

To the Editor
Myeloid differentiation primary response 88 gene (MYD88) encodes an essential adaptor protein connecting Toll-like receptor (TLR) and IL-1 receptor (IL-1R) signaling to activation of IL-1 receptor-associated kinases (IRAKs). Upon receptor stimulation, MyD88 oligomerizes causing recruitment and activation of IRAKs to form the myddosome (i.e. MyD88-signaling complex),1 ultimately triggering activation of NF-κB and/or interferon regulatory factor 7.2 Germline loss-of-function mutations in MYD88 lead to immunodeficiency with recurrent pyogenic infections.3 Somatic gain-of-function mutations contribute to certain B cell malignancies.4,5,6 Here we report an individual with a de novo germline gain-of-function MYD88 mutation, who has severe destructive arthritis and an intermittent rash.
The subject was a 14-year old Caucasian female with arthritis in small-to-medium sized joints since the age of 2 years leading to severe bone destruction and periarticular growth arrest (Fig 1, A). The asymmetric arthritis often followed minor trauma, initially resembling septic arthritis, but synovial fluid cultures were negative and antibiotics ineffective. Two synovial biopsies of chronic arthritis revealed a marked neutrophilic infiltrate (not shown), unlike the lymphocytic pattern seen in juvenile idiopathic arthritis (JIA). MRIs revealed substantial synovial, periarticular, and intramedullary bone inflammation (Fig 1, B). The arthritis was unresponsive to NSAIDs, methotrexate, and corticosteroids, while biologics had been declined. Intermittent rashes began around the onset of her arthritis and were erythematous and maculo-papular, generally lasting 1-2 weeks with spontaneous resolution (Fig 1, C). A recent skin biopsy from a dorsal hand lesion revealed interstitial granulomatous dermatitis. Whether this explains previous rashes is unclear. There was no history of unexplained fever, recurrent infection, or growth delay.
Fig 1.
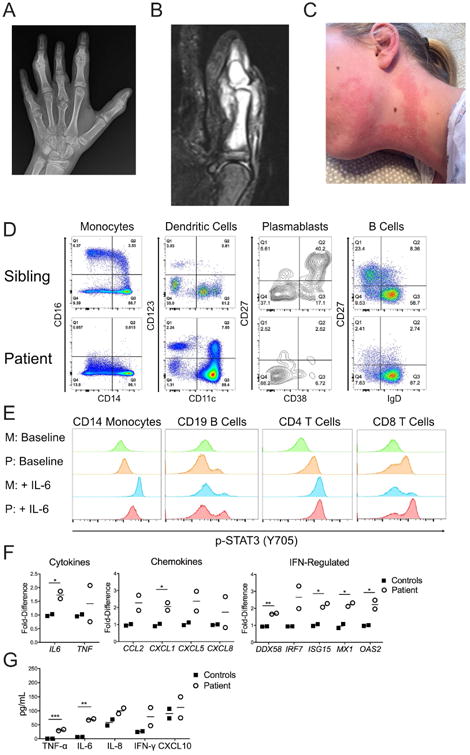
Clinical and immunological phenotype of patient with germline MyD88 S222R mutation. A, Left-hand radiograph. B, STIR MRI of left thumb. C, Appearance of recurrent rash. D, Representative immunophenotyping of monocytes, dendritic cells, plasmablasts, and B cells. Similar results were obtained 3 times over a span of 27 months, with sibling, mother, and an unrelated individual as controls. E, Representative histograms of baseline and IL-6 induced (50 ng/mL for 20 minutes) phosphorylation of STAT3 (p-STAT3, Y705) in patient (P) and mother (M) peripheral mononuclear cells by flow cytometry. Similar results were obtained twice, 6 months apart, with mother and unaffected sibling as controls. F, Baseline peripheral monocyte gene expression determined by qPCR. G, Cytokine secretion from 22 hour whole blood cultured without stimulation. For F and G, similar results were obtained twice, 1 year apart, using mother and unrelated healthy subject as controls. Lines represent the means. *, p<0.05; **, p<0.01; ***, p<0.001.
The subject's C-reactive protein has been intermittently elevated (but <7 mg/L; normal 0-4.99 mg/L), as has osteocalcin (113.8-201.6 ng/mL; normal 7.3-38.5 ng/mL), but with normal erythrocyte sedimentation rate, complete blood count, and serum immunoglobulins including IgG subsets. Lymphocyte subsets (CD3, CD4/CD3, CD8/CD3, CD19, and NK cells) were normal and no autoantibodies have been detected. For a summary of clinical findings, please see Table E1 in this article's Online Repository at www.jacionline.org.
The severe phenotype and inconsistencies with typical polyarticular JIA led us to pursue whole exome sequencing, which revealed a heterozygous missense mutation in MYD88 (c.666T>G, p.Ser222Arg or S222R) in the patient, but not other family members, also confirmed by Sanger sequencing (see Fig E1, A, in this article's Online Repository at www.jacionline.org). The mutation was present in ∼50% of reads from patient whole blood, CD14+ monocytes, cultured dermal fibroblasts, and patient-derived B lymphoblastoid cells (EBV-LCL) supporting a strong likelihood that it is germline (see Fig E1, B).
Immunophenotyping revealed monocytes lacking CD16, which is known to shed during TLR activation,E1 and identified a previously unreported CD123+CD11c+ dendritic cell population (Fig 1, D) that was negative for CD1c/BDCA-1, CD303/BDCA-2, CD141/BDCA-3, and the activated basophil marker CD203c (see Fig E2 in this article's Online Repository at www.jacionline.org). Additionally, CD19+CD20-CD27+CD38+ plasmablasts were absent and CD20+CD19+IgD-CD27+ memory B cells were decreased (Fig 1, D). Please see Table E1 for complete immunophenotyping data in this article's Online Repository at www.jacionline.org. There was a striking increase in Y705 STAT3 phosphorylation in unstimulated CD4+ and CD8+ T lymphocytes, a smaller increase in CD14+ monocytes, and a subpopulation of p-STAT3+ CD19+ B lymphocytes, as has been reported in gain-of-function MYD88 mutation-positive malignancies.4,5 STAT3 phosphorylation was similar to control cells after IL-6 stimulation, except for a population of highly phosphorylated CD8+ T lymphocytes in the patient (Fig 1, E). Neutrophil surface CD11b, CD66b, and CD62L expression was similar to controls (data not shown), possibly due to chronic homeostatic changes in rates of surface antigen shedding to production or cell death/clearance. Peripheral monocyte gene expression revealed an interferon-regulated signature (NanoString dataset available upon request, Fig 1, F), and IL6 expression was also elevated, but not TNF (Fig 1, F). Whole blood production of TNF-α and IL-6 from unstimulated patient cells was higher than controls (Fig 1, G), while the differences did not persist after TLR-stimulation (data not shown).
Amino acid residue 222 is located on the surface of the MyD88 TIR domain, which is required for oligomerization and myddosome formation.1 Oligomerization occurs via the BB-loop, the alpha E helix, and the C-terminus of the alpha helix C.7 Using molecular dynamics modeling, we found that the S222R mutation is predicted to introduce a novel R222-E245 salt bridge that together with steric effects between R222 and F248 side chains, induces a significant tilt of the alpha C helix (see Fig E3, A, in this article's Online Repository at www.jacionline.org). Similar to what is seen in the L265P activating mutation,7 this shift of the CD loop may promote MyD88 TIR-TIR symmetrical interaction. Since the patient was heterozygous for S222R, we re-expressed equal amounts of wild-type (WT) and/or S222R MyD88 proteins with different epitope tags within the same MyD88-deficient (MyD88-KO) cell (see Fig E3, B). Unstimulated cells containing S222R MyD88 in conjunction with WT display 6-7-fold higher NF-κB activity compared to cells containing only WT or only S222R MyD88 (see Fig E3, C), consistent with previous findings.8 Using a proximity ligation assay, S222R exhibited a 1.8-fold enhanced interaction with WT (using either tag combination) compared to interactions between two WT or two S222R-containing proteins (see Fig E3, D and E). These results demonstrate that the S222R mutation increases MyD88 interaction with WT MyD88, indicating that heterozygosity is sufficient for, and may actually maximize, MyD88 S222R gain-of-function effects.
We investigated MyD88 pathway activation in patient dermal fibroblasts, finding significantly elevated gene expression of several neutrophil-attracting chemokines (NanoString dataset available upon request). Elevations in CXCL1, CXCL5, and CXCL8 were confirmed by qPCR (Fig 2, A), and at the protein level for CXCL1 and IL-8 (see Fig E4, A, in this article's Online Repository at www.jacionline.org). Patient fibroblast-conditioned media attracted healthy donor neutrophils to a greater extent than control-conditioned media (see Fig E4, B). MyD88 knockdown caused a significant reduction in CXCL8 (Fig 2, B), which was not seen with knockdown of the upstream TIR-containing adaptor protein (TIRAP) (Fig 2, C). Downstream, the IRAK4 inhibitor AS2444697 caused a dose-dependent reduction in baseline CXCL8, with little effect in control cells (Fig 2, D). Lastly, knockdown of NF-κB p65 subunit significantly reduced CXCL8 to levels approaching control cells (Fig 2, E). Similar results were seen with CXCL1 and CXCL5 (not shown).
Fig 2.
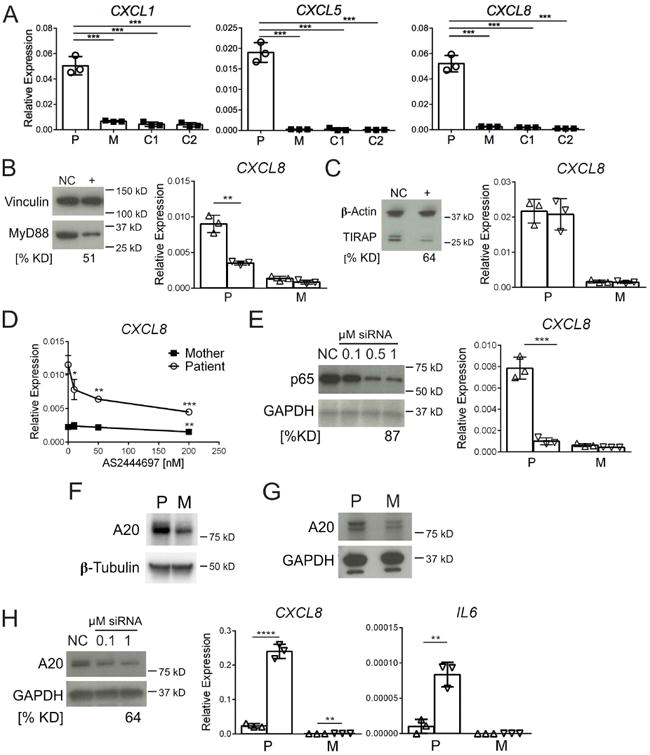
Enhanced neutrophil-attracting chemokine expression. A, Chemokine gene expression in dermal fibroblasts from patient (P), mother (M), and 2 unrelated healthy controls (C1 and C2). B, Effect of MyD88 knockdown on CXCL8 gene expression. C, Effect of TIRAP knockdown on CXCL8 gene expression. D, Effect of IRAK4 inhibitor (AS2444697) on CXCL8 gene expression. Asteriks represent p-values comparing AS2444697 concentrations vs. baseline (0 nM) for each subject. E, Effect of NF-κB p65 knockdown on CXCL8 gene expression. F, Baseline A20 protein expression in EBV-LCLs from patient (P) and mother (M). G, Baseline A20 protein expression in fibroblasts. H, A20 knockdown immunoblot in patient fibroblasts and resultant CXCL8 and IL6 gene expression. For knockdown blots, NC, negative control siRNA; (+), specific target knockdown; % KD, percent decrease from NC siRNA protein expression. Vinculin, β-Actin, GAPDH and β-Tubulin used as blot loading controls. For knockdown graphs, (Δ) negative control siRNA and (∇) specific target siRNA. For all graphs, each data point represents the median of 3 replicates, with results from 3 independent experiments (n=3) shown. Error bars represent ± SD. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
A20 (TNFAIP3), which negatively regulates the NF-κB pathway,E2 was upregulated in patient-derived EBV-LCL (Fig 2, F) and fibroblasts (Fig 2, G). It has been previously reported that A20 upregulation in murine B lymphocytes containing MyD88 L265P limits NF-κB activation and proliferation.E3 Knockdown of patient fibroblast A20 significantly increased CXCL8 (10.1-fold), as well as CXCL1 and CXCL5 (not shown) and caused an emergence of IL6 expression (Fig 2, H). These results suggest that A20 constrains – albeit incompletely - the increased activity of MyD88 S222R-regulated pathways producing pro-inflammatory mediators.
The S222R mutation has been reported in a single DLBCL cell line (SUDHL2),4 where it leads to overproduction of IL-6.E4 We confirmed and extended the SUDHL2 result to include IL-8 (see Fig E5, A, in this article's Online Repository at www.jacionline.org), and demonstrate that patient-derived EBV-LCL with MyD88 S222R also overexpress IL-6/IL-8 at the mRNA and protein level in an IRAK4-dependent manner (see Fig E5, B). SUDHL2, like other MYD88 mutation containing DLBCL cells, overexpress and hyper-phosphorylate STAT3 (Fig E5, C), which is believed to be due to autocrine IL-6.E4 Similarly, patient-derived EBV-LCL exhibit increased p-STAT3 (Y705) (see Fig E5, D), although STAT3 expression is not increased. To directly assess the role of autocrine IL-6, we first treated DLBCL U2932 cells (expressing wild-type MyD88) with SUDHL2-conditioned media, which caused increased p-STAT3 that could be partially blocked by the IL-6 receptor antagonist tocilizumab (TCZ) (see Fig E5, E). Similarly, we exposed unrelated healthy control EBV-LCL to patient or mother EBV-LCL-condition media. Like SUDHL2 media, patient-conditioned media caused increased p-STAT3 in these cells, that could also be partially blocked with TCZ, demonstrating bioactive IL-6 secreted from patient B cells (see Fig E5, F). Both SUDHL2 and patient's EBV-LCL also overproduced IL-10 compared to control cells (data not not shown), which could also be influencing Y705 p-STAT3 levels.
This is the first report of a germline gain-of-function mutation in MYD88 (S222R) in a subject with destructive polyarthritis and rash. We show that S222R activates MyD88 confirming and extending previous observations,4,7,8 and provide novel evidence for enhanced interaction with WT MyD88 as a plausible mechanism. Increased production of neutrophil-attracting chemokines and IL-6 are likely to contribute to the joint and skin phenotype, and the patient exhibited immunological perturbations consistent with increased MyD88 signaling, such as increased leukocyte p-STAT3, shedding of CD16, and increased whole blood secretion of IL-6 and TNF-α. Interestingly, our subject lacked evidence of lymphoproliferation. It is unlikely that S222R alone is sufficient to cause lymphoproliferation as the S222R-containing SUDHL2 cell line also harbors bialleilic functional deletions of A20 and its re-expression arrests proliferation.E5 Similarly, ectopic expression of S209R-MyD88 (the murine ortholog of S222R) in murine B cells does not induce proliferation.E3 Further delineating the mechanism(s) by which MyD88 S222R leads to the clinical and immunological phenotype will require additional studies including animal modeling.
Supplementary Material
Fig E1. Genotype of pedigree and patient CD14+ monocytes, dermal fibroblasts, and EBV-LCLs. A, Sanger sequencing chromatograms of peripheral leukocyte DNA. P, patient; M, mother; F, father; S, sister. B, Sanger sequencing chromatograms demonstrating the presence of c.666T>G mutation in patient CD14+ monocytes, dermal fibroblasts, and EBV B lymphoblastoids.
Fig E2. Immunophenotyping of peripheral blood dendritic cells. CD1c (BDCA-1), CD303 (BDCA-2), CD141 (BDCA-3), and CD203c surface expression for CD123+CD11c- (Q1), CD123+CD11c+ (Q2), and CD123-CD11c+ (Q3) dendritic cells. Positive control CD203c+ cells were gated from HLA-DR+ myeloid fraction of peripheral leukocytes.
Fig E3. Molecular dynamics modeling and effect of S222R on MyD88 self-association. A, Molecular dynamics cluster analysis of wild type (left) and S222R (right) MyD88 TIR domains. Images show superposition of the representative structure for the most populated cluster for each of 4 independent replicate simulations. B, Representative immunoblot showing relative expression of MyD88-AU1 and MyD88-GFP in THP-1 transductants, with vinculin as a loading control. C, NF-κB activity measured by secreted alkaline phosphatase activity as reporter gene. Activity is relative to cells containing WT-MyD88-AU1 & WT-MyD88-GFP. Each data point represents the average of the median of 3 replicates from 3 independent experiments. D, Representative confocal images of proximity ligation assay (PLA). Image 1, MyD88-KO; 2, WT-MyD88-AU1 and WT-MyD88-GFP; 3, WT-MyD88-AU1 and S222R-MyD88-GFP; 4, S222R-MyD88-AU1 and WT-MyD88-GFP; and 5, S222R-MyD88-AU1 and S222R-MyD88-GFP. Blue, DAPI and red, PLA event using anti-AU1 and anti-GFP antibodies. E, Average PLA events per cell from 3 independent experiments. Error bars represent ± SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Fig E4. Dermal fibroblast neutrophil-attracting chemokine secretion and function. A, unstimulated patient and control fibroblast secretion of CXCL1 and IL-8 over 24 hours. B, unrelated health donor neutrophil chemotaxis over 4 hours in response to patient and mother fibroblast-conditioned media. P, patient; M, mother. RLU, relative light units. For A and B, each data point represents the median of 3 replicates, with results from 3 independent experiments (n=3) shown. Error bars represent ± SD. *, p<0.05; ****, p<0.0001.
Fig E5. SUDHL2 and patient EBV-LCL IL-6 and IL-8 expression, and STAT3 phosphorylation. A, Chemokine/cytokine gene and protein expression from U2932 and SUDHL2 cell lines. B, Chemokine/cytokine protein and gene expression from EBV-LCLs, without (Control) and with AS2444697 (200 nM for 16 hours) as indicated. P, Patient; M, mother; HC, unrelated healthy control. C, Levels of P-STAT3 and total STAT3 in SUDHL2 and U2932 cells. Representative immunoblot (left) with quantification (right), from 4 independent experiments. Due to marked overexpression of total STAT3 in SUDHL2 cells, blot was run using 1:10 dilution of SUDHL2 protein lysate. D, Levels of P-STAT3 and total STAT3 in EBV-LCLs from patient (P) and mother (M). E, STAT3 phosphorylation in U2932 cells in response to 2 hour incubation with conditioned media from SUDHL2 or U2932 cells. Conditioned media was used alone, or after human IgG (IgG) or tocilizumab (TCZ; 200 ng/mL) pre-treatment. Relative STAT3 phosphorylation (P-STAT3) was determined as for D. F, STAT3 phosphorylation in EBV-LCLs from healthy control (HC) in response to conditioned media from HC, mother (M), or patient (P). Conditioned media was used alone or after IgG or TCZ pre-treatment, and relative STAT3 phosphorylation determined as described in E. For D, E, and F, GAPDH used as a loading control. Error bars represent ± SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Table E1. Summary of subject's laboratory values and peripheral leukocyte immunophenotyping
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Z01-AR041184), Pediatric Translational Research Branch, of the National Institutes of Health.
Footnotes
The authors report no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. 2009;284(37):25404–25411. doi: 10.1074/jbc.M109.022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guven-Maiorov E, Keskin O, Gursoy A, VanWaes C, Chen Z, Tsai CJ, et al. The architecture of the TIR domain signalosome in the Toll-like receptor-4 signaling pathway. Sci Rep. 2015;5:13128. doi: 10.1038/srep13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321(5889):691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med. 2012;367(9):826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 7.Vyncke L, Bovijn C, Pauwels E, Van Acker T, Ruyssinck E, Burg E, et al. Reconstructing the TIR Side of the Myddosome: a Paradigm for TIR-TIR Interactions. Structure. 2016;24(3):437–447. doi: 10.1016/j.str.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Avbelj M, Wolz OO, Fekonja O, Benčina M, Repič M, Mavri J, et al. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood. 2014;124(26):3896–3904. doi: 10.1182/blood-2014-05-573188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig E1. Genotype of pedigree and patient CD14+ monocytes, dermal fibroblasts, and EBV-LCLs. A, Sanger sequencing chromatograms of peripheral leukocyte DNA. P, patient; M, mother; F, father; S, sister. B, Sanger sequencing chromatograms demonstrating the presence of c.666T>G mutation in patient CD14+ monocytes, dermal fibroblasts, and EBV B lymphoblastoids.
Fig E2. Immunophenotyping of peripheral blood dendritic cells. CD1c (BDCA-1), CD303 (BDCA-2), CD141 (BDCA-3), and CD203c surface expression for CD123+CD11c- (Q1), CD123+CD11c+ (Q2), and CD123-CD11c+ (Q3) dendritic cells. Positive control CD203c+ cells were gated from HLA-DR+ myeloid fraction of peripheral leukocytes.
Fig E3. Molecular dynamics modeling and effect of S222R on MyD88 self-association. A, Molecular dynamics cluster analysis of wild type (left) and S222R (right) MyD88 TIR domains. Images show superposition of the representative structure for the most populated cluster for each of 4 independent replicate simulations. B, Representative immunoblot showing relative expression of MyD88-AU1 and MyD88-GFP in THP-1 transductants, with vinculin as a loading control. C, NF-κB activity measured by secreted alkaline phosphatase activity as reporter gene. Activity is relative to cells containing WT-MyD88-AU1 & WT-MyD88-GFP. Each data point represents the average of the median of 3 replicates from 3 independent experiments. D, Representative confocal images of proximity ligation assay (PLA). Image 1, MyD88-KO; 2, WT-MyD88-AU1 and WT-MyD88-GFP; 3, WT-MyD88-AU1 and S222R-MyD88-GFP; 4, S222R-MyD88-AU1 and WT-MyD88-GFP; and 5, S222R-MyD88-AU1 and S222R-MyD88-GFP. Blue, DAPI and red, PLA event using anti-AU1 and anti-GFP antibodies. E, Average PLA events per cell from 3 independent experiments. Error bars represent ± SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Fig E4. Dermal fibroblast neutrophil-attracting chemokine secretion and function. A, unstimulated patient and control fibroblast secretion of CXCL1 and IL-8 over 24 hours. B, unrelated health donor neutrophil chemotaxis over 4 hours in response to patient and mother fibroblast-conditioned media. P, patient; M, mother. RLU, relative light units. For A and B, each data point represents the median of 3 replicates, with results from 3 independent experiments (n=3) shown. Error bars represent ± SD. *, p<0.05; ****, p<0.0001.
Fig E5. SUDHL2 and patient EBV-LCL IL-6 and IL-8 expression, and STAT3 phosphorylation. A, Chemokine/cytokine gene and protein expression from U2932 and SUDHL2 cell lines. B, Chemokine/cytokine protein and gene expression from EBV-LCLs, without (Control) and with AS2444697 (200 nM for 16 hours) as indicated. P, Patient; M, mother; HC, unrelated healthy control. C, Levels of P-STAT3 and total STAT3 in SUDHL2 and U2932 cells. Representative immunoblot (left) with quantification (right), from 4 independent experiments. Due to marked overexpression of total STAT3 in SUDHL2 cells, blot was run using 1:10 dilution of SUDHL2 protein lysate. D, Levels of P-STAT3 and total STAT3 in EBV-LCLs from patient (P) and mother (M). E, STAT3 phosphorylation in U2932 cells in response to 2 hour incubation with conditioned media from SUDHL2 or U2932 cells. Conditioned media was used alone, or after human IgG (IgG) or tocilizumab (TCZ; 200 ng/mL) pre-treatment. Relative STAT3 phosphorylation (P-STAT3) was determined as for D. F, STAT3 phosphorylation in EBV-LCLs from healthy control (HC) in response to conditioned media from HC, mother (M), or patient (P). Conditioned media was used alone or after IgG or TCZ pre-treatment, and relative STAT3 phosphorylation determined as described in E. For D, E, and F, GAPDH used as a loading control. Error bars represent ± SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Table E1. Summary of subject's laboratory values and peripheral leukocyte immunophenotyping