

Abstract
We investigated the feasibility of a combination therapy comprising fasudil, a Rho-kinase inhibitor, and DETA NONOate (diethylenetriamine NONOate, DN), a long-acting nitric oxide donor, both loaded in liposomes modified with a homing peptide, CAR (CARSKNKDC), in the treatment of pulmonary arterial hypertension (PAH). We first prepared and characterized unmodified and CAR-modified liposomes of fasudil and DN. Using individual drugs alone or a mixture of fasudil and DN as controls, we studied the efficacy of the two liposomal preparations in reducing mean pulmonary arterial pressure (mPAP) in monocrotaline (MCT) and SUGEN-hypoxia-induced PAH rats. We also conducted morphometric studies (degree of muscularization, arterial medial wall thickness, and collagen deposition) after treating the PAH rats with test and control formulations. When the rats were treated acutely and chronically, the reduction in mPAP was more pronounced in the liposomal formulation-treated rats than in plain drug-treated rats. CAR-modified liposomes were more selective in reducing mPAP than unmodified liposomes of the drugs. Both drugs, formulated in CAR-modified liposomes, reduced the degree of muscularization, medial arterial wall thickness, and collagen deposition more than the combination of plain drugs did. As seen with the in vivo data, CAR-modified liposomes of fasudil or DN increased the levels of the vasodilatory signaling molecule, cGMP, in the smooth muscle cells of PAH-afflicted human pulmonary arteries. Overall, fasudil and DN, formulated in liposomes, could be used as a combination therapy for a better management of PAH.
Keywords: pulmonary arterial hypertension, nitric oxide donor, fasudil, CAR peptide, peptide-conjugated liposomes, inhalation delivery
Graphical Abstract

1. INTRODUCTION
The current practice for the treatment of pulmonary arterial hypertension (PAH) entails the use of two or more drugs so that drugs with varying mechanisms of action can target various pathological signaling hubs, produce additive effects, improve efficacy, reduce the dose, and ease the transition from the injectable to oral therapy.1 For PAH therapy, endothelin, nitric oxide, and prostacyclin are the three major pathways of PAH pathogenesis that are chiefly targeted.2 As such, possible drug combinations are prostacyclin analogues plus endothelin receptor antagonists (ERAs), ERAs plus phosphodiesterase type 5 (PDE-5) inhibitors, and prostacyclin analogues plus PDE-5 inhibitors.3
However, clinical trials, conducted in Europe and the USA,4,5 and a series of meta-analyses6,7 suggest that the combination therapy using two or more drugs only marginally improves the clinical deterioration and pulmonary hemodynamics in PAH patients, and that the patient mortality has been no better in the combination arms than in patients treated with a single drug therapy.8 The patient outcomes have not improved much because current medications cannot cure the underlying pathophysiology, pulmonary vascular remodeling, and right heart enlargement.9 Thus, there is a need to better the existing therapy, explore new drug classes, identify superior combination therapy, and develop efficient carriers for a more selective delivery of anti-PAH drugs.
In the pathogenesis of PAH, the RhoA/Rho kinase (ROCK) pathways are known to be implicated at various stages of the disease development and progression.10 Rho, a small GTP-binding protein, after binding with the kinase domain of the enzyme, activates ROCK, suppresses myosin phosphatase activity, increases the amount of phosphorylated myosin light chain, and finally causes vascular smooth muscle cell (VSMC) contraction.11 Also, increased ROCK activity downregulates the endothelial nitric oxide synthase (eNOS) and thus increases the proliferation and migration of VSMC.12,13 The use of ROCK inhibitors has brought about therapeutic benefits in various diseases, including PAH.14 Of the various ROCK inhibitors, fasudil (Figure 1A) has emerged as a new drug for the treatment of PAH.15,16 We and others have shown that plain fasudil and fasudil formulated in liposomes, micelles, and erythrosomes produce pulmonary preferential vasodilation in PAH rats and eliminate/reduce the systemic side effects, such as peripheral vasodilation.17–20
Figure 1.

Structure of (A) Fasudil, (B) DETA NONOate, and (C) Cyclic CAR peptide.
The therapeutic benefit of a possible combination therapy comprising fasudil, especially the inhaled form of the drug, and other anti-PAH drugs has not yet been studied, except in a recent study wherein oral fasudil was used in combination with oral sildenafil.21 Like sildenafil, DETA NONOate (diethylenetriamine NONOate, DN; Figure 1B), a long-acting nitric oxide (NO) donor, targets the guanylate cyclase/cyclic guanosine monophosphate pathway. In a recent study, we have shown that DN can be used as an anti-PAH drug, and DN formulated in peptide-modified liposomes can be substituted for the ultrashort-acting inhaled NO in PAH therapy.20,22 The vasodilatory effect of NO at least, in part, stems from its inhibitory effect on the ROCK activity.23 Fasudil (Figure 1C), on the other hand, by inhibiting RhoA-mediated down-regulation of eNOS, can increase the availability of NO.24 Thus, the combination of fasudil and DN can presumably produce a synergistic effect by acting on both Rho-kinase and NO signaling.
As such, here we wish to test the hypothesis that fasudil and DN, when formulated in liposomes and administered as an admixture of two formulated drugs, additively reduce mean pulmonary arterial pressure (mPAP) and ameliorate pathological changes that occur in the hypertensive pulmonary arteries. We have prepared two liposomal formulations, plain liposomes and peptide-modified liposomes, to encapsulate the two drugs separately. We have used a cyclic peptide, CARSKNKDC (CAR, Figure 1C), to modify the liposomes because CAR homes onto PAH lesions and causes the drug formulations to accumulate in the diseased pulmonary arteries.18 CAR-modified liposomes are highly selective for the PAH-afflicted pulmonary arteries and thus elicit anti-PAH effects on the diseased pulmonary vasculature.18,20,25 By administering the formulations acutely and chronically to PAH rats, we have assessed the pulmonary hemodynamics, conducted morphometric studies, and determined the relative efficacies of the formulations in attenuating right ventricular hypertrophy (RVH), muscularization of arteries, and collagen deposition.26 By incubating the formulations with PAH-afflicted pulmonary arterial smooth muscle cells (PAH-SMCs), we have also evaluated the effect of the formulation on cGMP levels released by the cells.
2. MATERIALS AND METHODS
Lipids were procured from Avanti Polar Lipids, Inc. (Alabaster, AL), and N-succinimidyl 1,3-(2-pyridyldithio)propionate (SPDP) was from Molecular Biosciences (Boulder, CO). Diethylenetriamine NONOate (DN) was obtained from Cayman Chemical (Ann Arbor, MI). Fasudil hydrochloride was purchased from LC Laboratories (Woburn, MA). The CAR peptide (currently licensed by Vascular BioSciences, Durham, NC) was synthesized and supplied by LifeTein, LLC (South Plainfield, NJ). Other chemicals were procured from Fisher Scientific (Pittsburgh, PA) and Sigma-Aldrich, Inc. (St. Louis, MO). All chemicals were of analytical grade and used without further purification.
2.1. Preparation of the CAR-Conjugated Liposomes
On the basis of our previous studies,20,22 we used a film hydration method to prepare unmodified liposomes comprising cholesterol (CH), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-disteroyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE).20,22 We used ammonium sulfate-based active loading20 and sodium hydroxide-based passive loading22 to encapsulate fasudil and DN, respectively, into the liposomes. Unencapsulated fasudil and DN were removed by passing the formulations through a PD-10 column (Sephadex-25, GE Healthcare, Piscataway, NJ) containing phosphate-buffered saline (PBS, pH 7.4).
CAR peptide was conjugated onto the surface of the liposomes by using a previously established SPDP chemical cross-linking method.20,22 In short, SPDP was dissolved in 30 μL of dimethylformamide (DMF) and incubated with liposomes at room temperature to functionalize the amine groups of the phospholipid. Excess SPDP was removed from the liposomes by ultracentrifugation and sedimentation. Liposomes were resuspended in PBS and incubated with 1 mg of CAR peptide, dissolved in 100 μL of PBS, for 1 h. The unconjugated peptide was further removed by ultracentrifugation. We then evaluated the liposomal formulations for size, zeta potential, encapsulation efficiency, and in vitro release profiles, which were similar to those of the formulations used in our previous studies.20,22
2.2. Development of PAH in Rats
We have studied the acute and chronic effects of the two drugs and their formulations in monocrotaline-27 and SUGEN5415/hypoxia-induced25 PAH rats. In the acute studies, wherein rats were treated with a single dose of the drugs or formulations, we induced PAH by injecting 50 mg/kg MCT, subcutaneously, into adult male Sprague-Dawley (SD) rats (250–300 g) and allowed 4 weeks to develop PAH. These PAH rats received various treatments, listed below, 28 days after the MCT injection. For chronic studies, when the rats received various formulations every 48 h for 21 days, we administered 20 mg/kg of SUGEN-5416 (Bio-Techne, Minneapolis, MO), as a single subcutaneous injection, to 250–300 g male adult SD rats, and housed them in a hypoxic chamber (BioSpherix, Lacona, NY) at 10% oxygen for 3 weeks. After 21 days of hypoxia, rats were moved from the hypoxic chamber to normoxia and were treated with various formulations as described below. Both MCT- and Sugen-injected rats and those placed in normoxia had free access to food and water. All studies were carried out in agreement with NIH Guidelines for the Care and Use of Laboratory Animals under a protocol (AM-10012) that was approved by the Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee.
2.3. Hemodynamic Measurements
We used a Power-Lab 16/30 with LabChart Pro 7.0 software (ADInstruments, Inc., Colorado Springs, CO) to record the pulmonary hemodynamics of PAH rats treated with two drugs and their formulations. Rats, anesthetized with a cocktail of ketamine (90 mg/kg) and xylazine (10 mg/kg), were catheterized via the right carotid and pulmonary arteries to measure the mean systemic arterial pressure (mSAP) and mPAP, respectively. Making a small incision in the right ventral neck, we exposed the right jugular vein and then the right carotid artery, wherein we inserted a PE-50 (BD Intramedic, Sparks, MD) catheter to measure mSAP. By maneuvering through the right jugular vein and then through the right ventricle, we inserted a PV-1 (Tygon, Lima, OH) catheter, curved at a 60–65° angle,28 into the pulmonary artery. Memscap SP844 physiological pressure transducers (Memscap AS, Scoppum, Norway) and bridge amplifiers, connected to the PowerLab system, were used to record mPAP and mSAP. In measuring mPAP, we confirmed the placement of the catheter in the pulmonary artery by the visually inspecting the characteristic peaks for PAP, displayed by PowerLab system.29
2.4. Treatments of PAH Rats with the Drugs and Formulations
Using a microsprayer (Penn-Century Micro-Sprayer, Philadelphia, PA) for small animals, we administered the drugs and formulations directly into the lungs. For single dose studies, three groups (n = 3 or 4) of MCT-induced PAH rats30,31 received 3 mg/kg of fasudil and 1 mg/kg of DN, administered intra-tracheally, in the following forms: (i) a mixture of fasudil and DN in saline, (ii) an admixture containing fasudil-filled and DN-filled unmodified liposomes, and (iii) an admixture containing CAR-modified liposomes of fasudil and DN. All liposomal formulations were prepared freshly on the day of dosing or were used within 4 weeks after preparation, the period when liposomes remain stable upon refrigeration. For these groups of rats, we started recording mPAP and mSAP soon after administration of the drugs or formulations and continued recording both pressures for 6 h after the dosing. In the case of chronic studies, six groups of (n = 3 or 4) Sugen/hypoxia-induced PAH rats received six different treatments, every 48 h, for 3 weeks, when rats were in normoxia, via the intra-tracheal route: (i) saline, (ii) plain fasudil, (iii) plain DN, (iv) plain fasudil plus plain DN in saline, (v) an admixture of unmodified liposomal formulations of fasudil and DN, and (vi) an admixture of CAR-modified liposomes of fasudil and DN. An additional group of healthy rats with no PAH, the seventh group, received intra-tracheal saline and was used as a sham control. mPAP and mSAP in these groups were recorded for 30 min on the 21st day of normoxia (42nd day considering the length of time before the treatment: 21 days in hypoxia and 21 days in normoxia).
2.5. Preparation of Heart and Lung Samples and Measurement of RVH
To measure RVH and conduct the morphometric analysis, we collected the hearts and lungs from the Sugen/hypoxia/normoxia-induced PAH rats. At the end of the 21-day treatment period, we anesthetized the PAH rats and measured pulmonary hemodynamics, as described above. After recording the mPAP, we sacrificed the animals, opened the chests, and excised the hearts and lungs. The hearts were washed with saline; the lungs were inflated, fixed with 10% formalin, and removed en bloc from the chest cavity, and the block was stored in paraformaldehyde for a day.29 The hearts and lungs were later separated from each other to measure RVH and conduct morphometric analyses, respectively. For the RVH measurement, we separated the right ventricle (RV) from the left ventricle (LV) and associated septum (S), weighed and calculated the degree of RVH from the ratio of the weight of the right ventricle to that of the left ventricle plus septum (RV/ LV+S).
2.6. Staining of Lung Sections for α-Smooth Muscle Actin and the von-Willebrand Factor
Using a Leica Microtome RM2255 (Leica Biosystems, Buffalo Grove, IL), we made formalin-fixed and paraffin-embedded 5 μm slices of left lungs, deparaffinized with CitriSolv (Fisher Scientific, Pittsburgh, PA), and then rehydrated with a series of gradient ethanol. Sections were boiled in antigen unmasking solution; blocked using 10% horse serum (Vector Laboratories, Burlingame, CA); incubated overnight first with a mouse monoclonal α-smooth muscle actin (α-SMA) antibody, clone 1A4 (Sigma-Aldrich, St. Louis, MO), and then with peroxidase-conjugated anti-mouse/anti-rabbit secondary antibody (RTU Vectastatin Universal, Vector Laboratories, Burlingame, CA); and finally stained with peroxidase sensitive ImmPACT diaaminobenzidine substrate (Vector Laboratories, Burlingame, CA). Similarly, the von-Willebrand factor (vWF) was stained with rabbit polyclonal anti-vWF antibody (Sigma-Aldrich, St. Louis, MO) and peroxidase sensitive ImmPACT VIP substrate (Vector Laboratories, Burlingame, CA).32 The nuclei were counterstained with methyl green (Vector Laboratories, Burlingame, CA), and slides were viewed at 200× using the Olympus IX81 microscope (Olympus Scientific Solutions Americas Corp., Waltham, MA). ImageJ software analyzed the images, determined the degree of muscularization, and measured the medial wall thickness (MWT).
2.7. Degree of Muscularization and MWT
Pulmonary blood vessels that were <100 μm in diameter and had positive α-SMA were used to determine the degree of muscularization.27 On the basis of the α-SMA staining, vessels were categorized as fully muscularized (100-75%), partially muscularized (<75%), and nonmuscularized. The degree of muscularization was represented as the % ratio of the number of fully muscularized blood vessels to the number of total vessels. The MWT was determined using 10 muscularized arteries (<50 μm in diameter). To measure MWT, we measured the perpendicular lumen radius and thickness of vessels and then calculated MWT from the ratio of the average of vessel thickness to the average of vessel radius.
2.8. Extent of Collagen Deposition
We measured the extent of collagen deposition in muscularized pulmonary artery using Mallory’s trichrome staining.27 For this, first we washed the deparafinized lung slices with deionized water, which was stained in sequence with Groat’s iron hematoxylin, acid fuchsin, and aniline blue. After the slides were rinsed with tap water, treated with DPX mounting media (VWR International, Randor, PA), and finally covered with coverslips, we then visualized them under an Olympus IX81 microscope (Waltham, MA). ImageJ software analyzed the images and determined the extent of collagen deposition in arteries.
2.9. Effect of CAR-Modified Liposomes on cGMP Secretion by Pulmonary Arterial Smooth Muscle Cells
Primary PAH-SMCs, collected from a female PAH patient and obtained from the Pulmonary Arterial Hypertension Breakthrough Initiative (PHBI) cell bank (www.ipahresearch.org), were used in this experiment. We have used SMC growth medium (Lonza Inc., Walkersville, MD) for the cell growth. Cells were cultured in a 0.2% gelatin-coated T-flask and after 90% confluence, cells were seeded into 6-well gelatin-coated dishes at a concentration of 1.2 × 105 cells/well. All of the cells were used in a passage between 5 and 7 for this experiment. For the fasting and treatment, we have used SMC basal medium. The PAH cells were cultured and incubated during the treatment in a CO2 incubator at 37 °C and 5% CO2 flow.
To study the influence of plain drugs or the formulation on cGMP secreted by PAH-SMCs, we first treated the cells with (i) plain fasudil, (ii) plain DN, and (iii) a combination of plain fasudil plus DN. In each case, the drug concentration was 0.01 μM, which was selected based on pilot studies for identifying the concentrations that fall within the linear segment of the dose–response curve. Later, using CAR-modified or unmodified liposomes containing 0.01 μM of either drug, we treated the cells with (i) fasudil-loaded CAR-modified liposomes, (ii) DN-loaded CAR-modified liposomes, or (iii) an admixture of CAR-modified liposomes containing fasudil plus CAR-modified liposomes containing DN. We performed a parallel set of control studies by treating the cells with (i) blank (no drug) CAR-modified liposomes, (ii) blank unmodified liposomes, or (iii) no treatment. We used three replicates per experiment and the data were generated from three independent experiments.
For measuring cGMP levels, we used an ELISA kit (R&D Systems, Inc., Minneapolis, MN). First, we lysed the cells using the lysis buffer (composed of 20 mM Tris, 15% glycerol, 1% tritron X-100, 8 mM MgSO4, 150 mM NaCl, and 1 mM EDTA supplemented with 0.02% β-glycerophosphate, protease inhibitor, phosphatase inhibitor, and NaF) and quantified the total amount of protein using the BCA protein assay kit (Thermo Fisher Scientific Inc., Waltham, MA). For the cGMP analysis, we used 100 μg of protein for each treatment group and calculated the cGMP concentrations according to the manufacturer’s instructions.
2.10. Statistical Analysis
The data are presented in mean ± SD and were analyzed by one-way ANOVA followed by Fisher LSD post hoc analysis using Origin Pro 2015 (OriginLab Corporation, Northampton, MA). The data presented in Figure 2 were analyzed by a two-way repeated-measure ANOVA followed by Bonferroni’s post hoc test. p < 0.05 is considered to be statistically significant.
Figure 2.

Changes in mPAP and mSAP in MCT-induced PAH rats when a single dose of fasudil and DN was administered intra-tracheally in various forms: (A) a mixture of fasudil and DN in saline, (B) an admixture of liposomal formulations of fasudil and DN, and (C) an admixture of CAR-conjugated liposomal formulations of fasudil and DN. (D) Lung targeting indices (LTI) for different formulations (n = 3–4, *p < 0.05, §p < 0.005, Z̶p < 0.001, ¥p < 0.0001).
3. RESULTS
3.1. Properties of CAR-Modified Liposomes of Fasudil and DN
The diameters of CAR-modified liposomes of fasudil and DN were 202.3 ± 6.5 and 171.1 ± 4 nm, respectively, and the drug entrapment efficiency of liposomal fasudil was 78% and that of liposomal DN was 46%. Both formulations released the drugs for 8–12 h. CAR-modified liposomes of fasudil and DN did not undergo any major changes in vesicle size when stored at 4 °C for 28 days, although the drug concentration declined upon storage: 10% fasudil and 25% DN leached out of the liposomal formulations upon storage for 28 days. The physical properties and storage stability of unmodified liposomes (liposomes with no CAR) of both drugs were very similar to those of CAR-modified liposomes (Table 1). Importantly, the size, zeta potential, drug loading, stability upon storage and nebulization, and release properties of these liposomes were consistent with the properties of liposomal formulations used in our previous studies.20,22
Table 1.
Properties of Unmodified and CAR-Modified Liposomal Formulations of Fasudil and DN and Percent Reduction in mPAP Produced by the Formulations When Administered as a Monotherapy (One Drug Formulation) in Earlier Studies20,22
| liposomes | size (nm) | zeta potential (mV) | entrapment efficiency (%) | drug loss upon storage | reduction in mPAP (% from baseline) |
|---|---|---|---|---|---|
| fasudil in unmodified liposomes | 198.8 ± 5.2 | −33.4 ± 3.1 | 83 ± 5.6 | <10% (~7%) after 28 days at 4 °C | ~30a |
| DN in unmodified liposomes | 168.1 ± 3.0 | −32.8 ± 2.1 | 47 ± 5.5 | ~25% after 28 days at 4 °C | ~25–30a |
| fasudil in CAR-modified liposomes | 202.3 ± 6.5 | −35.6 ± 2.5 | 78 ± 5.7 | <10% after 28 days at 4 °C | ~40a |
| DN in CAR-modified liposomes | 171.1 ± 4.0 | −37 ± 3.8 | 46 ± 5.0 | ~25% after 28 days at 4 °C | ~40a |
3.2. CAR-Modified Liposomes of Fasudil and DN Reduce mPAP after a Single Dose
Previously, we have evaluated the effect of a monotherapy using CAR-modified liposomes of fasudil and DN along with their unmodified liposomal formulations in MCT-induced PAH rats.20 In our published studies, CAR-modified liposomes of fasudil and DN, given separately, reduced mPAP by 40% (Table 1), when administered intra-tracheally at the same doses as in this study, 3 mg/kg of fasudil and 1 mg/kg of DN, which were selected based on previous studies.20,22 The vasodilatory duration for fasudil and DN formulations was 250 and 180 min, respectively.
As described in the published report, 4 weeks after MCT was injected into the rats, the mPAP was 37 ± 10 mmHg, a 3-fold increase from the mPAP in healthy rats, indicating that the animals have developed PAH. Plain forms of fasudil plus DN reduced the mPAP by 35%, which lasted for ~60 min; mSAP also declined ~30%, which took ~150 min to return to the initial value (Figure 2A). Fasudil and DN loaded in unmodified liposomes, given intra-tracheally, produced a similar reduction in mPAP (30%) for ~150 min (Figure 2B); however, the reduction in mSAP was less than that of the plain drug combinations group (~17%), which returned to the initial value in >90 min. Compared with fasudil plus DN in saline (Figure 2A), fasudil and DN formulated in unmodified liposomes evoked a much more pronounced reduction in mPAP, which may have resulted in part from the amount of free drugs that got into the systemic circulation and that were released, slowly and locally, from the liposomal formulations onto the respiratory epithelium. The differences between the extents of reduction, both time and magnitude, in mPAP and in mSAP were statistically significant, as determined by repeated-measure ANOVA, which we performed based on the assumption that both drug and time after dosing may independently affect mPAP/mSAP, the treatment outcomes. Consistent with our previous studies with CAR-modified liposomes, the magnitude and duration of mPAP reduction were more prominent in rats treated with CAR-modified liposomes of fasudil and DN than in rats treated with the plain drug mixtures or mixtures of unmodified liposomes of two drugs. In the CAR-modified liposome-treated group, mPAP was reduced by 40% and the reduced mPAP was sustained for 6 h, but mSAP remained unchanged from the start to end of the experiments (Figure 2C). The lung targeting index (LTI), calculated from the ratio of the area above the pressure/time curve (AAC) for mPAP and mSAP using the equation LTI = AAC of mPAP/AAC of mSAP,20,22 of CAR-modified liposomes was 2-fold greater than that for plain liposomes, indicating that CAR-modified liposomes were more lung specific than the unmodified counterparts (Figure 2D).
3.3. Admixture of Fasudil- and DN-Filled CAR-Modified Liposomes Reduced mPAP without Reducing mSAP after Chronic Administration
The goal of chronic studies, performed in SUGEN/hypoxia-induced PAH rats, was to determine the effect of combination therapy in reducing the mPAP and improving various cellular and biochemical changes that occur in PAH-afflicted pulmonary vasculatures and right ventricles. Thus, before sacrificing the rats for various histological studies, we first studied the effect of the formulations on the pulmonary hemodynamics of rats that received various drugs and formulations every 48 h for 21 days of normoxia. We chose a dosing interval of 48 h to reduce the total number of dosing and so to alleviate the stress that animals may experience due to repeated dosing. Further, unlike in vitro release patterns, liposomal formulations deposited on the lung may produce a depot-like effect and release the drugs for more than 24 h.
On the 22nd day of normoxia, the mPAPs in sham rats (healthy rats with no PAH) were 13.91 ± 0.98 mmHg, but PAH rats treated with saline (no drug) showed an mPAP of 56.25 ± 4.11 mmHg, indicating the development of a severe form of PAH in these animals. However, mPAPs in rats treated with the drugs and formulations were significantly lower than that in rats treated with the vehicle (Figure 3A). The mPAPs decreased from 55 mmHg in vehicle-treated control PAH rats to 30–25 mmHg, a 45–60% reduction, in PAH rats treated with either a combination of plain drugs or combinations of the formulations. CAR-modified liposomal formulations elicited the greatest drop in mPAP (Figure 3B), which was statistically significant, when compared with mPAPs in other treatment groups. In contrast, mSAP remained practically unchanged in all of the treatment groups (Figure 3B).
Figure 3.
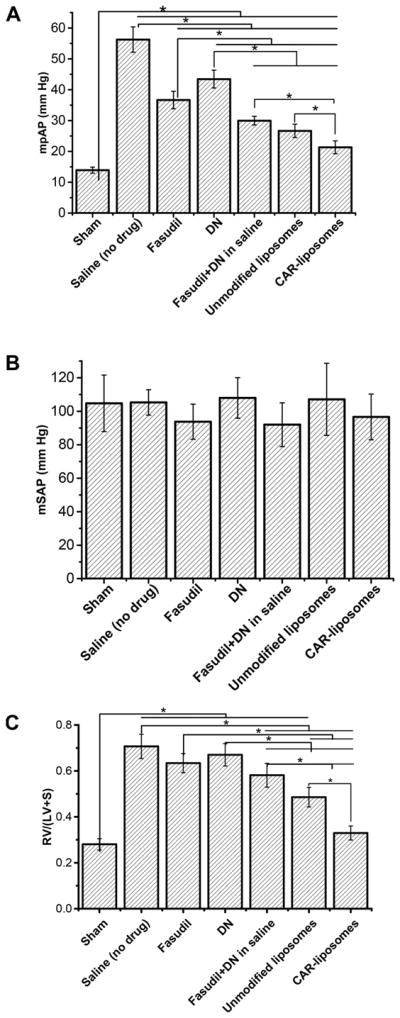
Influence of fasudil and DN, administered in various forms, for 21 days on (A) mPAP, (B) mSAP, and (C) RVH of SUGEN/ hypoxia-induced PAH rats. The various formulations were administered to PAH rats every 48 h for 3 weeks, when rats were kept in normoxia. Data represent mean ± SD (n = 3 or 4, *p < 0.05).
3.4. Admixture of Fasudil- and DN-Filled CAR-Modified Liposomes Reduced the RVH in PAH Rats
Because RV-triggered heart failure is a major cause of death in PAH patients, we investigated the effect of CAR-modified liposomes of fasudil and DN in reducing RVH. In vehicle-treated PAH rats, the RV/(LV+S) ratio (0.71 ± 0.05) was significantly greater than that of healthy animals, confirming that the PAH caused RVH (Figure 3C). The plain drug combination reduced RVH more than the vehicle or DN treatment did. Compared with the single or plain drug combination-treated PAH animals, the improvement in RVH was statistically significant in PAH rats treated with the mixture of the unmodified liposomes of two drugs (Figure 3C). However, like the hemodynamics data, the RV/(LV+S) dropped from 0.71 ± 0.05 in non-drug-treated rats to 0.33 ± 0.03, a > 50% decline, in rats treated with the admixture of CAR-modified liposomes of fasudil and DN. The regression in RVH was more pronounced in the CAR-modified liposome-treated group than in the unmodified liposome-treated group (Figure 3A). Indeed, statistically, the RV/(LV+S) ratio in CAR-modified liposome-treated rats was no different from that of healthy non-PAH rats.
3.5. Admixture of CAR-Modified Liposomes of Fasudil and That of DN Slows Arterial Remodeling
Because one of the major goals of PAH therapy is to arrest pulmonary arterial remodeling, a hallmark of the disease progression,26 we have investigated the effect of the combination therapy in ameliorating vascular remodeling by measuring the arterial MWT, degree of muscularization, and extent of collagen deposition.33,34
3.5.1. Arterial MWT
In PAH, small pulmonary arteries undergo muscularization as evidenced by the increased expression of α-SMA.35 The levels of vWF, a marker for vascular injury and dysfunction, increase in PAH patients because of endothelial cell dysfunction in the pulmonary arteries.36 We have evaluated the expression of α-SMA, vWF, and MWT in rats treated with various forms of drugs and formulations. In the absence of any drug treatments, vehicle-treated PAH rats showed an increased expression of α-SMA and vWF (Figure 4A), but the expression of these markers was reduced in rats treated with either CAR-modified liposomes or unmodified liposomes (Figure 4A). These data reiterate the evidence that chronic treatment with the CAR-modified liposomes reduces mPAP and reverses the structural changes in the pulmonary arterioles.
Figure 4.

Changes in pulmonary vascular morphometry upon chronic treatments in SUGEN/hypoxia-induced PAH rats with fasudil and DN given in various forms. (A) Representative photomicrographs of pulmonary arteries <100 μm in diameter indicated by arrows where brown and purple-brown colors show α-SMA and vWF, respectively. (B) MWT of pulmonary arteries <100 μm in diameter; the percentage of (C) nonmuscularized, (D) partially muscularized, and (E) fully muscularized small pulmonary arteries <100 μm in diameter. Data represent mean ± SD (n = 3 or 4, *p < 0.05).
As seen in the data presented in Figure 4A, the MWT of small pulmonary arteries (≤50 μm in diameter) increased from 0.14 ± 0.01 in healthy animals to 0.27 ± 0.02 in the vehicle treated PAH rats. The MWT was reduced to 0.18 ± 0.01 in PAH rats treated with CAR-modified liposomes of fasudil and DN. One-way ANOVA revealed that MWT in drug-treated groups was less than that in the non-drug-treated PAH group. Of all of the treatment groups, CAR-modified liposomes of fasudil and DN elicited the greatest reduction in MWT (Figure 4B).
3.5.2. Degree of Muscularization
Increased expression of α-SMA in PAH-afflicted arteries makes them more muscular, and thus pulmonary arteries and arterioles stiffen with the progression of the disease.26 PAH medications relax stiff vessels by reducing α-SMA expression and consequently decrease the amount of muscularized pulmonary arterioles.37–39 Consistent with the data on the pulmonary hemodynamics after chronic administration, the percent of nonmuscularized arteries in vehicle-treated PAH rats was greater than that of healthy rats (Figure 4C). The number of nonmuscularized vessels in PAH rats treated with unmodified liposomes and CAR-modified liposomes of fasudil and DN was greater than that of vehicle-treated PAH rats. Unlike nonmuscularized vessels, the percent of partially and fully muscularized arteries increased in the vehicle-treated group (Figure 4D,E). The differences between the percent of fully muscularized vessels in rats treated with liposomal combination therapy and that in vehicle-treated PAH rats were statistically significant, although there was no statistically significant difference between unmodified and CAR-modified liposome-treated groups. Together, these results show that liposomal combinations are effective in reducing arterial muscularization, a major contributing factor to the hypertension and right-heart remodeling in PAH patients.
3.5.3. Extent of Collagen Deposition
Like muscularization of arteries, deposition of extracellular matrix proteins, including collagen fibers, fibronectin, and elastin, is a pathogenic feature in PAH.40 Consistent with this fact, the amount of collagen in the pulmonary arteries of the PAH rats was greater than that in healthy animals (Figure 5A), and it was reduced in PAH rats treated with plain drugs or formulations. The combination of plain drugs and liposomal formulations reduced collagen deposition more than the individual drugs did. The percent collagen deposition, calculated for the whole microscopic field (Figure 5C), also showed that CAR-modified liposomes caused a more pronounced reduction in collagen deposition than plain drugs did. One-way ANOVA indicated that the drug treatment led to a significant reduction in collagen deposition, and that the amount of collagen in the treatment groups was no different than that in sham animals. On the basis of the fact that the amount of collagen deposition in liposome-treated groups was the same as that in sham animals, it is apparent that liposomal formulations reversed collagen deposition in PAH. However, like the data from muscularization studies, these data did not show superiority of CAR-modified liposomal formulation of the drugs over unmodified liposomal formulations.
Figure 5.

Extent of collagen deposition in pulmonary arterioles of SUGEN/hypoxia-induced PAH rats treated with various forms of fasudil and DN. (A) Representative photomicrographs of pulmonary arteries showing collagen (stained in blue as indicated by arrows). (B) Percent collagen deposition in individual pulmonary arteries. (C) Percent collagen deposition in the whole microscopic field (n = 3 or 4, *p < 0.05). Lung sections were stained with trichrome to visualize collagen deposition in arteries.
3.6. CAR-Modified Liposomes of Fasudil and DN Increase the Secretion of cGMP, a Vasodilatory Marker, in PAH-SMCs
Fasudil and DN, when treated as a single agent, increased the intracellular cGMP concentrations (Figure 6A), although liposomes without drug, when used as a negative control, had no effect on the cGMP levels. This is consistent with the fact that lipids used to prepare liposomes have no therapeutic effect. However, the level of cGMP markedly increased when cells were treated with a combination of fasudil and DN. Both fasudil and DN-loaded CAR liposomes showed a surge in the intracellular cGMP level, which elevated further when the cells were treated with an admixture of liposomes (Figure 6B). However, the mixture of unmodified liposomes of fasudil and DN did not increase the cGMP level higher than that of the single use of liposomes containing fasudil and DN (Figure 6C). On the contrary, the mixture of CAR-modified liposomes of fasudil and DN produced an increase in the cGMP level 2-fold higher than that of the unmodified liposome-treated groups and thus indicated a preferential accumulation of CAR-modified liposomes in PAH-SMCs.
Figure 6.

Levels of intracellular cGMP in PAH-SMCs treated with various forms of fasudil and DN. (A) Single plain drugs vs the combination of plain drugs. (B) Individual CAR liposomes formulation of the drugs or the admixture of two CAR-modified liposomes of fasudil and DN. (C) CAR-modified liposomal combination vs unmodified liposomes containing either drugs or admixture of two formulations. *p < 0.05.
4. DISCUSSION
In this study, we tested the hypothesis that drugs loaded in CAR-modified liposomes preferentially accumulate in the lungs, and that chronic administration of the admixture CAR-modified liposomes containing DN and fasudil improves the pulmonary hemodynamics and slows disease progression. Further, because of the progressive nature of the disease and lack of response to monotherapies, the therapeutic intervention in PAH involves an upfront combination therapy that recommends taking two or more drugs right after diagnosis of the disease.41,42 In line with the existing practice, we have evaluated the effects of an upfront combination therapy, upon chronic administration, comprising various forms of fasudil and DN, plain versus two liposomal formulations (no-CAR liposomes and CAR-modified liposomes). Further, we have used two different models for assessing the acute versus chronic effects of the formulations. While both models are widely used, the MCT model is better suited for acute studies because this model consistently reproduces the chief clinical symptoms of PAH, elevated mPAP. However, the MCT model can only partially recapitulate the histopathological changes that occur in the pulmonary arteries of PAH patients. Because of high mortality, the MCT model is not robust enough for chronic studies. The SUGEN5415/hypoxia model, on the other hand, closely resembles human PAH with plexiform lesions and severe RVH. This model allows studying the extent of muscularization, arterial wall thickening, RVH, and other histopathological features without the loss of any animals during the treatment period.43 As such, we used the MCT and SUGEN5415/hypoxia models for acute and chronic studies, respectively.
In the acute study, the rapid reductions in mPAP and mSAP may have resulted from the fast absorption of both drugs from the lungs, after aerosolization, and that from the strong vasodilation in the pulmonary and systemic vasculatures, as reported by us and others.17,19,20,22 The reason for a more pronounced reduction in mPAP was that liposomal formulations, unlike plain drugs, were pulmonary vasculature selective because of the liposomal entrapment and localized release of the drug in the lungs. Indeed, the vasodilatory duration for the combination therapy was >1.4-fold longer than that for monotherapy in our previous studies, indicating that the combination of the formulations additively extended the duration of action of the drugs.20,22 In addition, the differences between the mPAP and mSAP continued to be statistically significant for the duration of the study (360 min), suggesting a strong pulmonary selectivity. The prolongation of the vasodilatory duration in rats treated with CAR-modified liposomes, administered as an admixture of two separate formulations of the drugs, is the result of the additive effects of two vasodilators and the propensity of CAR peptides to accumulate in PAH lesions, as shown by us and others.19,44
Although two of the currently approved anti-PAH therapeutics are available for inhalational delivery (iloprost, Ventavis, and treprostinil, Tyvaso), little is known about the mechanism by which an inhaled targeted formulation produces pulmonary specific vasodilation. One hypothesis is that,20,22 after entering the systemic circulation, CAR-modified liposomes of the two drugs travel back from the systemic circulation to the PAH-afflicted pulmonary vasculature, and then they complex with the excess cell-surface heparan sulfate for internalization by the pulmonary arterial smooth muscle cells (PASMCs).45 The prevailing assumption is that increased expression of TGF-β in PASMCs, due to the vascular remodeling in PAH, induces cell-surface heparan sulfate expression.46–48 The drugs that are released from CAR-modified liposomes bind with the cell surface or are internalized by the cells, and subsequently they reduce mPAP to a greater extent than plain drugs, which enter the circulation much faster. Moreover, plain drugs are not equipped with a homing peptide for preferential accumulation in the pulmonary vasculature, nor can plain drugs act as a depot for a continuous release. CAR liposomal formulations caused little or no reduction in mSAP, because only a fraction of the two drugs gets into the circulation where, because of a short half-life, the drugs do not persist for long. Moreover, the larger LTI of CAR-modified liposomes also substantiates a greater drop in mPAP but a modest fall in mSAP. On the basis of these data, we assume that CAR-modified liposomes can be used for prolonging pulmonary selective vasodilation and arresting progression of PAH.
The goal of chronic studies was to determine the effect of the combination therapy in reducing mPAP and improving various cellular and biochemical changes that occur in PAH-afflicted pulmonary vasculatures and right ventricles. The CAR-modified liposomal formulations caused the greatest drop in mPAP, which was statistically significant, when compared with mPAPs in other treatment groups. These data echo our hypothesis that CAR-modified liposomes accumulate in PAH lesions of pulmonary arteries and release drug for an extended period. In contrast, no major changes in mSAP were observed in any of the treatment groups. The combination of CAR-modified liposomes of fasudil and DN, as the data suggest, can possibly be administered at a longer dosing interval in PAH patients, who require a life-long therapy. Considering that the current anti-PAH drugs are administered multiple times a day for the lifetime of PAH patients,49 the liposomal therapy, if it can be successfully translated into clinical application, will enhance patient compliance and improve the overall quality of life. However, in this study, we treated the rats with drug formulations for only 21 days, and the number of animals in each group was ~4. Moreover, Sugen/hypoxia-induced PAH is known to develop a more severe form of PAH, when rats are kept in normoxia for ≤12 weeks.50,51 Thus, we have to delve farther into the influence of chronic administration on the mortality of PAH rats and confirm whether the proposed combination therapy extends survival time by improving various pathological features in PAH.
For the evaluation of the RVH, the combination therapy using liposomal formulations not only slowed down arterial smooth muscle layer thickening and endothelial dysfunction but also reduced mPAP and bettered the structural changes in the pulmonary artery, such as reduced muscularization and collagen deposition. By decreasing MWT, the number of fully muscularized arteries, and the mean percent of collagen deposition, CAR-modified liposomes of fasudil and DN, when administered as an admixture, support our hypothesis that the combination of two vasodilators with lung-targeted delivery produces additive effects in reversing the disease. Cellular studies in PAH-SMCs showed that the combination of CAR-modified liposomal therapy increases the cellular cGMP, the common molecular regulator for both fasudil- and DN-mediated vasodilation via the Rho-kinase and NO pathways,24 more than any of the other drugs. CAR-modified liposomal combination of fasudil and DN, although it releases drugs slowly, can effectively modulate the vasodilatory signaling pathway in diseased PASMCs.
However, this preclinical study may not have adequate statistical power because we have used 3 or 4 animals (n = 3 or 4) in each treatment group, which we determined based on a pilot study instead of conventional power analysis. Further, all studies were performed in male animals, and as such, sex has not been used as a biological variable in this study. Because PAH is known to have a sex bias, the use of animals of both sexes would have provided more rigorous data. Importantly, if these formulations were to be used in a clinical setting, formulation should be optimized to extend the shelf-life for at least 6 months. Studying the distribution of both fasudil and DN onto organs other than the lungs may also give new insights into the safety of these two investigational drugs.
5. CONCLUSIONS
This study demonstrates that fasudil and DN, formulated in unmodified and CAR-modified liposomes, can be used as an upfront combination therapy in PAH. A combination therapy comprising these drugs decreases mPAP and extends vasodilatory duration in PAH rats in an additive fashion. Importantly, CAR-modified liposomes selectively reduce mPAP without reducing mSAP, a major side effect of current anti-PAH drugs. Human PAH-derived PASMCs showed a significant increase in vasodilatory signaling in response to the CAR-modified liposomal combination treatment. Chronic administration of the combination of the liposomal formulations of fasudil and DN mitigated the major pathological changes that occurred in the PAH rats. CAR-modified liposomes of fasudil and DN were most effective in reversing pulmonary hypertension and RVH.
Acknowledgments
This work was supported in part by two NIH Grants (R15HL103431 and R01HL114677) awarded to Fakhrul Ahsan. We obtained human PAH cells from the Pulmonary Hypertension Breakthrough Initiative (PHBI) cell bank. The PHBI is supported by a NHLBI R24 Grant (R24HL123767) and the Cardiovascular Medical Research and Education Fund (CMREF). The cellular studies were supported by a CMREF Grant awarded to Dr. Ahsan. We thank Dr. F. Alam for her help in cellular studies.
Footnotes
The authors declare no competing financial interest.
References
- 1.Channick RN. Combination therapy in pulmonary arterial hypertension. Am J Cardiol. 2013;111:16C–20C. doi: 10.1016/j.amjcard.2013.01.320. [DOI] [PubMed] [Google Scholar]
- 2.Latus H, Delhaas T, Schranz D, Apitz C. Treatment of pulmonary arterial hypertension in children. Nat Rev Cardiol. 2015;12:244. doi: 10.1038/nrcardio.2015.6. [DOI] [PubMed] [Google Scholar]
- 3.O’Callaghan DS, Savale L, Jais X, Natali D, Montani D, Humbert M, Simonneau G, Sitbon O. Evidence for the use of combination targeted therapeutic approaches for the management of pulmonary arterial hypertension. Respir Med. 2010;104:S74–80. doi: 10.1016/j.rmed.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Sitbon O, Sattler C, Bertoletti L, Savale L, Cottin V, Jais X, De Groote P, Chaouat A, Chabannes C, Bergot E, Bouvaist H, Dauphin C, Bourdin A, Bauer F, Montani D, Humbert M, Simonneau G. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J. 2016;47:1727–36. doi: 10.1183/13993003.02043-2015. [DOI] [PubMed] [Google Scholar]
- 5.Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 6.Fox BD, Shtraichman O, Langleben D, Shimony A, Kramer MR. Combination Therapy for Pulmonary Arterial Hypertension: A Systematic Review and Meta-analysis. Can J Cardiol. 2016;32:1520. doi: 10.1016/j.cjca.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Lajoie AC, Lauziere G, Lega JC, Lacasse Y, Martin S, Simard S, Bonnet S, Provencher S. Combination therapy versus monotherapy for pulmonary arterial hypertension: a meta-analysis. Lancet Respir Med. 2016;4:291–305. doi: 10.1016/S2213-2600(16)00027-8. [DOI] [PubMed] [Google Scholar]
- 8.Bai Y, Sun L, Hu S, Wei Y. Combination therapy in pulmonary arterial hypertension: a meta-analysis. Cardiology. 2011;120:157–65. doi: 10.1159/000334431. [DOI] [PubMed] [Google Scholar]
- 9.Galie N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2008;30:394–403. doi: 10.1093/eurheartj/ehp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doe Z, Fukumoto Y, Takaki A, Tawara S, Ohashi J, Nakano M, Tada T, Saji K, Sugimura K, Fujita H, et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ J. 2009;73:1731–1739. doi: 10.1253/circj.cj-09-0135. [DOI] [PubMed] [Google Scholar]
- 11.Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discovery. 2005;4:387–398. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- 13.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol. 2008;155:444–54. doi: 10.1038/bjp.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P, Zhang HM, Tang YJ, Sheng CF, Liu JX, Zeng YJ. Influence of Rho kinase inhibitor fasudil on late endothelial progenitor cells in peripheral blood of COPD patients with pulmonary artery hypertension. Bratisl Lek Listy. 2015;116:150–3. doi: 10.4149/bll_2015_030. [DOI] [PubMed] [Google Scholar]
- 15.Fujita H, Fukumoto Y, Saji K, Sugimura K, Demachi J, Nawata J, Shimokawa H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels. 2010;25:144–9. doi: 10.1007/s00380-009-1176-8. [DOI] [PubMed] [Google Scholar]
- 16.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91:391–392. doi: 10.1136/hrt.2003.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, Wang YF, Zhao QH, Jiang R, Wu Y, Peng FH, Xu XQ, Wang L, He J, Jing ZC. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: a randomized, controlled, crossover study. Int J Cardiol. 2014;177:61–5. doi: 10.1016/j.ijcard.2014.09.101. [DOI] [PubMed] [Google Scholar]
- 18.Gupta N, Al-Saikhan FI, Patel B, Rashid J, Ahsan F. Fasudil and SOD packaged in peptide-studded-liposomes: Properties, pharmacokinetics and ex-vivo targeting to isolated perfused rat lungs. Int J Pharm. 2015;488:33–43. doi: 10.1016/j.ijpharm.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta N, Ibrahim HM, Ahsan F. Peptide-micelle hybrids containing fasudil for targeted delivery to the pulmonary arteries and arterioles to treat pulmonary arterial hypertension. J Pharm Sci. 2014;103:3743–53. doi: 10.1002/jps.24193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nahar K, Absar S, Gupta N, Kotamraju VR, McMurtry IF, Oka M, Komatsu M, Nozik-Grayck E, Ahsan F. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol Pharmaceutics. 2014;11:4374–84. doi: 10.1021/mp500456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elias-Al-Mamun M, Satoh K, Tanaka S, Shimizu T, Nergui S, Miyata S, Fukumoto Y, Shimokawa H. Combination therapy with fasudil and sildenafil ameliorates monocrotaline-induced pulmonary hypertension and survival in rats. Circ J. 2014;78:967–76. doi: 10.1253/circj.cj-13-1174. [DOI] [PubMed] [Google Scholar]
- 22.Nahar K, Rashid J, Absar S, Al-Saikhan FI, Ahsan F. Liposomal Aerosols of Nitric Oxide (NO) Donor as a Long-Acting Substitute for the Ultra-Short-Acting Inhaled NO in the Treatment of PAH. Pharm Res. 2016;33:1696–1710. doi: 10.1007/s11095-016-1911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mills TM, Chitaley K, Lewis RW, Webb RC. Nitric oxide inhibits RhoA/Rho-kinase signaling to cause penile erection. Eur J Pharmacol. 2002;439:173–4. doi: 10.1016/s0014-2999(02)01408-5. [DOI] [PubMed] [Google Scholar]
- 24.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–77. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta N, Rashid J, Nozik-Grayck E, McMurtry IF, Stenmark KR, Ahsan F. Cocktail of Superoxide Dismutase and Fasudil Encapsulated in Targeted Liposomes Slows PAH Progression at a Reduced Dosing Frequency. Mol Pharmaceutics. 2017;14:830–841. doi: 10.1021/acs.molpharmaceut.6b01061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S13–S24. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 27.Gupta V, Gupta N, Shaik IH, Mehvar R, Nozik-Grayck E, McMurtry IF, Oka M, Komatsu M, Ahsan F. Inhaled PLGA particles of prostaglandin E(1) ameliorate symptoms and progression of pulmonary hypertension at a reduced dosing frequency. Mol Pharmaceutics. 2013;10:1655–67. doi: 10.1021/mp300426u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stinger RB, Iacopino VJ, Alter I, Fitzpatrick TM, Rose JC, Kot PA. Catheterization of the pulmonary artery in the closed-chest rat. J Appl Physiol. 1981;51:1047–50. doi: 10.1152/jappl.1981.51.4.1047. [DOI] [PubMed] [Google Scholar]
- 29.Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–97. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 30.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302:L363–9. doi: 10.1152/ajplung.00212.2011. [DOI] [PubMed] [Google Scholar]
- 31.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 32.Van Rheen Z, Fattman C, Domarski S, Majka S, Klemm D, Stenmark KR, Nozik-Grayck E. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol. 2011;44:500–8. doi: 10.1165/rcmb.2010-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones R, Zapol WM, Reid L. Pulmonary artery remodeling and pulmonary hypertension after exposure to hyperoxia for 7 days. A morphometric and hemodynamic study. Am J Pathol. 1984;117:273–85. [PMC free article] [PubMed] [Google Scholar]
- 34.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–72. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones R, Jacobson M, Steudel W. alpha-smooth-muscle actin and microvascular precursor smooth-muscle cells in pulmonary hypertension. Am J Respir Cell Mol Biol. 1999;20:582–94. doi: 10.1165/ajrcmb.20.4.3357. [DOI] [PubMed] [Google Scholar]
- 36.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. doi: 10.1146/annurev.biochem.67.1.395. [DOI] [PubMed] [Google Scholar]
- 37.Saito Y, Nakamura K, Akagi S, Sarashina T, Ejiri K, Miura A, Ogawa A, Matsubara H, Ito H. Epoprostenol sodium for treatment of pulmonary arterial hypertension. Vasc Health Risk Manage. 2015;11:265–270. doi: 10.2147/VHRM.S50368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schermuly RT, Kreisselmeier KP, Ghofrani HA, Samidurai A, Pullamsetti S, Weissmann N, Schudt C, Ermert L, Seeger W, Grimminger F. Antiremodeling effects of iloprost and the dual-selective phosphodiesterase 3/4 inhibitor tolafentrine in chronic experimental pulmonary hypertension. Circ Res. 2004;94:1101–1108. doi: 10.1161/01.RES.0000126050.41296.8E. [DOI] [PubMed] [Google Scholar]
- 39.Schermuly RT, Yilmaz H, Ghofrani HA, Woyda K, Pullamsetti S, Schulz A, Gessler T, Dumitrascu R, Weissmann N, Grimminger F, Seeger W. Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am J Respir Crit Care Med. 2005;172:358–63. doi: 10.1164/rccm.200502-296OC. [DOI] [PubMed] [Google Scholar]
- 40.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 41.Sitbon O, Jais X, Savale L, Cottin V, Bergot E, Macari EA, Bouvaist H, Dauphin C, Picard F, Bulifon S, Montani D, Humbert M, Simonneau G. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43:1691–7. doi: 10.1183/09031936.00116313. [DOI] [PubMed] [Google Scholar]
- 42.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65:1976–1997. doi: 10.1016/j.jacc.2015.03.540. [DOI] [PubMed] [Google Scholar]
- 43.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 44.Urakami T, Jarvinen TA, Toba M, Sawada J, Ambalavanan N, Mann D, McMurtry I, Oka M, Ruoslahti E, Komatsu M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am J Pathol. 2011;178:2489–95. doi: 10.1016/j.ajpath.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue X, Li X, Nguyen HT, Chin DR, Sullivan DE, Lasky JA. Transforming growth factor-beta1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J Biol Chem. 2008;283:20397–407. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Botney MD, Bahadori L, Gold LI. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol. 1994;144:286–295. [PMC free article] [PubMed] [Google Scholar]
- 47.Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44:14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarvinen TA, Ruoslahti E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc Natl Acad Sci U S A. 2010;107:21671–6. doi: 10.1073/pnas.1016233107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humbert M, Oleg VE, Stasch J. Pharmacotherapy of Pulmonary Hypertension. Vol. 576 Springer; New York: 2013. [Google Scholar]
- 50.Sakao S, Tatsumi K. The effects of antiangiogenic compound SU5416 in a rat model of pulmonary arterial hypertension. Respiration. 2011;81:253–261. doi: 10.1159/000322011. [DOI] [PubMed] [Google Scholar]
- 51.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–38. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]