

Abstract
Nephrotic syndrome can be caused by various diseases, from primary kidney diseases to systemic diseases. A kidney biopsy is useful for confirming the causes of nephrotic syndrome and in its management. We herein describe a case of nephrotic syndrome with thrombocytopenia, lymphadenopathy, systemic inflammation, splenomegaly, kidney enlargement, and progressive renal insufficiency. A kidney biopsy showed endothelial swelling with mild interstitial fibrosis and tubular atrophy. This case met the diagnostic criteria for TAFRO syndrome. Little is known about TAFRO syndrome, especially in relation to the associated kidney pathophysiology. The accumulation of a greater number of cases in which the kidney biopsy findings are investigated is needed to clarify the pathogenesis of kidney involvement in this condition.
Keywords: nephrotic syndrome, renal dysfunction, small vessel lesions, TAFRO syndrome, idiopathic multicentric Castleman Disease
Introduction
Nephrotic syndrome can be caused by various diseases, from primary (idiopathic) kidney diseases to systemic diseases. TAFRO syndrome is a systemic inflammatory disease; the defining characteristics include thrombocytopenia (T), anasarca (A), fever (F), reticulin myelofibrosis (R), and organomegaly (O). This syndrome was first reported in 2010 (1). The diagnostic criteria were proposed in 2016 and 2017 (2,3). While TAFRO syndrome can cause progressive kidney insufficiency or proteinuria (4,5), little is known about the kidney involvement in TAFRO syndrome. We present a case of biopsy-proven nephrotic syndrome, which was later diagnosed as TAFRO syndrome.
Case Report
A 54-year-old woman was admitted to a nearby hospital for severe edema (12 kg weight gain over one month), pleural effusion, dyspnea, and fever. She had no relevant medical history, but she had smoked for approximately 35 years. Dyspnea forced her to quit smoking. Her father and brother suffered from colon cancer; her mother suffered from breast cancer. She had proteinuria (4.2 g/day), hematuria [Red Blood Cells (RBC) 50-90/high power field (HPF)], granular casts, hypoalbuminemia (2.2 g/dL), slight thrombocytopenia (platelet 9.2×104/μL), and systemic inflammation [C-reactive protein (CRP) 5.7 mg/dL], with a serum creatinine level of 0.6 mg/dL. Based on these findings, she was diagnosed with nephrotic syndrome. At two weeks after her admission to another hospital, she was referred to our hospital for further examination and treatment. Her initial vital signs at our facility included: body temperature, 38.4℃; blood pressure, 154/94 mmHg; heart rate, 103 bpm; and oxygen saturation, 98% at 2 L/min of oxygen via nasal cannula. She complained of orthopnea. A physical examination revealed the absence of breath sounds over the bilateral lower lung fields, abdominal swelling due to ascites, and pitting edema in both legs. The laboratory results revealed thrombocytopenia (platelet count 6.9×104/μL), normocytic anemia (hemoglobin 9.7 g/dL), hypoalbuminemia (albumin 2.5 g/dL), systemic inflammation (CRP 7.0 mg/dL), slightly alkaline phosphatase elevation (598 IU/L), slight renal dysfunction [serum creatinine 1.1 mg/dL; estimated glomerular filtration rate (eGFR) 40.7 mL/min/1.73 m2], proteinuria [3.2 g/g creatinine (Cr)], hematuria (dysmorphic RBC 12/HPF), tubular dysfunction [urine N-acetyl-beta-D-glucosaminidase (NAG) 35.8 U/L and beta-2 microglobulin (B2MG) 3,831 μg/L], with granular casts, epithelial cell casts, and fatty casts (Table). Her immunoglobulin (Ig) levels were within the normal range, and her IgG4 level was 10.0 mg/dL. Neither M-protein nor cryoglobulins were detected. The patient was negative for antinuclear antibody (ANA), anti-double stranded DNA antibody, anti-Sm antibody, myeloperoxidase-antineutrophil cytoplasmic antibody (ANCA), and proteinase-3 ANCA, matrix metalloproteinase-3, anti-citrullinated protein antibody, anti-ribonucleoprotein antibody, anti-SS-A antibody, anti-SS-B antibody, and anti-topoisomerase antibody. Her soluble-interleukin (IL) 2 receptor level was 1,580 U/mL, and her IL-6 level was 8.2 pg/mL (normal range 0-4 pg/mL). Her pleural fluid had exudative characteristics, and was sterile without malignant cells (Table). A chest X-ray film showed pleural effusion; computed tomography (CT) showed pleura effusion, cardiac effusion, ascites, kidney enlargement, splenomegaly, and systemic lymphadenopathy (Fig. 1). Positron emission tomography-CT with [18F]-fluorodeoxyglucose (FDG) revealed splenomegaly, pleural effusion, and lymphadenopathy in deep cervical, paratracheal, subaortic, splenic hilar, and splenic artery lymph nodes, the uptake of FDG in all of these lymph nodes was strong. The slight uptake of FDG was observed in her axillary lymph nodes and her enlarged spleen (Fig. 2). The maximum diameter of these subaortic lymph nodes was 35 mm, while the diameter of most other lymph nodes was approximately 10 mm.
Table.
The Laboratory Tests on Admission.
| White Blood Cells | 116.6×102/μL | IgG | 1,022mg/dL |
| Neu | 79.3% | IgA | 154mg/dL |
| Ly | 11.7% | IgM | 52mg/dL |
| Mon | 7.6% | IgG4 | 10.0mg/dL |
| Eo | 1.1% | Cryoglobulins | - |
| Ba | 0.3% | C3 | 128mg/dL |
| Red Blood Cells | 356×104/μL | C4 | 20mg/dL |
| Hb | 9.7 g/dL | ANA | <40 |
| Ht | 29.1% | ds-DNA | <10IU/mL |
| Platelet | 6.9×104/μL | Sm Ab | ≤7.0U/mL |
| PT-INR | 1.3 | MMP-3 | 25.1ng/mL |
| APTT | 43.5 s | CCP Ab | 1.2U/mL |
| Fibrinogen | 529.0mg/dL | RNP Ab | ≤7.0U/mL |
| D-dimer | 11.2μg/mL | SS-A Ab | ≤7.0U/mL |
| SS-B Ab | ≤7.0U/mL | ||
| Pleural fluid | Scl-70 Ab | 13.2U/mL | |
| LDH | 102IU/L | s-IL2 R | 1,580U/mL |
| TP | 4.1g/dL | IL-6 | 8.2pg/mL |
| Alb | 2.4g/dL | PA-IgG | 152ng/107cells |
| ADA | 8.6IU/L | Antiplatelet Ab | - |
| Culture | - | MPO-ANCA | <10U/mL |
| Cytology | Class II | PR3-ANCA | <10U/mL |
| TP | 4.9 g/dL | HBs Ag | 0.01 IU/mL |
| Alb | 2.5 g/dL | HCV-Ab | - |
| BUN | 29.1 mg/dL | EB VCA IgG | + |
| Cr | 1.11mg/dL | EB VCA IgM | - |
| eGFR | 40.7mL/min/1.73m2 | EB EBNA IgG | + |
| UA | 5.9 mg/dL | Anti-Cytomegalovirus pp65 Ab | - |
| AST | 14 IU/L | HIV-Ab | - |
| ALT | 8 IU/L | Beta D glucan | ≤5.0pg/mL |
| ALP | 598 IU/L | Blood culture | - |
| T-Bil | 0.8mg/dL | ||
| γGTP | 102 IU/L | Urinalysis | |
| LDH | 285 IU/L | Protein | 3.2g/ gCr |
| TG | 148mg/dL | Sugar | - |
| LDL-C | 80mg/dL | Red Blood Cells | 12 /HPF |
| Na | 135mEq/L | White Blood Cells | 11.7/HPF |
| K | 5.0mEq/L | Granular casts | + |
| CL | 100mEq/L | Epithelial cell casts | + |
| Ca | 7.2mg/dL | fatty casts | + |
| P | 5.3mg/dL | NAG | 35.8 U/L |
| CRP | 7.0mg/dL | β2MG | 3,831μg/L |
| HbA1c(NGSP) | 5.8% | Bence Jones protein | - |
| Fecal occult blood | - |
Ab: antibody, ADA: adenosine deaminase activity, Ag: antigen, Alb: albumin, ALP: alkaline phosphatase, ALT: alanine aminotransferase, ANA: antinuclear antibody, ANCA: antineutrophil cytoplasmic antibody, APTT: activated partial thromboplastin time, AST: asparate aminotransferase, Ba: basophils, β2MG: beta-2 microglobulin, BUN: blood urea nitrogen, C: Complement Component, CCP: anti-citrullinated protein, Cr: creatinine, CRP: C-reactive protein, ds: double stranded, EBNA: Epstein-Barr nuclear antigen, EBV: Epstein-Barr Virus, Eo: eosinophils, γGTP: gamma-glutamyl transpepidase, Hb: hemoglobin, HBs: hepatitis B surface, HCV: hepatitis C virus, HIV: human immunodeficiency virus, Ht: hematocrit, Ig: Immunoglobulin, LDH: lactate dehydrogenase, LDL-C: low-density lipoprotein cholesterol, Ly: lymphocytes, MMP: matrix metalloproteinase-3, Mon: monocytes, MPO: myeloperoxidase, NAG: N-acetyl-beta-D-glucosaminidase, Neu: neutrophils, NGSP: National Glycohemoglobin Standardization Program, PA-IgG: platelet-associated immunoglobulin, PT-INR: prothrombin time-international normalized ratio, RBC: red blood cells, RNP: anti-ribonucleoprotein, Scl-70: anti-centromere, s-IL2R: soluble-interleukin 2 receptor, TG: triglycerides, T-Bil: total-bilirubin, TP: total protein, UA: uric acid, VCA: viral-capsid antigen, WBC: white blood cells
Figure 1.

Chest X-ray and computed tomography at the initial admission to our hospital. A chest X-ray film showed bilateral pleural effusion (a). Computed tomography showed cardiac effusion, pleura effusion (b), ascites, splenomegaly (c), and kidney enlargement (d).
Figure 2.

Multiple lymphadenopathy and splenomegaly were observed on positron emission tomography-computed tomography. Positron emission tomography-computed tomography with [18F]-fluorodeoxyglucose (FDG) revealed lymphadenopathy with the strong uptake of FDG in the deep cervical lymph nodes (a arrows), the paratracheal lymph nodes (b arrows), the subaortic lymph nodes (c arrow), the splenic hilar and the arterial lymph nodes (d arrowhead). The maximum diameter of the subaortic lymph nodes was 35 mm (c arrow). The slight uptake of FDG was also observed in the enlarged spleen (d arrow) and in the axillary lymph nodes (e arrows).
A left axillary lymph node biopsy revealed atrophic germinal centers with the expansion of the interfollicular zone (Fig. 3). The proliferation of highly dense endothelial venules was seen in both the germinal centers and the interfollicular zone; relatively few mature plasma cells were seen (Fig. 4). The test for human herpesvirus-8 was not available. No other diseases, such as infection, collagen diseases, and malignancies were detected. A bone marrow aspiration sample was rather hypocellular. There was no evidence of thrombocytosis, hyperplastic change, or the infiltration of neoplastic cells (Fig. 5).
Figure 3.
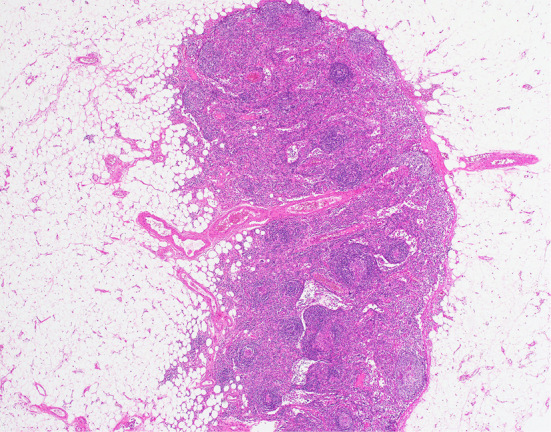
The histological findings in the left axillary lymph node (×40). The germinal centers were atrophic with the expansion of the interfollicular zone (×40, Hematoxylin and Eosin staining).
Figure 4.
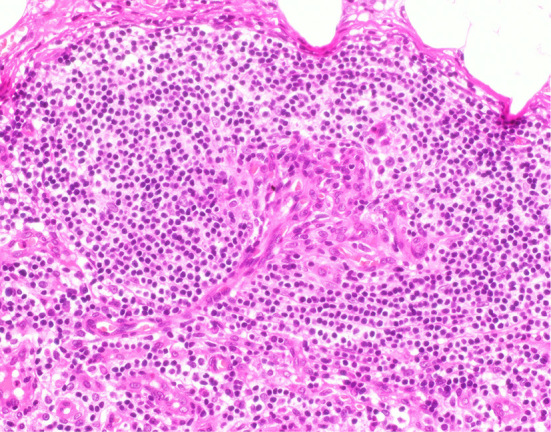
The histological findings in the left axillary lymph node (×400). The proliferation of highly dense endothelial venules was seen in both the germinal centers and in the interfollicular zone. Relatively few mature plasma cells were seen (×400 Hematoxylin and Eosin staining).
Figure 5.
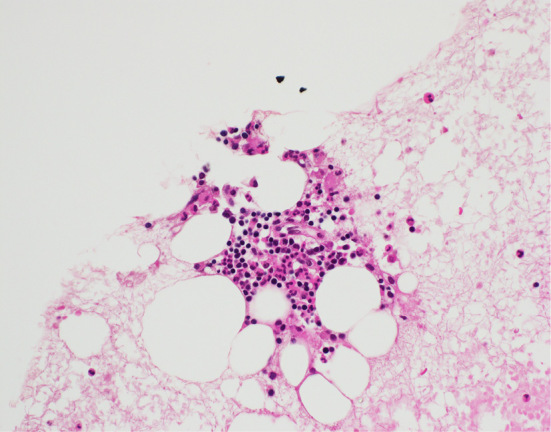
Bone marrow sample (×400). The bone marrow sample was rather hypocellular. There was no evidence of thrombocytosis, hyperplastic change, or the infiltration of neoplastic cells. The number of nucleated cells was 3.2 ×104 /μL; the proportion of megakaryocytes was 0.2%.
A histological analysis of the kidney revealed 23 glomeruli, 3 of which showed global sclerosis. The analysis showed endothelial swelling, mild interstitial inflammation, interstitial fibrosis and tubular atrophy (Fig. 6, 7). No other specific changes, such as endotheliosis, thrombotic microangiopathy, double contour, subendothelial accumulation, mesangial cell proliferation, mesangial matrix expansion, or mesangiolysis were evident. An immunofluorescence analysis revealed no deposits of IgG, IgA, IgM, Complement Component (C) 3, C4, or fibrinogen. Electron microscopy showed endothelial swelling (Fig. 8) and slight tubule epithelial changes. No particular basement membrane changes, dense deposits, or fibrils were found.
Figure 6.

The histological findings in the kidney (×40). Light microscopy showed some glomeruli with mild interstitial inflammation and fibrosis (Hematoxylin and Eosin staining).
Figure 7.
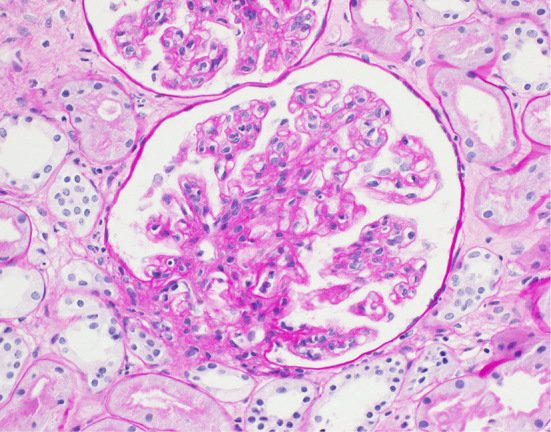
The histological findings in the kidney (×400). The endothelial cells appeared swollen in each glomerulus (periodic acid-Schiff staining).
Figure 8.
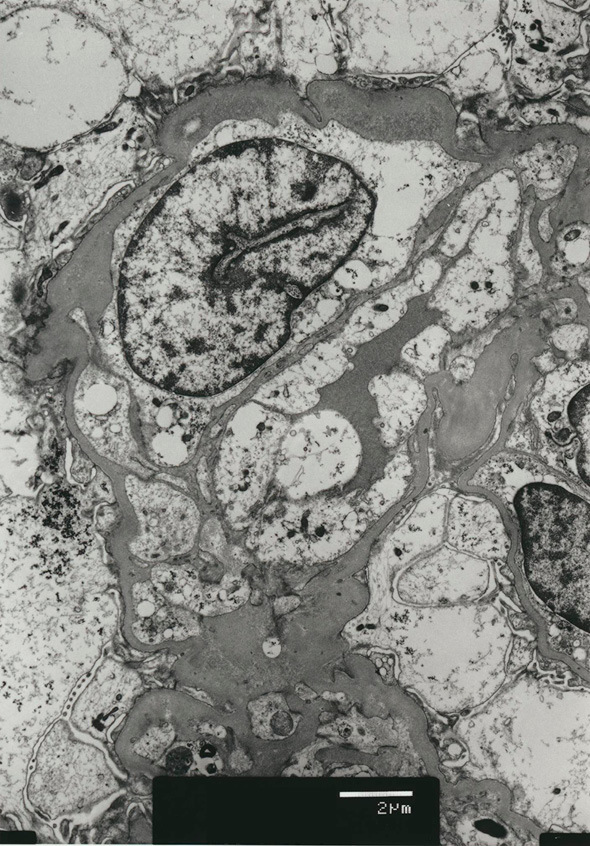
The histological findings in the kidney (electron microscopy). Electron microscopy also showed endothelial swelling. Other changes, such as changes in the glomerular basement membrane and foot processes, were not prominent.
Her nephrotic syndrome was persistent with other general symptoms, including fatigue and fever; her serum creatinine level reached 1.67 mg/dL (eGFR 26.0 mL/min/1.73 m2). Oral prednisolone therapy (40 mg/day, 0.8 mg/kg/day) was initiated. Her urinary protein level decreased to 0.7 g/gCr after a week, and many of her other problems were relieved: anasarca was reduced, her CRP level decreased to 0.05 mg/dL, her platelet count was 8.9×104/μL, and her serum creatinine returned to 0.43 mg/dL (eGFR 114.9 mL/min/1.73 m2), without urine casts. Still, her blood pressure was approximately 150/90 mmHg with the administration of olmesartan medoxomil (an antihypertensive agent). Cilnidipine was added, and her blood pressure fell to around 125/60 mmHg. The dose of prednisolone was gradually reduced to 2.5 mg. Her urinary protein level decreased to 0.3 g/gCr, and her urinary RBC count fell to 4/HPF without casts. Moreover, her platelet count increased to 13×104/μL, her hypoalbuminemia returned to normal (4.7 g/dL), and her kidney function was maintained (serum creatinine 0.61 mg/dL) without systemic inflammation (CRP 0.03 mg/dL).
Discussion
The diagnostic criteria for TAFRO syndrome were not published when the patient in the present case developed nephrotic syndrome and underwent biopsy. According to the 2015 diagnostic criteria for TAFRO syndrome (2), this case met all of the three major categories (anasarca, thrombocytopenia, and systemic inflammation) and three of the four minor categories [Castleman disease (CD)-like features on lymph node biopsy, mild organomegaly, and progressive renal insufficiency]. Iwaki et al. proposed the diagnostic criteria for TAFRO-iMCD based on their clinicopathological analysis of TAFRO syndrome (6). Kidney involvement was not mentioned in the criteria. However, this case fulfilled all of the major criteria: four of the five TAFRO symptoms (thrombocytopenia, anasarca, fever, and organomegaly), the absence of hypergammaglobulinemia, and small volume lymphadenopathy in addition to the histopathological criteria and one of the two minor criteria: high levels of serum alkaline phosphatase without markedly elevated serum transaminase.
On the other hand, renal insufficiency was defined as a glomerular filtration rate of <60 mL/min/1.73 m2 in the 2015 diagnostic criteria for TAFRO syndrome (2). Based on this classification, the case was classified as grade 3: slightly severe. In the consensus diagnostic criteria for idiopathic multicentric CD (iMCD), proteinuria (150 mg/24 h, or 10 mg/100 mL) is included with renal dysfunction (eGFR<60 mL/min/1.73 m2) (3).
Little is known about kidney pathophysiology in TAFRO syndrome. Kidney biopsy specimens have rarely been obtained from patients with the condition. Kawashima et al. reported the kidney biopsy results in one case, which consisted of the mild-to-moderate proliferation of mesangial cells, a thickened basement membrane with double contour, and spike formation, along with mild to moderate positivity for IgA, IgM, and C3 tracing the glomerulocapillary in an immunohistochemical examination (4). On the other hand, although a kidney biopsy was not performed, Kubokawa et al. assumed that the pathophysiology of their case was interstitial nephritis due to the presence of mild proteinuria and mildly elevated urinary NAG and B2MG without hematuria (5). In our case, a kidney biopsy showed endothelial cell swelling and mild interstitial inflammation without any signs in an immunohistochemical analysis. To the best of our knowledge, this is the first reported case of nephrotic syndrome with TAFRO syndrome.
Small-vessel lesions are suggested to be the most frequent kidney manifestation in CD (7,8). Meanwhile, vascular endothelial growth factor (VEGF) was thought to be involved in the pathogenesis of both CD (7,8) and TAFRO syndrome (2-5). The production of VEGF by podocytes seems to have an essential role in the glomerular endothelium (9). Elevated systemic serum levels of VEGF have been shown both in patients with CD (7) and in those with TAFRO syndrome (1-5). However, the glomerular expression of VEGF is reported to be downregulated in patients with small vessel lesions caused by CD (7). Thus, the glomerular expression of VEGF seems to be more important to the function of the glomerular endothelium than the presence of systemic VEGF. This might explain the small vessel lesions that are observed in patients receiving anti-VEGF agents and in those with preeclampsia (7). Unfortunately, we did not check the systemic level of VEGF or the glomerular expression of VEGF in this case. Additionally, the distinct mechanisms that cause each kidney biopsy finding (i.e., thrombotic microangiopathy, crescentic glomerulonephritis, and endothelial swelling) are not known. We could only hypothesize that VEGF might have caused the nephrotic syndrome in the present case. The accumulation of a greater number of studies is needed, and the kidney biopsy findings of such cases should be investigated in order to determine the pathogenesis of the kidney involvement.
Corticosteroids were often used as a first-line therapy; 47.8% responded well to initial corticosteroid treatment, while some other cases required additional treatments, such as cyclosporine, tocilizumab, and rituximab (6). At present there is no way to predict the patients who are likely to be refractory to corticosteroid therapy.
It is possible that the patient's blood pressure had some influence on her urinary protein level. At the onset of her symptoms, her blood pressure was continuously high, while prednisolone improved her proteinuria and urinary casts. We assume that her proteinuria was mainly caused by inflammation from TAFRO syndrome, and not by hypertension.
We did not observe reticulin fibrosis in the bone marrow samples in this case. A bone marrow biopsy would have been needed to observe reticulin fibrosis.
In summary, we presented a case of nephrotic syndrome that was later diagnosed as TAFRO syndrome. A kidney biopsy showed endothelial swelling, which could have been due to small vessel lesions. The accumulation of a greater number of case studies is needed, and the kidney biopsy findings of such cases should be investigated in order to clarify the pathogenesis of kidney involvement.
This article does not contain any studies with human participants or animals.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
Authors thank Professor Jean Louise Mahar for providing instruction on professional and technical writing.
References
- 1.Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki 51: 320-325, 2010. (in Japanese, Abstract in English). [PubMed] [Google Scholar]
- 2.Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol 103: 686-692, 2016. [DOI] [PubMed] [Google Scholar]
- 3.Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 129: 1646-1657, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawashima M, Usui T, Okada H, et al. TAFRO syndrome: 2 cases and review of the literature. Mod Rheumatol 1-5, 2015. (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 5.Kubokawa I, Yachie A, Hayakawa A, et al. The first report of adolescent TAFRO syndrome, a unique clinicopathologic variant of multicentric Castleman's disease. BMC Pediatr 14: 139, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol 91: 220-226, 2016. [DOI] [PubMed] [Google Scholar]
- 7.El Karoui K, Vuiblet V, Dion D, et al. Renal involvement in Castleman disease. Nephrol Dial Transplant 26: 599-609, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Xu D, Lv J, Dong Y, et al. Renal involvement in a large cohort of Chinese patients with Castleman disease. Nephrol Dial Transplant 27 (Suppl 3): iii119-iii125, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Ollero M, Sahali D. Inhibition of the VEGF signalling pathway and glomerular disorders. Nephrol Dial Transplant 30: 1449-1455, 2015. [DOI] [PubMed] [Google Scholar]