 genotype on the myelin phenotype are uncoupled from the amyloid cascade hypothesis but do not preclude additional impact on Aβ transport and processing.
Our study does not consider the effects of tauopathy, the second hallmark of AD, on OL pathology. This is in no way meant to imply that we dismiss the importance of tau as a component of the etiology of Alzheimer’s disease. We would note, however, that the earliest tau pathology (increased CSFtau) occurs after the initial amyloid aggregation [2, 86], and the spread of hyperphosphorylated tau at different Braak stages does not match the anatomical progression of myelin loss in the human brain (from neocortex to entorhinal regions) [87]. Further, tau pathology was not detected in most cases of the plaque-free DEM group. These observations suggest that the OL degeneration observed in the frontal cortex is unlikely to be the direct result of either an overt or cryptic tauopathy. Nevertheless, a recent report revealed that WMH are highly correlated with hyperphosphorylated tau in postmortem brain [88]. The final explanation of AD mechanisms, therefore, will no doubt include pathologic forms of tau. Indeed, tau-driven axonal problems might be expected to exert negative effects on the ensheathing myelinating oligodendrocytes.
4.4 Conclusion
Recent clinical trial data suggest that the progression of Alzheimer’s dementia, if not its initiation, requires explanations beyond the amyloid cascade hypothesis [4]. Invoking myelin loss via an age-related accumulation of DNA damage offers a ‘non-canonical’ pathway to dementia and to Alzheimer’s disease that is solidly rooted in the genetics and biology of both aging and Alzheimer’s.
Supplementary Material
RESEARCH IN CONTEXT.
1. Systematic review
White matter abnormalities increase with age and constitute an early pathology of Alzheimer’s disease (AD). As myelin loss occurs years before amyloid-cascade begin, it may be a pathology hallmark for preclinical AD. From literatures (PubMed and Scopus), the authors identified age-associated DNA damage as the likely causative insults.
2. Interpretation
This study indicates that myelin degeneration are highly correlated with cognitive decline, but independent of the amyloid aggregations. Instead, it is DNA damage of post-mitotic oligodendrocyte that drives ectopic cell cycle re-entry and myelin pathology. These findings are consistent with the hypothesis that genomic DNA damage is the major driver of brain ageing.
3. Future directions
Future studies on the mechanism(s) of cycle-associated cell death of oligodendrocytes will provide a better insight of myelin loss in ageing. Translational study may explore the use of oligodendrocyte/myelin damage as the diagnostic and therapeutic target for preclinical AD.
Acknowledgments
The present work was generously supported by Research Grants Council, Hong Kong Special Administrative Region (AoE/M-604/16, GRF16124916, GRF16101315 and GRF660813), and the National Key Basic Research Program of China (2013CB530900). We acknowledge the kind support from the staff at NeuroBioBank of National Institutes of Health (NIH) and National Alzheimer’s Coordinating Center (NACC).
All human tissue in this study was obtained from the NIH Neurobiobank at the University of Maryland, Baltimore, MD and the Human Brain and Spinal Fluid Resource Center, CA.
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIAfunded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie-Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron. 2014;84:608–22. doi: 10.1016/j.neuron.2014.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–9. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 5.Herrup K, Carrillo MC, Schenk D, Cacace A, Desanti S, Fremeau R, et al. Beyond amyloid: getting real about nonamyloid targets in Alzheimer’s disease. Alzheimers Dement. 2013;9:452–8 e1. doi: 10.1016/j.jalz.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grydeland H, Walhovd KB, Tamnes CK, Westlye LT, Fjell AM. Intracortical myelin links with performance variability across the human lifespan: results from T1- and T2-weighted MRI myelin mapping and diffusion tensor imaging. J Neurosci. 2013;33:18618–30. doi: 10.1523/JNEUROSCI.2811-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters A, Sethares C. Aging and the myelinated fibers in prefrontal cortex and corpus callosum of the monkey. J Comp Neurol. 2002;442:277–91. doi: 10.1002/cne.10099. [DOI] [PubMed] [Google Scholar]
- 8.Bartzokis G, Beckson M, Lu PH, Nuechterlein KH, Edwards N, Mintz J. Age-related changes in frontal and temporal lobe volumes in men: a magnetic resonance imaging study. Arch Gen Psychiatry. 2001;58:461–5. doi: 10.1001/archpsyc.58.5.461. [DOI] [PubMed] [Google Scholar]
- 9.Raz N. Ageing and the Brain. eLS: John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 10.Bartzokis G, Cummings JL, Sultzer D, Henderson VW, Nuechterlein KH, Mintz J. White matter structural integrity in healthy aging adults and patients with Alzheimer disease: a magnetic resonance imaging study. Arch Neurol. 2003;60:393–8. doi: 10.1001/archneur.60.3.393. [DOI] [PubMed] [Google Scholar]
- 11.Salthouse TA. When does age-related cognitive decline begin? Neurobiol Aging. 2009;30:507–14. doi: 10.1016/j.neurobiolaging.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bender AR, Volkle MC, Raz N. Differential aging of cerebral white matter in middle-aged and older adults: A seven-year follow-up. NeuroImage. 2016;125:74–83. doi: 10.1016/j.neuroimage.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glasser MF, Goyal MS, Preuss TM, Raichle ME, Van Essen DC. Trends and properties of human cerebral cortex: correlations with cortical myelin content. NeuroImage. 2014;93(Pt 2):165–75. doi: 10.1016/j.neuroimage.2013.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salami M, Itami C, Tsumoto T, Kimura F. Change of conduction velocity by regional myelination yields constant latency irrespective of distance between thalamus and cortex. Proc Natl Acad Sci U S A. 2003;100:6174–9. doi: 10.1073/pnas.0937380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calabrese M, Agosta F, Rinaldi F, Mattisi I, Grossi P, Favaretto A, et al. Cortical lesions and atrophy associated with cognitive impairment in relapsing-remitting multiple sclerosis. Arch Neurol. 2009;66:1144–50. doi: 10.1001/archneurol.2009.174. [DOI] [PubMed] [Google Scholar]
- 16.Tobin WO, Popescu BF, Lowe V, Pirko I, Parisi JE, Kantarci K, et al. Multiple sclerosis masquerading as Alzheimer-type dementia: Clinical, radiological and pathological findings. Multiple sclerosis. 2016;22:698–704. doi: 10.1177/1352458515604382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tse KH, Herrup K. Re-imagining Alzheimer’s disease - the diminishing importance of amyloid and a glimpse of what lies ahead. J Neurochem. 2017 doi: 10.1111/jnc.14079. [DOI] [PubMed] [Google Scholar]
- 18.Vaidya A, Mao Z, Tian X, Spencer B, Seluanov A, Gorbunova V. Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 2014;10:e1004511. doi: 10.1371/journal.pgen.1004511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 20.Chow HM, Herrup K. Genomic integrity and the ageing brain. Nat Rev Neurosci. 2015;16:672–84. doi: 10.1038/nrn4020. [DOI] [PubMed] [Google Scholar]
- 21.Tse KH, Herrup K. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech Ageing Dev. 2017;161:37–50. doi: 10.1016/j.mad.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, et al. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–61. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Cohen ML, Lerner AJ, Yang Y, Herrup K. DNA damage and cell cycle events implicate cerebellar dentate nucleus neurons as targets of Alzheimer’s disease. Mol Neurodegener. 2010;5:60. doi: 10.1186/1750-1326-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex. 2009;19:1273–93. doi: 10.1093/cercor/bhn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–8. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Chen J, Vinters HV, Gatti RA, Herrup K. Stable brain ATM message and residual kinase-active ATM protein in ataxia-telangiectasia. J Neurosci. 2011;31:7568–77. doi: 10.1523/JNEUROSCI.0778-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Herrup K. Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. J Neurosci. 2005;25:2522–9. doi: 10.1523/JNEUROSCI.4946-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–78. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- 29.Koenning M, Jackson S, Hay CM, Faux C, Kilpatrick TJ, Willingham M, et al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J Neurosci. 2012;32:12528–42. doi: 10.1523/JNEUROSCI.1069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartzokis G, Lu PH, Mintz J. Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 2007;3:122–5. doi: 10.1016/j.jalz.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behrendt G, Baer K, Buffo A, Curtis MA, Faull RL, Rees MI, et al. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia. 2013;61:273–86. doi: 10.1002/glia.22432. [DOI] [PubMed] [Google Scholar]
- 32.Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nat Neurosci. 2003;6:309–15. doi: 10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- 33.Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, et al. The National Alzheimer’s Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 34.Lamb BT, Call LM, Slunt HH, Bardel KA, Lawler AM, Eckman CB, et al. Altered metabolism of familial Alzheimer’s disease-linked amyloid precursor protein variants in yeast artificial chromosome transgenic mice. Hum Mol Genet. 1997;6:1535–41. doi: 10.1093/hmg/6.9.1535. [DOI] [PubMed] [Google Scholar]
- 35.Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harbor protocols. 2013;2013:854–68. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- 36.Luo F, Zhang J, Burke K, Miller RH, Yang Y. The Activators of Cyclin-Dependent Kinase 5 p35 and p39 Are Essential for Oligodendrocyte Maturation, Process Formation, and Myelination. J Neurosci. 2016;36:3024–37. doi: 10.1523/JNEUROSCI.2250-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikula S, Binding J, Denk W. Staining and embedding the whole mouse brain for electron microscopy. Nat Methods. 2012;9:1198–201. doi: 10.1038/nmeth.2213. [DOI] [PubMed] [Google Scholar]
- 38.Luo F, Burke K, Kantor C, Miller RH, Yang Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3beta signaling. J Neurosci. 2014;34:10415–29. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shim YS, Yang DW, Roe CM, Coats MA, Benzinger TL, Xiong C, et al. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement Geriatr Cogn Disord. 2015;39:92–104. doi: 10.1159/000366411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blair JA, Wang C, Hernandez D, Siedlak SL, Rodgers MS, Achar RK, et al. Individual Case Analysis of Postmortem Interval Time on Brain Tissue Preservation. PLoS One. 2016;11:e0151615. doi: 10.1371/journal.pone.0151615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouw AA, Seewann A, Vrenken H, van der Flier WM, Rozemuller JM, Barkhof F, et al. Heterogeneity of white matter hyperintensities in Alzheimer’s disease: post-mortem quantitative MRI and neuropathology. Brain. 2008;131:3286–98. doi: 10.1093/brain/awn265. [DOI] [PubMed] [Google Scholar]
- 43.Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, et al. Olig2-dependent developmental fate switch of NG2 cells. Development. 2012;139:2299–307. doi: 10.1242/dev.078873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 45.Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, et al. Microglial derived tumor necrosis factor-alpha drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol Dis. 2014;62:273–85. doi: 10.1016/j.nbd.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci. 2008;28:10786–93. doi: 10.1523/JNEUROSCI.2441-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–84. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamb BT, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, et al. Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci. 1999;2:695–7. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- 50.Hock BJ, Lattal KM, Kulnane LS, Abel T, Lamb BT. Pathology associated memory deficits in Swedish mutant genome-based amyloid precursor protein transgenic mice. Curr Aging Sci. 2009;2:205–13. doi: 10.2174/1874609810902030205. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, et al. Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci U S A. 2012;109:16480–5. doi: 10.1073/pnas.1117943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schoenemann PT, Sheehan MJ, Glotzer LD. Prefrontal white matter volume is disproportionately larger in humans than in other primates. Nat Neurosci. 2005;8:242–52. doi: 10.1038/nn1394. [DOI] [PubMed] [Google Scholar]
- 54.Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S, et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol. 2006;63:246–50. doi: 10.1001/archneur.63.2.246. [DOI] [PubMed] [Google Scholar]
- 55.Bartzokis G, Lu PH, Geschwind DH, Tingus K, Huang D, Mendez MF, et al. Apolipoprotein E affects both myelin breakdown and cognition: implications for age-related trajectories of decline into dementia. Biol Psychiatry. 2007;62:1380–7. doi: 10.1016/j.biopsych.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 56.Bartzokis G, Lu PH, Heydari P, Couvrette A, Lee GJ, Kalashyan G, et al. Multimodal magnetic resonance imaging assessment of white matter aging trajectories over the lifespan of healthy individuals. Biol Psychiatry. 2012;72:1026–34. doi: 10.1016/j.biopsych.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 57.Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804–11. doi: 10.1212/01.wnl.0000319691.50117.54. [DOI] [PubMed] [Google Scholar]
- 58.Erten-Lyons D, Woltjer R, Kaye J, Mattek N, Dodge HH, Green S, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013;81:977–83. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kandel BM, Avants BB, Gee JC, McMillan CT, Erus G, Doshi J, et al. White matter hyperintensities are more highly associated with preclinical Alzheimer’s disease than imaging and cognitive markers of neurodegeneration. Alzheimers Dement (Amst) 2016;4:18–27. doi: 10.1016/j.dadm.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Compe E, Malerba M, Soler L, Marescaux J, Borrelli E, Egly JM. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci. 2007;10:1414–22. doi: 10.1038/nn1990. [DOI] [PubMed] [Google Scholar]
- 61.McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, et al. Motor skill learning requires active central myelination. Science. 2014;346:318–22. doi: 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, et al. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci. 2016;19:1210–7. doi: 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jeffries MA, Urbanek K, Torres L, Wendell SG, Rubio ME, Fyffe-Maricich SL. ERK1/2 Activation in Preexisting Oligodendrocytes of Adult Mice Drives New Myelin Synthesis and Enhanced CNS Function. J Neurosci. 2016;36:9186–200. doi: 10.1523/JNEUROSCI.1444-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–98. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith TW, Lippa CF. Ki-67 immunoreactivity in Alzheimer’s disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995;54:297–303. doi: 10.1097/00005072-199505000-00002. [DOI] [PubMed] [Google Scholar]
- 66.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999;52:525–7. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 67.Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–85. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grinspan JB, Reeves MF, Coulaloglou MJ, Nathanson D, Pleasure D. Re-entry into the cell cycle is required for bFGF-induced oligodendroglial dedifferentiation and survival. J Neurosci Res. 1996;46:456–64. doi: 10.1002/(SICI)1097-4547(19961115)46:4<456::AID-JNR7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 69.Muir DA, Compston DA. Growth factor stimulation triggers apoptotic cell death in mature oligodendrocytes. J Neurosci Res. 1996;44:1–11. doi: 10.1002/(SICI)1097-4547(19960401)44:1<1::AID-JNR1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 70.Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply 49–62. [DOI] [PubMed] [Google Scholar]
- 71.Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65. doi: 10.1002/glia.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–53. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilkinson BL, Cramer PE, Varvel NH, Reed-Geaghan E, Jiang Q, Szabo A, et al. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol Aging. 2012;33:197 e21–32. doi: 10.1016/j.neurobiolaging.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goris ED, Ansel KN, Schutte DL. Quantitative systematic review of the effects of non-pharmacological interventions on reducing apathy in persons with dementia. Journal of advanced nursing. 2016;72:2612–28. doi: 10.1111/jan.13026. [DOI] [PubMed] [Google Scholar]
- 75.Ballard C, Orrell M, Sun Y, Moniz-Cook E, Stafford J, Whitaker R, et al. Impact of antipsychotic review and non-pharmacological intervention on health-related quality of life in people with dementia living in care homes: WHELD-a factorial cluster randomised controlled trial. International journal of geriatric psychiatry. 2016 doi: 10.1002/gps.4572. [DOI] [PubMed] [Google Scholar]
- 76.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–9. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 77.Matias-Guiu JA, Oreja-Guevara C, Cabrera-Martin MN, Moreno-Ramos T, Carreras JL, Matias-Guiu J. Amyloid Proteins and Their Role in Multiple Sclerosis. Considerations in the Use of Amyloid-PET Imaging. Front Neurol. 2016;7:53. doi: 10.3389/fneur.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–21. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275–83. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- 81.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011;10:187–98. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci. 2016;19:995–8. doi: 10.1038/nn.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ryan L, Walther K, Bendlin BB, Lue LF, Walker DG, Glisky EL. Age-related differences in white matter integrity and cognitive function are related to APOE status. Neuroimage. 2011;54:1565–77. doi: 10.1016/j.neuroimage.2010.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Juurlink BH, Thorburne SK, Hertz L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998;22:371–8. doi: 10.1002/(sici)1098-1136(199804)22:4<371::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 85.Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–24. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McAleese KE, Firbank M, Dey M, Colloby SJ, Walker L, Johnson M, et al. Cortical tau load is associated with white matter hyperintensities. Acta neuropathologica communications. 2015;3:60. doi: 10.1186/s40478-015-0240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.