

New donkey genome reference unveils chromosomal rearrangements potentially involved in equine speciation and hybrid sterility.
Abstract
Donkeys and horses share a common ancestor dating back to about 4 million years ago. Although a high-quality genome assembly at the chromosomal level is available for the horse, current assemblies available for the donkey are limited to moderately sized scaffolds. The absence of a better-quality assembly for the donkey has hampered studies involving the characterization of patterns of genetic variation at the genome-wide scale. These range from the application of genomic tools to selective breeding and conservation to the more fundamental characterization of the genomic loci underlying speciation and domestication. We present a new high-quality donkey genome assembly obtained using the Chicago HiRise assembly technology, providing scaffolds of subchromosomal size. We make use of this new assembly to obtain more accurate measures of heterozygosity for equine species other than the horse, both genome-wide and locally, and to detect runs of homozygosity potentially pertaining to positive selection in domestic donkeys. Finally, this new assembly allowed us to identify fine-scale chromosomal rearrangements between the horse and the donkey that likely played an active role in their divergence and, ultimately, speciation.
INTRODUCTION
The equid family flourished during the last 55 million years and counts more than a dozen genera described in the paleontological record (1). Today, however, it is solely comprised of a single genus, Equus, which includes three zebra and three ass species, as well as the horse. The most recent common ancestor of the genus lived some 4.0 to 4.5 million years (Ma) ago (2). The first divergence within the genus separated the caballine lineage on the one hand, including the ancestor of the modern horse, and the stenonine lineage on the other hand. The stenonine lineage experienced additional splits between 1.7 and 2.0 Ma ago, giving rise to the ass and zebra lineages, and the Asiatic and African ass groups further emerged around 1.5 to 1.75 Ma ago.
Two members of the Equus genus, the horse and the donkey, have been successfully domesticated. Both domestication processes deeply affected human history, mainly by offering unprecedented means of transportation over long distances. Although extensive genomic work has been carried out to study the process of horse domestication (3, 4), this process is not as well documented in donkeys. The donkey is believed to have been domesticated from the African wild ass in Egypt approximately 5000 years ago (5), possibly following environmental shifts to drier climate in the region. Earlier studies have proposed that domestication occurred twice, probably from Nubian and Somali wild ass subspecies, according to patterns of mitochondrial DNA variation observed in both ancient and modern animals (6, 7). Extant subspecies of wild asses, as well as certain donkey breeds, are endangered and attract substantial conservation efforts (8).
Equids are known for their exceptional karyotypic plasticity, as shown by extensive centromere repositioning and multiple chromosome fusion and fission events (9). It has been traditionally postulated that karyotype rearrangements and low recombination help maintain genomic islands of speciation (10, 11), which promote postzygotic isolation despite postdivergence population contact (12).
On the basis of their karyotypic plasticity, equids should thus exhibit low interbreeding rates. Equid hybrids are nevertheless well known and are even commercially bred. Donkeys and horses produce hinnies or mules, the latter of which representing an example of hybrid vigor, displaying higher cognitive ability and stamina than the parent species. Previous genomic work also revealed that significant gene flow took place between different members of the Equus genus, including species with extremely divergent numbers of chromosomes (9). Because of their capacity to produce viable hybrids (almost always sterile) despite extensive karyotypic changes, the horse and the donkey not only represent an excellent model to study parallel evolutionary processes, such as domestication within the same taxonomic family, but could also help evaluate predictions drawn by speciation models, if a high-quality assembly for the donkey genome was available.
Transforming relatively short reads into longer scaffolds has continuously challenged genomic assemblies. Novel sequencing technologies producing longer reads, such as Oxford Nanopore and Pacific Biosciences, often come with elevated error rates, in as much as ~15% of the sites. To correct for these error rates while remaining cost-effective, single-molecule sequencing is typically combined with short reads generated by Illumina platforms, generating so-called hybrid de novo assemblies (13).
Alternative approaches use long-range chromatin interactions to capture read pairs located far apart in the genome, such as the so-called Chicago libraries that, coupled with a bespoke pipeline for assembly (HiRise), have been shown to produce long scaffolds, at the subchromosomal level, with low error rates (14).
Here, we used the Chicago HiRise assembly technology to produce a high-quality genome assembly for the donkey. This new genomic resource comes from a single male individual, Willy, belonging to the Equus asinus species. The new assembly provides scaffolds of much higher quality and length than currently available donkey genome assemblies, opening for further research on selective breeding, equine evolution (including both speciation and domestication), and conservation. We illustrate the utility of this new donkey assembly by identifying runs of homozygosity (ROHs), resulting from low effective population sizes and the relatedness of Willy’s progenitors, and by exploring chromosomal rearrangements and their impact on the patterns of distance existing between the donkey and the horse.
RESULTS
Genome assembly
Two Chicago HiRise libraries (14) were prepared from blood DNA extracts of Willy, a donkey jack that was born at the Copenhagen Zoo on 26th June 1997. Following paired-end sequencing on Illumina HiSeq platforms, we generated ~365 million reads that were processed through the HiRise assembler, yielding a total of 9021 scaffolds. Finally, we realigned all paired-end Illumina data previously generated from the same individual (2), as well as four complementary Illumina PCR-free DNA libraries, to identify DNA sequence variants. Further details are found in Materials and Methods.
The de novo assembly generated as part of this study was found to show an average depth of coverage of 61.2× and N50 of 15.4 Mb (Table 1). The latter represents a substantial improvement both in contig and scaffold length compared to two previous assemblies (Fig. 1). Furthermore, our new assembly contains a smaller fraction of missing base pairs (often represented by undetermined bases, N). The total length of the assembled genome was ~3% less than that by Huang et al. (15). The average GC content was 41.34%, which is on par with the base composition of the horse genome (16) and previously reported estimates for the donkey (15).
Table 1. Quality metrics for this assembly compared to previous donkey genome assemblies.
The number of annotated genes (lower than that in previous assemblies) shows a better homologous correspondence with the horse gene set (see Gene annotation).
| This study | Huang et al. (15) | Orlando et al. (2) | |
| N50 contigs | 140.3 kb | 66.7 kb | 6.38 kb |
| N50 scaffolds | 15.4 Mb | 3.8 Mb | 100.94 kb |
| Coverage | 61.2× | 42.4× | 12.4× |
| Total bases | 2.320 Gb | 2.391 Gb | 2.293 Gb |
| Largest scaffold | 84.20 Mb | 17.06 Mb | 1.09 Mb |
| Unresolved bases per 100 kb | 1121.61 | 1384.93 | 4128.43 |
| Total number of predicted protein-coding genes | 18,984 | 23,850* | 24,156 |
*Calculated using one isoform per gene and 42,247 total transcripts.
Fig. 1. Distribution of the cumulative scaffold length compared to previously published genome assemblies.
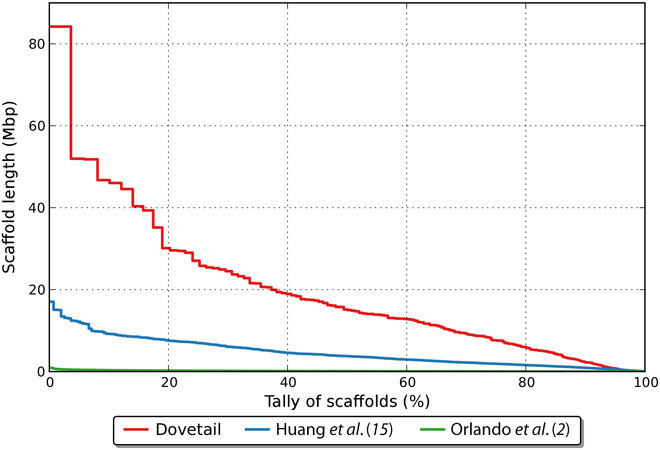
The red line represents the genome assembly obtained in this work using the Chicago HiRise technology. It shows that the greater N50 value of our new assembly is not simply due to a few longer scaffolds than two previously reported assemblies. Mbp, million base pairs.
Gene annotation
We predicted a total of 18,984 protein-coding genes, a number significantly lower (~80%) than that in previous assemblies (15). Using a single transcript as representative of every predicted protein-coding gene revealed that only a moderate fraction of the protein-coding genes annotated in the horse genome remain unassigned to our predictions in the new donkey assembly [3486 of 22,654 (~15.4%); fig. S1]. Reciprocally, the number of predicted donkey transcripts showing no pairing to the horse gene set [1985 of 18,984 (~10.4%)] is lower than that in previous assemblies [fig. S2 for the Venn diagram corresponding to annotations present in the study of Huang et al. (15)]. Furthermore, the total combined length for all annotated genes was of 505 Mb, including 29.8 Mb of exons, which assigns approximately ~1.3% of the total assembled length to protein-coding and untranslated regions.
We next used the annotations available for the horse to predict the functions of their 16,999 identified donkey orthologs. These were significantly enriched in housekeeping cellular functions, such as cell cycle (adjusted P = 0), DNA repair (adjusted P = 0), and proteolysis (adjusted P = 0), which indicates a bias for highly conserved genes in our horse-donkey ortholog assignments. Notably, no functional categories are over-represented in the subset of 3486 horse transcripts showing no pairing in the donkey genome. This suggests that our gene annotations are conservative and may not describe the complete donkey gene set.
Heterozygosity rate
Our new donkey assembly provides a phylogenetically closer reference than the horse to which sequence reads from all other extant equine species can be mapped to. This is expected to reduce mapping biases and the impact of unidentified/missing copy number variants (CNVs) and, thus, to provide more accurate patterns of nucleotide heterozygosity across the Equus genus.
To assess this heterozygosity, we realigned available shotgun sequencing data (9) against the new genomic reference. The data include Illumina paired-end sequencing reads for three ass species—a Somali wild ass (E. africanus somaliensis), an Onager (E. hemionus onager), and a Tibetan Kiang (E. kiang)—and for three zebra species—a Burchell’s plains zebra (E. quagga burchellii), a Grévy’s zebra (E. grevyi), and a Hartmann’s mountain zebra (E. zebra hartmannae). Overall, we found similar trends, as reported by Jónsson and colleagues (9), with the Somali wild ass genome being less heterozygous than the domestic donkey (here, represented by Willy; table S5). This is in line with the extremely limited population size of the Somali wild ass, its “critically endangered” conservation status, and the extreme inbreeding level predicted from the pedigree of the analyzed individual, described by Jónsson et al. (9). Heterozygosity is higher for the Grévy’s zebra and the Hartmann’s Mountain zebra and, especially, for the Onager and the Burchell’s zebra.
The depth of coverage achieved for each individual following mapping against our new de novo assembly increased by an average of 4.2% across the entire genome compared to the previous mapping against the horse reference genome (9). Given that the new assembly provided a reference genome phylogenetically closer to each individual species investigated, we expected the higher coverage to yield higher rates of heterozygosity by facilitating the alignment of the most divergent reads. In contrast, we found that our heterozygosity estimates are systematically lower than those reported by Jónsson et al. (9), particularly for species such as the Onager, the Kiang, and the Somali wild ass that are phylogenetically closer to the donkey (Fig. 2 and table S5). We believe that this finding could reflect the fact that CNVs specific to noncaballine equine species could map against the same region of horse genome and artifactually increased previous heterozygosity estimates.
Fig. 2. Heterozygosity rates for various equine species.
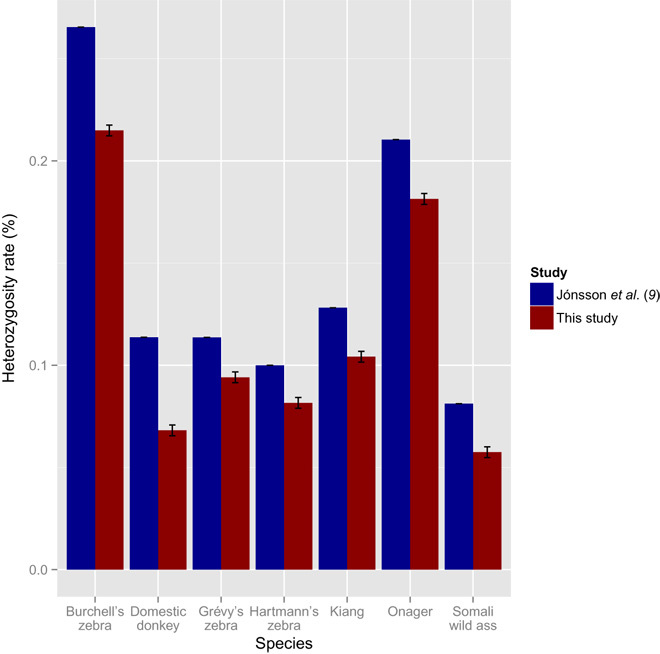
The heterozygosity estimates were computed using the same data aligned both to the horse genome (EquCab2.0) from a previous study and to the donkey reference presented in this study.
Estimates of effective population sizes (Ne) over time also depend on the underlying levels of heterozygosity. We, thus, revisited Ne estimates of most noncaballine species by leveraging on genome-wide read alignments to the new donkey reference (Fig. 3). Pairwise Sequentially Markovian Coalescent (PSMC) modeling reveals an equal effective population size between asses and zebras up to ~1.8 million years, when profiles diverged from an ancestral population of ~10,000 individuals, assuming a site-wise mutation rate of 7.242 × 10−9 per generation (3) and a generation time of 8 years. The comparable Ne for these species, at ~2 Ma ago, is consistent with previous estimates of their split time, based on phylogenetic tree inference (9). However, the estimate in effective population size over time by Jónsson and colleagues (fig. S8) suggests a slightly more recent split, around ~1.7 Ma ago, likely owing to discrepancies in the levels and patterns of heterozygosity (table S5). According to our PSMC, the Onager diverged about ~1.7 Ma ago, followed by a split of the Somali wild ass and the domestic donkey lineages around 700 thousand years (ka) ago. After the former split, the Onager population expanded, whereas both the Somali wild ass and domestic donkeys slightly declined.
Fig. 3. Demographic trajectories of zebras and asses during the last ~2.5 million years (Ma).

(A and B) PSMC reconstruction of the effective population size over time, for different ass species (A) and zebra species (B). The first 100 ka are highlighted for the ass and zebra species.
The PSMC profiles for the three zebra species, E. grevyi, E. zebra hartmannae, and the extinct E. quagga burchellii, start to diverge approximately ~1.3 Ma ago, from an ancestral population size of ~13,000 individuals. After the split, and in agreement with their heterozygosity rate, the Burchell’s zebra experienced a rapid population expansion, whereas the effective population size remained relatively low for Grévy’s and the Hartmann’s Mountain zebra, going down to less than about 10,000 individuals. The last glacial maximum (LGM), which occurred ~19 to 26 ka ago, probably had a greater impact on the availability of vegetation in Asia compared to Africa (17, 18). Because the Onager and the Kiang are Asiatic species, the effect of the LGM is reflected as an oscillatory Ne profile, with a notable drop in effective population size around 30 ka ago, followed by an expansion. The Kiang demographic trajectory does not show an expansion after the LGM, potentially due to the harshness of the Tibetan plateau. These oscillatory Ne changes are slightly different for African species, where the drop starts at ~90 ka ago, followed by a rapid expansion at ~30 ka ago. These observations are consistent with increases before ~70 ka ago and drops of temperatures after ~ 50 ka ago in Africa during the middle Pleistocene (19).
Runs of homozygosity
To detect inbreeding and/or selection signatures pertaining to the domestication process, we calculated the heterozygosity levels within 50-kb genomic sliding windows of the new donkey assembly. To represent local estimates of heterozygosity, we first sorted donkey scaffolds and oriented them according to horse chromosomes (hereafter, labeled as ECA). Although using the horse does not guarantee synteny in the original donkey genome due to rearrangement events, large genomic stretches showing very low heterozygosity levels were observed within several scaffolds (fig. S6).
Our procedure resulted in the identification of a total of 7374 ROHs in our new genome reference for the domestic donkey. With an average length of 98 kb, this accounted for ~724 Mb. This is significantly less than that in the genome of the Somali wild ass sequenced by Jónsson et al. (9), where we could identify 3660 ROHs using the same procedure, representing a total and average length of 910 Mb and 249 kb, respectively (fig. S9). The latter was characterized for an individual showing extreme inbreeding, with many related individuals within the last six generations, including a male individual who sired both the individual in question and his own mother (9).
Recombination is expected to rapidly break down ROHs. Consequently, ROHs originated during the early stages of the domestication process are expected to be shorter than those originating from inbreeding and/or recent demographic collapses. We thus assigned the set of ROHs into three categories, those between 100 and 500 kb, 500 kb to 1 Mb, and finally, those larger than 1 Mb. A total of 443, 114, and 192 genes overlapped these three categories. Smaller and medium-sized ROHs showed significant functional enrichment for human diseases involving muscle weakness (P = 2.7 × 10−6; adjusted P = 0.001). Genes involved in Down’s syndrome were also significantly found in smaller and medium-sized ROHs (P = 5.1 × 10−06; adjusted P = 0.001). The genes falling into muscle weakness and Down’s syndrome categories were no longer statistically significant when running the enrichment analysis on the genes found in the largest category of ROHs, which could potentially result from recent inbreeding (table S3).
Identifying Y chromosome scaffolds
Because of its repetitive nature, assembling the Y chromosome is particularly challenging. Nevertheless, the Y chromosome can provide useful insights into the evolutionary processes affecting the paternal lineage, which is particularly relevant for livestock because selective breeding often involves male specimens (4). The donkey individual underlying our new genome assembly was a jack (male), allowing for the identification of contigs/scaffolds belonging to the Y chromosome. Using a previous Y chromosome assembly from the horse (20–22), we detected three large donkey scaffolds that are likely located on the Y chromosome (ScCGjx6_630, ScCGjx6_695.2, and ScCGjx6_760). They encompass a total length of 822 kb. Among the five genes structurally annotated on those scaffolds, three were identified as TXLNGY, KDM5D, and AMELY. In humans, all three genes are linked to the Y chromosome, confirming the location of the three donkey scaffolds identified on the Y chromosome.
Alignment to the horse genome
The unprecedented scaffold size achieved in our new assembly allowed us to perform synteny comparisons to the horse genome. Despite the overall strong collinearity observed between both genomes, we identified multiple chromosomal rearrangements, such as translocations and inversions (Fig. 4 and table S1). It is important to bear in mind that these analyses might be affected by two important limitations. First, both the horse reference assembly and the contigs that form the donkey scaffolds are not necessarily well oriented and free of errors. For example, two proximal sections of the chromosome 12 in the horse (ECA12:12,396,945-13,988,621 and ECA12:14,886,333-16,777,588) were not completely covered by donkey scaffolds. Only the donkey scaffold ScCGjx6_285 was located inside this gap, aligning to ECA12:13,988,622-14,886,332 and showing a genetic distance to the horse (D) larger than flanking scaffolds (D = 0.0216 versus 0.0164 and 0.0119) and the average horse genome (95% confidence interval, 0.0115 to 0.0161; fig. S5). The remaining 3,482,931 uncovered sites are thus likely to reflect problems with the horse and donkey assemblies, rather than be large-scale deletions in the donkey genome or large-scale insertions in the horse genome. The annotation of 15 Ensembl genes as olfactory receptors within this region of the horse chromosome 12 confirmed the likely presence of an assembly artifact because this rapidly evolving multigene family, with a large number of tandemly repeated number of gene copies, is difficult to assemble.
Fig. 4. Dot plot showing the correspondence of unique 101-nucleotide oligomers from the donkey scaffolds to their location on the horse genome, using exact matches.
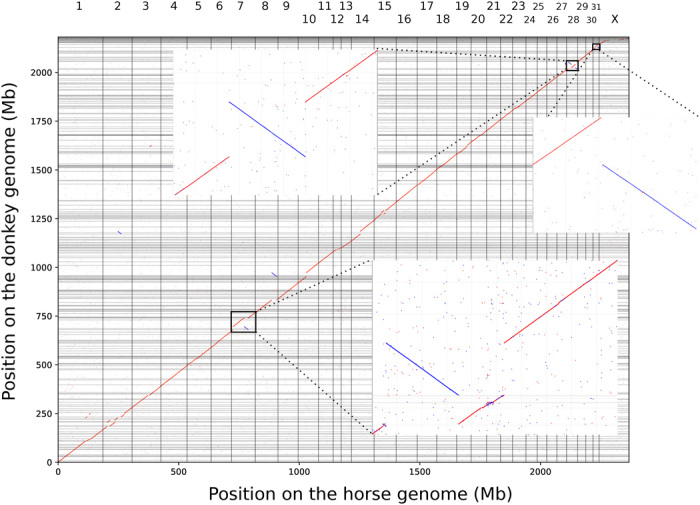
Because the orientation of the donkey scaffolds is unknown a priori, those were oriented using the strand that minimized the number and the size of inversions with respect to the horse chromosomes. The large inversions on the donkey scaffolds aligning to ECA7, ECA28, and ECA31 are enlarged for clarity. In the enlarged alignment to ECA7, donkey scaffold ScCGjx6_197 is not reverse-complemented consistently with the figures found in the Supplementary Materials.
The second limitation is due to the fact that the relative orientation between the donkey scaffolds was not known a priori. The donkey scaffolds were oriented as to minimize the number of inversion events and, thus, to maximize the collinearity to the horse. It follows that the fact that one inversion in the middle of the scaffold was considered to be more likely than two inversions on each terminus. In the event of an equal number of translocations, having 95% of the scaffold with the same orientation as the horse reference was preferable to having only 5% with the same orientation. Refer to Materials and Methods for further details.
The previous assumption entails an important implication. Because the orientation between donkey scaffolds is based on a parsimonious criterion as to minimize the number of chromosomal rearrangements events, only rearrangements occurring within donkey scaffolds are considered reliable and further investigated. However, we acknowledge that, in the absence of (i) high-quality genomes from (ii) phylogenetically close outgroup species (currently, the closest phylogenetic relatives, rhinos and tapirs, diverged more than 55 Ma ago), determining in which lineage these rearrangements occurred is unfeasible. Taking into account the above limitations, we identified nine chromosomal translocations between the horse and the donkey (table S1 and fig. S3), as well as six chromosomal inversions.
We focused on DNA inversions, given their key role as recombination suppressors in models of sympatric and parapatric speciation (23, 24). The suppression of recombination precludes the breakup of the inverted region such that the embedded allele combination is inherited as a linked DNA segment. In one possible mechanism, physical impairment of chromosome alignment during meiosis, or improper synapsis (25), prevents recombination even if so-called inversion loops are formed to restore correct orientation along the inverted region. In a second possible mechanism, the presence of an odd number of recombination events within inverted regions of heterokaryotypic individuals yields unbalanced gametes containing potentially harmful DNA duplications or deletions, resulting in prezygotic isolation. An even number of recombination events results instead in viable gametes, hampering prezygotic isolation and, therefore, speciation (23, 26). By conditioning the probability of physical impairing, as well as that of having an odd number of recombination events, the length of inversions is thus crucial to understand their potential role in speciation.
The seven inversions found can be classified as small (<1 Mb) or large (>10 Mb). The smallest inversion is 143 kb long and occurs within the donkey scaffold ScCGjx6_414, which aligns to the chromosome 31 of the horse (ECA31). ECA27 harbors another small inversion of 401 kb, involving three donkey scaffolds (ScCGjx6_121, ScCGjx6_380, and ScCGjx6_1). The last small inversion encompasses 586 kb in the donkey scaffold ScCGjx6_92, which aligns to ECA21. The presence of large inversions (>10 Mb) can be identified within donkey scaffolds aligning to ECA7, ECA28, and ECA31 (Fig. 4 and fig. S4, B, G, and E). The set of 282 protein-coding genes located within these three large inversions revealed no significant functional enrichment after correcting for multiple testing (table S2). However, the most represented categories were associated with metabolism and regulation of embryonic development, as well as cell division, especially related to centrosome functioning (table S2).
Speciation models based on suppressed recombination predict that inversions that contributed to reproductive isolation might exhibit elevated levels of sequence divergence, in comparison to the genome average (27). We quantified the divergence between the horse and the donkey, along large inversions, and found no evident variation along these mapping to ECA7 and ECA31 (fig. S7).
However, the last of the three large inversions, pertaining to ECA28, shows elevated genetic divergence around its breakpoints, with a D value ranking top 5% according to the genome-wide distribution (Fig. 5). The genetic divergence declines toward the middle of the inversion, suggesting a partial recovery of the normal recombination rate. This is on par with certain particularities near the inversion breakpoints, including difficulties in synaptonemal complex formation, differential recruitment of recombination suppressors, and double-recombination events toward the middle of large inversions, which generate balanced gametes (23, 26, 28).
Fig. 5. Genetic distance of the donkey scaffold to ECA28.
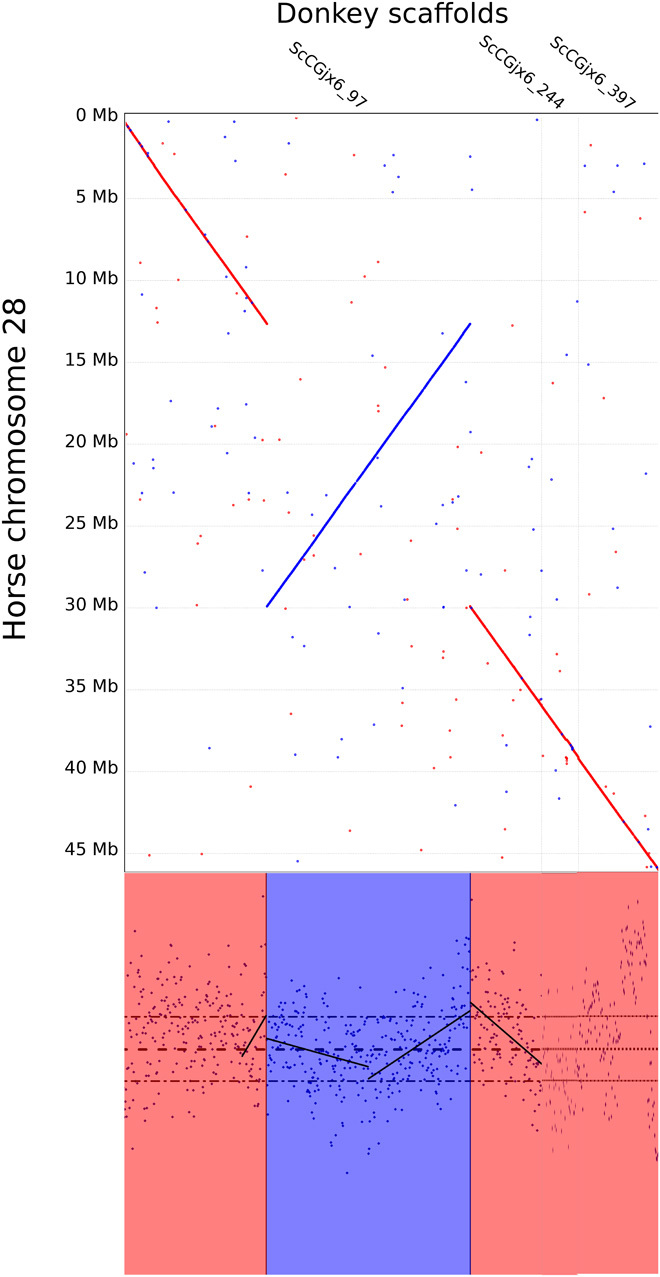
The middle part of the scaffold (~20 Mb) represents a good candidate for an inversion in either lineage and shows inflated level of divergence at the breakpoints. The dotted lines in the bottom panel represent the genomic average and the 95% confidence interval for the upper and lower divergence.
Partial recombination recovery toward the center of the region could suggest that this inversion was not maintained to prevent the separation of an epistatic combination of alleles. Nevertheless, the KITLG gene, a ligand for the receptor-type protein-tyrosine kinase KIT, is present within this inversion and under strong selection in diverse domesticated animals, including the horse (2). Among other phenotypes, mutations affecting KITLG produce variations on pigmentation patterns, in species as divergent as humans and the stickleback fish (29). KITLG has been also associated with male infertility (30). An x-ray–induced chromosome inversion of ~65.6 Mb in mice is known to have displaced distal regulatory regions from the targeted KITLG gene (31). The inversion observed in the donkey genome may have also induced KITLG transcriptional changes, with possible consequences on fertility and, ultimately, speciation. However, further work is needed to test this hypothesis. In addition to KITLG, the central region of the inversion contains the ALDH1L2 gene, which represents a candidate for positive selection during the domestication of the horse (3). In juvenile humans and mice, ALDH1L2 is expressed in multiple tissues, including testis, where it regulates the levels of retinoic acid indispensable for male fertility. Inhibition of other aldehyde dehydrogenase (ALDH) family members in testis is known to cause meiotic defects (32). We speculate that, following the hypothetical role proposed for KITLG, changes in the ALDH1L2 expression patterns may have resulted from the inversion observed in the donkey genome, again with possible consequences on speciation.
DISCUSSION
Using the Chicago HiRise technology, we have generated and described a new genome assembly for the domestic donkey. With an N50 of 15.4 Mb, this assembly contains scaffolds four times larger than the best previously published donkey assembly (15). Although the previous assembly had 13 scaffolds larger than 10 Mb, the assembly generated in this work has 75 scaffolds larger than 10 Mb. The N50 for the contigs (140.3 kb) is higher than that of the horse reference, EquCab2 (N50 for contigs of 112.4 kb). However, the N50 of the scaffolds remains smaller than that of EquCab2 (N50 for scaffolds of 47 Mb) (16) because a radiation hybrid map was built for the latter, after sequence assembling, to further orient and produce larger scaffolds.
There are a few horse chromosomal regions undetectable in our donkey assembly, an effect that could be explained by misassemblies in horse reference, EquCab2. This is the case for a region including an array of olfactory receptors in ECA12, a horse chromosome known to contain multiple CNVs associated with immunity genes and chemosensory genes (33). Other EquCab2 regions are only covered by short donkey scaffolds, such as the X chromosome. The horse X chromosome is covered by 352 donkey scaffolds, whereas only 308 are required to account for the rest of the genome. The assembly quality of the X chromosome may have been greater if a jennet individual was selected as a DNA donor, given that the expected depth of coverage would be doubled [however, the selection of a jack individual expanded our knowledge on the equine Y chromosome, for which only short contigs are available for the horse across a 13.6-Mb region (22); see below]. In addition, sex chromosomes tend to generally be more repetitive than autosomes, and the X chromosome in donkeys seems to have undergone several rearrangements (34), which have limited the N50 of the X chromosome scaffolds assembled here to 0.57 Mb.
We have also obtained three scaffolds mapping against the latest assembly of the horse Y chromosome [ECAY (22)]. These encompass a total of 822 kb and include five genes, three of which were characterized using orthologs. Although not completely covered, the nonrecombining nature of the Y chromosome implies that these Y contigs will be physically linked as a single marker, providing partially redundant information about the same evolutionary history.
Despite the few limitations listed above, our high-quality draft assembly opens opportunities to embrace the power of genomics in donkeys, something that was previously limited by the relatively low quality of previous assemblies. To illustrate these new possibilities, we conducted a series of analyses in zebras and asses, which benefited from the characterization of a high-quality genome assembly from a phylogenetically closer reference. Although only a single representative was characterized for each species, precluding a generalization to the entire species, we consistently estimated lower heterozygosity rates than those by Jónsson and colleagues (9), using the same sequencing data from the same individuals, especially for genomes from phylogenetically closer species. This reduced heterozygosity is concomitantly reflected in small PSMC incongruences, such as marginally lower Ne estimates in noncaballine equids. The low heterozygosity also appears to affect the split time between zebras and donkeys, which we estimated to be approximately 200 ka earlier than the PSMC by Jónsson and colleagues (9). This indicates that the pattern of heterozygosity in noncaballine equids is differentially affected when mapped along the horse reference, possibly as a by-product of undetected CNVs.
We investigated the distribution of ROHs. We found 7374 ROHs (98 kb long on average). ROHs resulting from recent inbreeding in captivity are expected to be longer and not present in other domesticated donkeys. In contrast, short ROHs shared between multiple donkeys are likely to reflect the molecular signature of positive selection, potentially following domestication. After excluding long ROHs resulting from recent breeding, we found ROH enrichment for genes associated with diseases involving muscle weakness in humans. Future comparisons to other donkey genomes will provide more robust estimates of genetic diversity within the species and will enable to further assess whether this enrichment was driven by selective pressures during domestication.
Equids have wide dispersal rates, and their distribution ranges have overlapped in the past. These patterns suggest that the horse-donkey speciation process occurred in the presence of gene flow, as evidenced by D statistics deviating from zero (9). Equids are well known for their hybridization, with some studies occasionally reporting reproductively viable mules (35). With two high-quality genome assemblies within the Equidae family, for the donkey and the horse, synteny-based analyses for large structural variants become feasible, opening for testing predictions drawn by rearrangement models of speciation, such as lower recombination and, therefore, higher divergence within chromosome inversions.
Of the three large chromosomal inversions, spanning more than 57 Mb in total, we only found a single molecular signature compatible with suppressed recombination on horse chromosome ECA28. The genetic distance between the horse and the donkey increases around the rearrangement breakpoints and asymptotically converges to the genomic background level toward the central region of the inversion. Recombination within the inversion is incompatible with the idea that inversions maintain epistatic combinations of alleles that promote isolation and speciation (36). However, the KITLG gene is located close to this inversion breakpoint. This inversion could have altered KITLG transcription, due to “position effects” or separation from distal cis-regulatory elements, located outside the inversion. Transcriptional changes of genes regulating spermatogenesis, such as for KITLG, can ultimately promote reproductive isolation (37).
It is also plausible that genetic incompatibilities were embedded within both inversion breakpoints because those are the only regions showing increased horse-donkey divergence. According to our annotations, five genes are located within 500 kb around the recombination breakpoints: MGAT4C, NTS, POLR3B, TCP11L2, and TMEM263. The MGAT4C gene was previously found to be deleted in human patients with Sertoli cell–only syndrome, a condition resulting in male infertility (38). In addition, TCP11L2 has been shown to be involved in sperm tail morphology and motility (39). If we explore 1.5 Mb on either side of breakpoints, further genes are associated with fertility and spermatogenesis. For example, CEP290, located 1.5 Mb away from the insertion breakpoint on donkey scaffold ScCGjx6_97, has been shown to play an essential role in sperm ciliogenesis in Drosophila (40). PRDM4 is abundantly expressed in developing spermatozoa and maturing oocytes, but its loss does not result in mice infertility (41). Another study looking at gene expression in testes in fertile versus infertile samples found TIMP3 to be significantly differentially expressed (42). The FBXO7 gene, located 0.8 Mb away from the inversion breakpoint on donkey scaffold ScCGjx6_97, was found to prevent mitochondrial disruptions, which lead to male sterility (43). Although we cannot establish which genes or set of genes are responsible, these observations suggest that further studies aiming at understanding the molecular pathways involved in the infertility of donkey/horse hybrids should focus on the chromosomal rearrangements identified here and the respective roles of the genes within and flanking large inversions.
Pending the availability of long-read data, this new donkey assembly provides an excellent reference for scientific community addressing fundamental and/or applied questions, including the role of chromosomal rearrangement in speciation.
MATERIALS AND METHODS
Chicago and PCR-free DNA libraries
Four aliquots of 100 μl of donkey blood (sample reference CGG_1_014644), stored frozen in heparin tubes, were extracted for genomic DNA using the DNeasy Blood & Tissue kit (Qiagen), following the manufacturer’s instructions. With the exception of the donkey, the data from other equids came from Jónsson et al. (9). The additional sequencing of material from the donkey followed all applicable laws and guidelines from Denmark/European Union/European Economic Area. After quantification using Qubit dsDNA HS Assay (Thermo Fisher Scientific), 1 μg of each DNA extract was fragmented using the Bioruptor (six cycles of 25-s on/90-s off; Diagenode). Fragment sizes were checked using the TapeStation 2200, High Sensitivity D1000 ScreenTape (Agilent). One Illumina sequencing library was built on each extract, using the TruSeq DNA PCR-free Kit (Illumina) but replacing the bead purification steps with MinElute column purifications (Qiagen). Libraries labeled “Willy 3” and “Willy 4” were size-selected using LabChip XT DNA 750 kit (Caliper). Final libraries were checked on a BioAnalyzer 2100 High Sensitivity DNA chip (Agilent). Further details about sequencing of the Chicago DNA libraries can be found in the Supplementary Materials.
Genome quality metrics
Various quality metric were computed, including N50 and largest scaffold. Further details are found in Supplementary Materials and Methods. To generate the genome-wide synteny plot, we first sampled ~1% of the 101-nucleotide oligomers present in the reference assembly [in this case, the horse reference, EquCab2 (16)] and kept only the 101-nucleotide oligomers that are unique. Each dot seen in Fig. 4 represents the presence in the donkey scaffolds of a unique horse 101-nucleotide oligomer. The methods for the per-horse-chromosome alignments found in the Supplementary Materials are detailed below.
Repeat masking for gene annotation
Before gene annotation and to quantify the amount of repeats in the newly assembled genome, repeat masking was performed. The scaffolds were masked using the procedure detailed in the Supplementary Materials. Two rounds of masking were performed, one with a generic mammalian database and a second round using a particular donkey database generated from the repetitive elements found in the assembly.
Gene annotation
To provide gene model annotations to the community and to compare them to known proteins in the horse, we performed a gene annotation of the new donkey genome. Ab initio gene annotation was conducted following a homology-based approach, with known protein sequences from humans, horses, and mice being used to prime the prediction. A tailored gene model, specifically trained for the horse, was used to further refine predictions. Additional details are found in the Supplementary Materials.
The resulting gene set was used to identify the corresponding homologs in the horse genome, which enabled us to assign gene symbols to the predicted donkey genes. Methodological details are described in the Supplementary Materials.
Heterozygosity estimates
To assess the improvement obtained using this new reference, especially for noncaballine species, we revisited estimates of heterozygosity and effective population size over time (9). The original DNA sequences, extracted from representatives of different equid species, were realigned to the new donkey reference. The resulting alignment was used to estimate the effective population size over time, as well as to compute local and global estimates of heterozygosity.
Because the local estimates of heterozygosity revealed large stretches of ROHs, the annotated genes within these were tested for functional enrichment. Again, details regarding the specific programs and the command line used are found in the Supplementary Materials.
Genome-wide alignments
To identify and characterize potential chromosomal rearrangements between the donkey and the horse genome, a series of genome-wide alignments were conducted. Because the orientation of donkey scaffolds to horse chromosomes is unknown, two criteria for their orientation were applied, where scaffolds were reverse-complemented if needed. The first criterion, used to generate the general synteny plot in Fig. 4, was based on minimizing alignments on different strands. The second criterion was based on minimizing the amount of chromosomal rearrangements and was used to generate the per-chromosome alignment plots found in fig. S4.
Furthermore, the homology between the different scaffolds and the horse chromosomes was not known beforehand. The plot found in Fig. 4 was used to establish this correspondence. To determine whether regions showing evidence of large rearrangements exhibited altered patterns of sequence divergence, genetic distance between the donkey and the horse genome was estimated, as described in the Supplementary Materials. Because the individual underlying EquCab2 was a mare (16), the donkey scaffolds potentially pertaining to the Y chromosome were identified by aligning donkey scaffolds against a recent and independent assembly of the horse Y chromosome (22). The exact procedure is found in the Supplementary Materials.
Supplementary Material
Acknowledgments
We thank the staff of the Danish National High-Throughput DNA Sequencing Center for the technical support, T. Korneliussen for the technical help with ANGSD (analysis of next-generation sequencing data), and M. Schubert for the access to Y chromosome scaffolds for the horse. We thank the Animal Health Trust and the Donkey Sanctuary for the support. We also thank M. Hartley and S. McWilson from Dovetail Genomics and finally, J. Clausen for help with the donkey samples. Funding: This work was supported by the Danish Council for Independent Research, Natural Sciences (grant 4002-00152B); the Danish National Research Foundation (grant DNRF94); Initiative d’Excellence Chaires d’attractivité, Université de Toulouse (OURASI); the Villum Fonden miGENEPI research project; the European Research Council (ERC-CoG-2015-681605); and the Donkey Sanctuary. G.R. was supported by a Marie Curie Intra-European fellowship (752657). Author contributions: L.O. conceived and coordinated the project. A.S.-O. extracted the DNA, built the Illumina PCR-free DNA libraries, and supervised the sequencing. B.P. performed the genome annotations, with input from G.R. and P.L. G.R., P.L., and L.O. designed and performed the analyses and wrote the manuscript. M.F.B., A.W., R.N., R.P., N.B., M.V., and L.O. provided the reagents and materials. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Sequencing data is available through https://www.ebi.ac.uk/ena/data/view/ERP106704. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession PSZQ00000000. The version described in this paper is version PSZQ01000000. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0392/DC1
Supplementary Materials and Methods
section S1. Supplementary Methods
fig. S1. Venn diagram of the protein-coding genes that were annotated in the donkey assembly versus the protein-coding gene annotation for the horse.
fig. S2. Venn diagram of the protein-coding genes that were annotated in the donkey assembly published by Huang et al. (15) versus the protein-coding gene annotation for the E. caballus genome (version EquCab2.0) using Ensembl genes (version 86).
fig. S3. Alignment of horse chromosomes to six donkey scaffolds with putative signs of translocations.
fig. S4. Alignment of donkey scaffolds to corresponding horse chromosomes.
fig. S5. Genetic distance between scaffolds spanning the gap on ECA12 versus the background.
fig. S6. Measured heterozygosity rates for the donkey scaffolds aligned to the various horse chromosomes.
fig. S7. Nei’s genetic distance by windows of 30 kb between donkey and horse chromosomes for scaffolds with signs of inversions.
fig. S8. Effective population size over time by aligning to the horse reference.
fig. S9. Measured heterozygosity rates for the African wild ass using the donkey scaffolds aligned to the horse chromosomes.
table S1. Translocations found between the donkey and horse scaffolds.
table S2. Gene ontologies of biological processes and enriched Reactome pathways associated with genes found in donkey scaffolds with signs of inversions when compared to the horse genome.
table S3. Human phenotypes, human diseases, and pathways associated with genes enriched in detected ROHs.
table S4. Horse sequences used for the detection of donkey scaffolds pertaining to the Y chromosome.
table S5. Heterozygosity rates for various species of asses and zebras computed when aligning to the donkey reference described in this study and recomputed on the basis of the data reported by Jónsson et al. (9), which were aligned to the horse reference.
table S6. Listing missing proteins in complete and partially complete Eukaryotic Orthologous Groups from the Core Eukaryotic Genes Mapping Approach.
table S7. Repeat elements and low-complexity DNA sequences masked in the donkey genome using RepeatMasker.
table S8. Repeat elements and low-complexity DNA sequences masked in the donkey genome using the second of the RepeatMasker using the model generated from RepeatModeler as custom library input on the previously masked genome.
table S9. Statistics of the completeness of the different versions of the donkey genome based on 248 Core Eukaryotic Genes.
REFERENCES AND NOTES
- 1.Woodburne M. O., MacFadden B. J., Fossil horses: Systematics, paleobiology, and evolution of the family Equidae. Syst. Biol. 43, 299–303 (1994). [Google Scholar]
- 2.Orlando L., Ginolhac A., Zhang G., Froese D., Albrechtsen A., Stiller M., Schubert M., Cappellini E., Petersen B., Moltke I., Johnson P. L. F., Fumagalli M., Vilstrup J. T., Raghavan M., Korneliussen T., Malaspinas A.-S., Vogt J., Szklarczyk D., Kelstrup C. D., Vinther J., Dolocan A., Stenderup J., Velazquez A. M. V., Cahill J., Rasmussen M., Wang X., Min J., Zazula G. D., Seguin-Orlando A., Mortensen C., Magnussen K., Thompson J. F., Weinstock J., Gregersen K., Røed K. H., Eisenmann V., Rubin C. J., Miller D. C., Antczak D. F., Bertelsen M. F., Brunak S., Al-Rasheid K. A. S., Ryder O., Andersson L., Mundy J., Krogh A., Gilbert M. T. P., Kjær K., Sicheritz-Ponten T., Jensen L. J., Olsen J. V., Hofreiter M., Nielsen R., Shapiro B., Wang J., Willerslev E., Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Schubert M., Jónsson H., Chang D., Der Sarkissian C., Ermini L., Ginolhac A., Albrechtsen A., Dupanloup I., Foucal A., Petersen B., Fumagalli M., Raghavan M., Seguin-Orlando A., Korneliussen T. S., Velazquez A. M. V., Stenderup J., Hoover C. A., Rubin C.-J., Alfarhan A. H., Alquraishi S. A., Al-Rasheid K. A. S., MacHugh D. E., Kalbfleisch T., MacLeod J. N., Rubin E. M., Sicheritz-Ponten T., Andersson L., Hofreiter M., Marques-Bonet T., Gilbert M. T. P., Nielsen R., Excoffier L., Willerslev E., Shapiro B., Orlando L., Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc. Natl. Acad. Sci. U.S.A. 111, E5661–E5669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Librado P., Gamba C., Gaunitz C., Der Sarkissian C., Pruvost M., Albrechtsen A., Fages A., Khan N., Schubert M., Jagannathan V., Serres-Armero A., Kuderna L. F. K., Povolotskaya I. S., Seguin-Orlando A., Lepetz S., Neuditschko M., Thèves C., Alquraishi S., Alfarhan A. H., Al-Rasheid K., Rieder S., Samashev Z., Francfort H.-P., Benecke N., Hofreiter M., Ludwig A., Keyser C., Marques-Bonet T., Ludes B., Crubézy E., Leeb T., Willerslev E., Orlando L., Ancient genomic changes associated with domestication of the horse. Science 356, 442–445 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Kimura B., Marshall F. B., Chen S., Rosenbom S., Moehlman P. D., Tuross N., Sabin R. C., Peters J., Barich B., Yohannes H., Kebede F., Teclai R., Beja-Pereira A., Mulligan C. J., Ancient DNA from Nubian and Somali wild ass provides insights into donkey ancestry and domestication. Proc. Biol. Sci 278, 50–57 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossel S., Marshall F., Peters J., Pilgram T., Adams M. D., O’Connor D., Domestication of the donkey: Timing, processes, and indicators. Proc. Natl. Acad. Sci. U.S.A. 105, 3715–3720 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beja-Pereira A., England P. R., Ferrand N., Jordan S., Bakhiet A. O., Abdalla M. A., Mashkour M., Jordana J., Taberlet P., Luikart G., African origins of the domestic donkey. Science 304, 1781 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Moehlman P. D., Kebede F., Yohannes H., The African wild ass (Equus africanus): Conservation status in the horn of Africa. Appl. Anim. Behav. Sci. 60, 115–124 (1998). [Google Scholar]
- 9.Jónsson H., Schubert M., Seguin-Orlando A., Ginolhac A., Petersen L., Fumagalli M., Albrechtsen A., Petersen B., Korneliussen T. S., Vilstrup J. T., Lear T., Myka J. L., Lundquist J., Miller D. C., Alfarhan A. H., Alquraishi S. A., Al-Rasheid K. A. S., Stagegaard J., Strauss G., Bertelsen M. F., Sicheritz-Ponten T., Antczak D. F., Bailey E., Nielsen R., Willerslev E., Orlando L., Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc. Natl. Acad. Sci. U.S.A. 111, 18655–18660 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levin D. A., Wilson A. C., Rates of evolution in seed plants: Net increase in diversity of chromosome numbers and species numbers through time. Proc. Natl. Acad. Sci. U.S.A. 73, 2086–2090 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush G. L., Case S. M., Wilson A. C., Patton J. L., Rapid speciation and chromosomal evolution in mammals. Proc. Natl. Acad. Sci. U.S.A. 74, 3942–3946 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feder J. L., Egan S. P., Nosil P., The genomics of speciation-with-gene-flow. Trends Genet. 28, 342–350 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Chakraborty M., Baldwin-Brown J. G., Long A. D., Emerson J. J., Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 44, e147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Putnam N. H., O’Connell B. L., Stites J. C., Rice B. J., Blanchette M., Calef R., Troll C. J., Fields A., Hartley P. D., Sugnet C. W., Haussler D., Rokhsar D. S., Green R. E., Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J., Zhao Y., Bai D., Shiraigol W., Li B., Yang L., Wu J., Bao W., Ren X., Jin B., Zhao Q., Li A., Bao S., Bao W., Xing Z., An A., Gao Y., Wei R., Bao Y., Bao T., Han H., Bai H., Bao Y., Zhang Y., Daidiikhuu D., Zhao W., Liu S., Ding J., Ye W., Ding F., Sun Z., Shi Y., Zhang Y., Meng H., Dugarjaviin M., Donkey genome and insight into the imprinting of fast karyotype evolution. Sci. Rep. 5, 14106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wade C. M., Giulotto E., Sigurdsson S., Zoli M., Gnerre S., Imsland F., Lear T. L., Adelson D. L., Bailey E., Bellone R. R., Blöcker H., Distl O., Edgar R. C., Garber M., Leeb T., Mauceli E., MacLeod J. N., Penedo M. C. T., Raison J. M., Sharpe T., Vogel J., Andersson L., Antczak D. F., Biagi T., Binns M. M., Chowdhary B. P., Coleman S. J., Della Valle G., Fryc S., Guérin G., Hasegawa T., Hill E. W., Jurka J., Kiialainen A., Lindgren G., Liu J., Magnani E., Mickelson J. R., Murray J., Nergadze S. G., Onofrio R., Pedroni S., Piras M. F., Raudsepp T., Rocchi M., Røed K. H., Ryder O. A., Searle S., Skow L., Swinburne J. E., Syvänen A. C., Tozaki T., Valberg S. J., Vaudin M., White J. R., Zody M. C.; Broad Institute Genome Sequencing Platform; Broad Institute Whole Genome Assembly Team, Lander E. S., Lindblad-Toh K., Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 326, 865–867 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills S. C., Barrows T. T., Telfer M. W., Fifield L. K., The cold climate geomorphology of the Eastern Cape Drakensberg: A reevaluation of past climatic conditions during the last glacial cycle in Southern Africa. Geomorphology 278, 184–194 (2017). [Google Scholar]
- 18.Ju L., Wang H., Jiang D., Simulation of the Last Glacial Maximum climate over East Asia with a regional climate model nested in a general circulation model. Palaeogeogr. Palaeoclimatol. Palaeoecol. 248, 376–390 (2007). [Google Scholar]
- 19.Ziegler M., Simon M. H., Hall I. R., Barker S., Stringer C., Zahn R., Development of Middle Stone Age innovation linked to rapid climate change. Nat. Commun. 4, 1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallner B., Vogl C., Shukla P., Burgstaller J. P., Druml T., Brem G., Identification of genetic variation on the horse y chromosome and the tracing of male founder lineages in modern breeds. PLOS ONE 8, e60015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippold S., Knapp M., Kuznetsova T., Leonard J. A., Benecke N., Ludwig A., Rasmussen M., Cooper A., Weinstock J., Willerslev E., Shapiro B., Hofreiter M., Discovery of lost diversity of paternal horse lineages using ancient DNA. Nat. Commun. 2, 450 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Wallner B., Palmieri N., Vogl C., Rigler D., Bozlak E., Druml T., Jagannathan V., Leeb T., Fries R., Tetens J., Thaller G., Metzger J., Distl O., Lindgren G., Rubin C.-J., Andersson L., Schaefer R., McCue M., Neuditschko M., Rieder S., Schlötterer C., Brem G., Y chromosome uncovers the recent oriental origin of modern stallions. Curr. Biol. 27, 2029–2035.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Rieseberg L. H., Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Kirkpatrick M., Barton N., Chromosome inversions, local adaptation and speciation. Genetics 173, 419–434 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davisson M. T., Akeson E. C., Recombination suppression by heterozygous Robertsonian chromosomes in the mouse. Genetics 133, 649–667 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anton E., Blanco J., Egozcue J., Vidal F., Sperm studies in heterozygote inversion carriers: A review. Cytogenet. Genome Res. 111, 297–304 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Faria R., Navarro A., Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends Ecol. Evol. 25, 660–669 (2010). [DOI] [PubMed] [Google Scholar]
- 28.P. A. Roberts, The genetics of chromosome aberration, in The Genetics and Biology of Drosophila, M. Ashburner, E. Novitski, Eds. (Academic Press, 1976), vol. 3d. [Google Scholar]
- 29.Miller C. T., Beleza S., Pollen A. A., Schluter D., Kittles R. A., Shriver M. D., Kingsley D. M., cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 131, 1179–1189 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galan J. J., De Felici M., Buch B., Rivero M. C., Segura A., Royo J. L., Cruz N., Real L. M., Ruiz A., Association of genetic markers within the KIT and KITLG genes with human male infertility. Hum. Reprod. 21, 3185–3192 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Guenther C. A., Tasic B., Luo L., Bedell M. A., Kingsley D. M., A molecular basis for classic blond hair color in Europeans. Nat. Genet. 46, 748–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levi B. P., Yilmaz Ö. H., Duester G., Morrison S. J., Aldehyde dehydrogenase 1a1 is dispensable for stem cell function in the mouse hematopoietic and nervous systems. Blood 113, 1670–1680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang W., Wang S., Hou C., Xing Y., Cao J., Wu K., Liu C., Zhang D., Zhang L., Zhang Y., Zhou H., Genome-wide detection of copy number variations among diverse horse breeds by array CGH. PLOS ONE 9, e86860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raudsepp T., Lear T. L., Chowdhary B. P., Comparative mapping in equids: The asine X chromosome is rearranged compared to horse and Hartmann’s mountain zebra. Cytogenet. Genome Res. 96, 206–209 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Rong R., Chandley A. C., Song J., McBeath S., Tan P. P., Bai Q., Speed R. M., A fertile mule and hinny in China. Cytogenet. Cell Genet. 47, 134–139 (1988). [DOI] [PubMed] [Google Scholar]
- 36.Felsenstein J., Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution 35, 124–138 (1981). [DOI] [PubMed] [Google Scholar]
- 37.Marquès-Bonet T., Cáceres M., Bertranpetit J., Preuss T. M., Thomas J. W., Navarro A., Chromosomal rearrangements and the genomic distribution of gene-expression divergence in humans and chimpanzees. Trends Genet. 20, 524–529 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Stouffs K., Gheldof A., Tournaye H., Vandermaelen D., Bonduelle M., Lissens W., Seneca S., Sertoli cell-only syndrome: Behind the genetic scenes. Biomed Res. Int. 2016, 6191307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y., Jiang M., Li C., Yang P., Sun H., Tao D., Zhang S., Ma Y., Human t-complex protein 11 (TCP11), a testis-specific gene product, is a potential determinant of the sperm morphology. Tohoku J. Exp. Med. 224, 111–117 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Basiri M. L., Ha A., Chadha A., Clark N. M., Polyanovsky A., Cook B., Avidor-Reiss T., A migrating ciliary gate compartmentalizes the site of axoneme assembly in Drosophila spermatids. Curr. Biol. 24, 2622–2631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bogani D., Morgan M. A. J., Nelson A. C., Costello I., McGouran J. F., , Kessler B. M., Robertson E. J., Bikoff E. K., The PR/SET domain zinc finger protein Prdm4 regulates gene expression in embryonic stem cells but plays a nonessential role in the developing mouse embryo. Mol. Cell. Biol. 33, 3936–3950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rockett J. C., Patrizio P., Schmid J. E., Hecht N. B., Dix D. J., Gene expression patterns associated with infertility in humans and rodent models. Mutat. Res. 549, 225–240 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Burchell V. S., Nelson D. E., Sanchez-Martinez A., Delgado-Camprubi M., Ivatt R. M., Pogson J. H., Randle S. J., Wray S., Lewis P. A., Houlden H., Abramov A. Y., Hardy J., Wood N. W., Whitworth A. J., Laman H., Plun-Favreau H., The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentley D. R., Balasubramanian S., Swerdlow H. P., Smith G. P., Milton J., Brown C. G., Hall K. P., Evers D. J., Barnes C. L., Bignell H. R., Boutell J. M., Bryant J., Carter R. J., Keira Cheetham R., Cox A. J., Ellis D. J., Flatbush M. R., Gormley N. A., Humphray S. J., Irving L. J., Karbelashvili M. S., Kirk S. M., Li H., Liu X., Maisinger K. S., Murray L. J., Obradovic B., Ost T., Parkinson M. L., Pratt M. R., Rasolonjatovo I. M., Reed M. T., Rigatti R., Rodighiero C., Ross M. T., Sabot A., Sankar S. V., Scally A., Schroth G. P., Smith M. E., Smith V. P., Spiridou A., Torrance P. E., Tzonev S. S., Vermaas E. H., Walter K., Wu X., Zhang L., Alam M. D., Anastasi C., Aniebo I. C., Bailey D. M., Bancarz I. R., Banerjee S., Barbour S. G., Baybayan P. A., Benoit V. A., Benson K. F., Bevis C., Black P. J., Boodhun A., Brennan J. S., Bridgham J. A., Brown R. C., Brown A. A., Buermann D. H., Bundu A. A., Burrows J. C., Carter N. P., Castillo N., Chiara M., Catenazzi E., Chang S., Neil Cooley R., Crake N. R., Dada O. O., Diakoumakos K. D., Dominguez-Fernandez B., Earnshaw D. J., Egbujor U. C., Elmore D. W., Etchin S. S., Ewan M. R., Fedurco M., Fraser L. J., Fuentes Fajardo K. V., Scott Furey W., George D., Gietzen K. J., Goddard C. P., Golda G. S., Granieri P. A., Green D. E., Gustafson D. L., Hansen N. F., Harnish K., Haudenschild C. D., Heyer N. I., Hims M. M., Ho J. T., Horgan A. M., Hoschler K., Hurwitz S., Ivanov D. V., Johnson M. Q., James T., Huw Jones T. A., Kang G.-D., Kerelska T. H., Kersey A. D., Khrebtukova I., Kindwall A. P., Kingsbury Z., Kokko-Gonzales P. I., Kumar A., Laurent M. A., Lawley C. T., Lee S. E., Lee X., Liao A. K., Loch J. A., Lok M., Luo S., Mammen R. M., Martin J. W., McCauley P. G., McNitt P., Mehta P., Moon K. W., Mullens J. W., Newington T., Ning Z., Ling Ng B., Novo S. M., O’Neill M. J., Osborne M. A., Osnowski A., Ostadan O., Paraschos L. L., Pickering L., Pike A. C., Pike A. C., Chris Pinkard D., Pliskin D. P., Podhasky J., Quijano V. J., Raczy C., Rae V. H., Rawlings S. R., Chiva Rodriguez A., Roe P. M., Rogers J., Rogert Bacigalupo M. C., Romanov N., Romieu A., Roth R. K., Rourke N. J., Ruediger S. T., Rusman E., Sanches-Kuiper R. M., Schenker M. R., Seoane J. M., Shaw R. J., Shiver M. K., Short S. W., Sizto N. L., Sluis J. P., Smith M. A., Ernest Sohna Sohna J., Spence E. J., Stevens K., Sutton N., Szajkowski L., Tregidgo C. L., Turcatti G., Vandevondele S., Verhovsky Y., Virk S. M., Wakelin S., Walcott G. C., Wang J., Worsley G. J., Yan J., Yau L., Zuerlein M., Rogers J., Mullikin J. C., Hurles M. E., McCooke N. J., West J. S., Oaks F. L., Lundberg P. L., Klenerman D., Durbin R., Smith A. J., Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurevich A., Saveliev V., Vyahhi N., Tesler G., QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.A. F. A. Smit, R. Hubley, P. Green, RepeatMasker (2017); www.repeatmasker.org/.
- 47.Holt C., Yandell M., MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parra G., Bradnam K., Korf I., CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Parra G., Bradnam K., Ning Z., Keane T., Korf I., Assessing the gene space in draft genomes. Nucleic Acids Res. 37, 289–297 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korf I., Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stanke M., Waack S., Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (suppl. 2), ii215–ii225 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Emms D. M., Kelly S., OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smedley D., Haider S., Durinck S., Pandini L., Provero P., Allen J., Arnaiz O., Awedh M. H., Baldock R., Barbiera G., Bardou P., Beck T., Blake A., Bonierbale M., Brookes A. J., Bucci G., Buetti I., Burge S., Cabau C., Carlson J. W., Chelala C., Chrysostomou C., Cittaro D., Collin O., Cordova R., Cutts R. J., Dassi E., Di Genova A., Djari A., Esposito A., Estrella H., Eyras E., Fernandez-Banet J., Forbes S., Free R. C., Fujisawa T., Gadaleta E., Garcia-Manteiga J. M., Goodstein D., Gray K., Guerra-Assunção J. A., Haggarty B., Han D. J., Han B. W., Harris T., Harshbarger J., Hastings R. K., Hayes R. D., Hoede C., Hu S., Hu Z. L., Hutchins L., Kan Z., Kawaji H., Keliet A., Kerhornou A., Kim S., Kinsella R., Klopp C., Kong L., Lawson D., Lazarevic D., Lee J. H., Letellier T., Li C. Y., Lio P., Liu C. J., Luo J., Maass A., Mariette J., Maurel T., Merella S., Mohamed A. M., Moreews F., Nabihoudine I., Ndegwa N., Noirot C., Perez-Llamas C., Primig M., Quattrone A., Quesneville H., Rambaldi D., Reecy J., Riba M., Rosanoff S., Saddiq A. A., Salas E., Sallou O., Shepherd R., Simon R., Sperling L., Spooner W., Staines D. M., Steinbach D., Stone K., Stupka E., Teague J. W., Dayem Ullah A. Z., Wang J., Ware D., Wong-Erasmus M., Youens-Clark K., Zadissa A., Zhang S. J., Kasprzyk A., The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 43, W589–W598 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korneliussen T. S., Albrechtsen A., Nielsen R., ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H., Durbin R., Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J., Duncan D., Shi Z., Zhang B., WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 41, W77–W83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.S. Turner, Annotated Manhattan plots and QQ plots for GWAS using R, Revisited (Nature Precedings, 2011); http://dx.doi.org/10.1038/npre.2011.6070.1.
- 59.Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., Salzberg S. L., Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Musilova P., Kubickova S., Vahala J., Rubes J., Subchromosomal karyotype evolution in Equidae. Chromosome Res. 21, 175–187 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Nei M., Genetic distance between populations. Am. Nat. 106, 283–292 (1972). [Google Scholar]
- 62.Kent W. J., BLAT—The BLAST-Like Alignment Tool. Genome Res. 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0392/DC1
Supplementary Materials and Methods
section S1. Supplementary Methods
fig. S1. Venn diagram of the protein-coding genes that were annotated in the donkey assembly versus the protein-coding gene annotation for the horse.
fig. S2. Venn diagram of the protein-coding genes that were annotated in the donkey assembly published by Huang et al. (15) versus the protein-coding gene annotation for the E. caballus genome (version EquCab2.0) using Ensembl genes (version 86).
fig. S3. Alignment of horse chromosomes to six donkey scaffolds with putative signs of translocations.
fig. S4. Alignment of donkey scaffolds to corresponding horse chromosomes.
fig. S5. Genetic distance between scaffolds spanning the gap on ECA12 versus the background.
fig. S6. Measured heterozygosity rates for the donkey scaffolds aligned to the various horse chromosomes.
fig. S7. Nei’s genetic distance by windows of 30 kb between donkey and horse chromosomes for scaffolds with signs of inversions.
fig. S8. Effective population size over time by aligning to the horse reference.
fig. S9. Measured heterozygosity rates for the African wild ass using the donkey scaffolds aligned to the horse chromosomes.
table S1. Translocations found between the donkey and horse scaffolds.
table S2. Gene ontologies of biological processes and enriched Reactome pathways associated with genes found in donkey scaffolds with signs of inversions when compared to the horse genome.
table S3. Human phenotypes, human diseases, and pathways associated with genes enriched in detected ROHs.
table S4. Horse sequences used for the detection of donkey scaffolds pertaining to the Y chromosome.
table S5. Heterozygosity rates for various species of asses and zebras computed when aligning to the donkey reference described in this study and recomputed on the basis of the data reported by Jónsson et al. (9), which were aligned to the horse reference.
table S6. Listing missing proteins in complete and partially complete Eukaryotic Orthologous Groups from the Core Eukaryotic Genes Mapping Approach.
table S7. Repeat elements and low-complexity DNA sequences masked in the donkey genome using RepeatMasker.
table S8. Repeat elements and low-complexity DNA sequences masked in the donkey genome using the second of the RepeatMasker using the model generated from RepeatModeler as custom library input on the previously masked genome.
table S9. Statistics of the completeness of the different versions of the donkey genome based on 248 Core Eukaryotic Genes.