

Abstract
Diseases such as Alzheimer's and cancer have been linked to metabolic dysfunctions, and further understanding of metabolic pathways raises hope to develop cures for such diseases. To broaden the knowledge of metabolisms in vitro and in vivo, methods are desirable for direct probing of metabolic function. Here, we are introducing a pulsed nuclear magnetic resonance (NMR) approach to generate hyperpolarized metabolites within seconds, which act as metabolism probes. Hyperpolarization represents a magnetic resonance technique to enhance signals by over 10 000‐fold. We accomplished an efficient metabolite hyperpolarization by developing an isotopic labeling strategy for generating precursors containing a favorable nuclear spin system to add para‐hydrogen and convert its two‐spin longitudinal order into enhanced metabolite signals. The transfer is performed by an invented NMR experiment and 20 000‐fold signal enhancements are achieved. Our technique provides a fast way of generating hyperpolarized metabolites by using para‐hydrogen directly in a high magnetic field without the need for field cycling.
Keywords: hyperpolarization, magnetic resonance, metabolite, para-hydrogen-induced polarization, sidearm hydrogenation
Probing and understanding metabolic pathways and dysfunction is an approach that leads to new insights, which may help to develop cures for a variety of diseases including Alzheimer's and cancer. Hyperpolarized nuclear magnetic resonance (HP‐NMR) is an emerging approach in the fields of chemistry, biochemistry, biological sciences, and medical diagnostics to probe chemical reactions or metabolic events in vitro and in vivo and correlate them to disease detection, progression, or treatment responses.1, 2, 3, 4, 5, 6 Various hyperpolarization methods exist, of which dynamic nuclear polarization (DNP),1, 2, 3, 4, 5, 6 spin exchange optical pumping (SEOP)7, 8, 9, 10 of noble gases, and the use of para‐hydrogen11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 are of relevance to medicine. In HP‐NMR, the signals of molecular tracers are boosted by more than 10 000‐fold compared to the thermal equilibrium values that are normally accessed in a typical laboratory. The hyperpolarization of 13C nuclei is of special interest, as they often possess longitudinal relaxation times that range typically between several tens of seconds to a few minutes, which define the enhanced polarization lifespan.3 For example, the signal of pyruvate can be monitored for 2–3 min if 13C‐labeled molecules are used.3 Currently, dissolution DNP is the most prominent methodology for hyperpolarizing metabolites. Despite enormous advancements, DNP remains a costly method that takes up to hours to polarize molecules of interest and it requires on‐site specialized equipment as well as the utilization of cryogenic liquid helium. para‐Hydrogen can be used as an alternative, simpler method for enhancing the NMR signal of molecules, and it can easily be generated in large quantities either on‐site or off‐site and stored for periods of weeks at room temperature, as the spin order resides in a long‐lived singlet state.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 Signal enhancements are achieved within a few seconds, allowing for rapid succession of experiments with potential new applications of measuring real‐time metabolic or treatment response upon a continuous replenishing of metabolites.
Two successful para‐hydrogen‐based hyperpolarization schemes have been devised to date: para‐hydrogen‐induced polarization (PHIP)11, 12, 13, 14, 15, 16, 17, 18, 19, 20 and signal amplification by reversible exchange (SABRE).21, 22, 23, 24, 25, 26, 27, 28, 29, 30 In the latter method, the singlet order of para‐hydrogen is converted into observable magnetization in a substrate of interest, mediated by a transition‐metal complex.21 para‐Hydrogen, the metal complex, and the substrate form a temporarily stable complex, during which polarization transfer to the nuclear spins occurs and the substrate remains unchanged.21 SABRE performs best in organic solvents, which has, so far, excluded in vivo experiments and work is on the way to make polarization accessible in biocompatible solvents.27, 28, 29, 30 PHIP is an approach in which an unsaturated bond in a precursor molecule is being hydrogenated with para‐hydrogen.11, 12, 13, 14, 15, 16, 17, 18, 19, 20 During the hydrogenation process, the symmetry of para‐hydrogen is broken and observable polarization is obtained.20 In vivo PHIP experiments have mainly been restricted to hyperpolarized hydroxyethyl propionate, succinate, and tetrafluoropropyl propionate.11, 12, 13, 14 Metabolites that lack unsaturated precursors, such as pyruvate, could not be polarized until the introduction of PHIP by means of a sidearm hydrogenation (PHIP‐SAH).15, 16, 17 In PHIP‐SAH, a 13C‐labeled metabolite is attached to a sidearm with an unsaturated bond that is hydrogenated with para‐hydrogen, and the resulting polarization is transferred from protons to the 13C nucleus. Finally, the sidearm is cleaved off, yielding the hyperpolarized metabolite.15 A similar approach has been investigated for hyperpolarizing ethanol and has recently been shown to be applicable in gas‐phase reactions.31, 32 Polarization transfer in PHIP‐SAH has been achieved so far by a field‐cycling procedure, in which a sample is shuttled into a magnetic field below the earth's field, followed by re‐magnetization to transfer proton polarization to 13C‐labeled nuclei.15, 16, 17 It was theoretically predicted that about 40 % of the initial two‐spin singlet order of para‐hydrogen could be transferred with this technique and about 5 % polarization was achieved experimentally.17 In this work, we extend the PHIP‐SAH methodology for generating hyperpolarized metabolites by introducing 1) a prototypical molecular system that acts as a metabolite precursor, based on a 13C and deuterium isotope labeling scheme at various positions; 2) a novel pulsed NMR experiment, which is operated at high field that can transfer 1H polarization originating from para‐hydrogen to the first 13C nucleus in the designed sidearm and, in a subsequent step, to the 13C nucleus in the metabolite. We show that the proposed pulse sequence can achieve a theoretical spin order transfer close to 95 % in the designed system. Finally, we demonstrate the cleavage of the precursor to release the hyperpolarized metabolite. The procedure is summarized in Figure 1, starting from an unsaturated, double 13C‐labeled cinnamyl ester (here: acetate or pyruvate). The chosen cinnamyl esters have a triple bond that result in the fast hydrogenation necessary for efficient competition with relaxation effects, resulting in high polarization. On the other hand, by utilizing a non‐terminal triple bond, the chosen catalyst selectively hydrogenates the compound in organic solvents (here: [D6]acetone) to the double bond only, as described in the literature.34 Moreover, it makes the starting molecules more hydrophobic, which is necessary for performing the reaction in organic solvent and may facilitate separation of the metabolite after its cleavage, for example, by extraction. Notably, all steps (para‐hydrogenation, coherent polarization transfer to the target 13C nuclei, and bond cleavage for metabolite release) are performed within the magnetic field of the NMR spectrometer. We demonstrate that our approach leads to over 23 000‐fold signal enhancement at 7 T or 13 % 13C polarization for para‐hydrogenation products of metabolite precursors within 20 s without the necessity for field cycling equipment.
Figure 1.
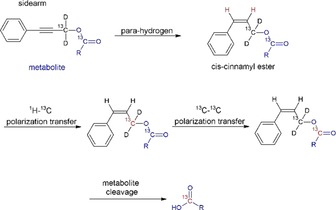
Steps of the pulsed polarization transfer from para‐hydrogen to a metabolite in a double‐labeled 13C precursor. Red indicates the polarized nucleus.
To efficiently polarize metabolites, we designed a novel NMR sequence and the labeled esters. The J‐connectivity of the molecular systems used in this work is depicted in Figure 2 a, and the NMR pulse sequence used for converting spin order from the para‐hydrogen 1H1‐*1H2 pair into heteronuclear in‐phase magnetization of the deuterated methylene 13C3 nucleus and of the carboxylate 13C4 nucleus is shown in Figure 2 b. The sequences are designed for spin‐order transfer (SOT) at high magnetic fields in weakly coupled systems, which may as well be a permanent magnet setup with, for example, B 0> 0.5 T.35 The sequence operation will be concisely described in the following paragraphs, whereas a more detailed analysis is presented in the Supporting Information. The initial density matrix of the added proton pair is described by the product operator ρ 0=I 1z I 2z, as hydrogenation occurs incoherently in high magnetic field (PASADENA‐style para‐hydrogenation).20 With reference to Figure 2 b, the longitudinal two‐spin order I 1z I 2z is firstly transferred from the para‐hydrogen protons to anti‐phase magnetization I 1z S 3x on the 1H2–13C3 pair. The sequence is then alternatively concatenated with the blocks in Figure 2 b1 or 2 b2 to obtain in‐phase magnetization on 13C3 or 13C4, respectively. In the one case, an INEPT block allows to direct the spin density operator I 1z S 3x to S 3y/2. In the other case, I 1z S 3x is firstly transformed into coherence involving only the two carbons S 3x S 4z and then to S 4y/2 for direct observation or to S 4z/2 for storage before the cleavage step. In linear J‐connected chains, the proposed sequences achieve 100 % coherent SOT from two‐spin longitudinal order of para‐hydrogen to carbon magnetization if relaxation is disregarded. Here, the efficiency of the transfer between an initial state ρ 0 at time t 0 and a given state ρ f at time t is intended as: φ=|<ρ f|U|ρ 0>|/(|ρ f∥ρ 0|), where U is the evolution operator between t 0 and t.
Figure 2.
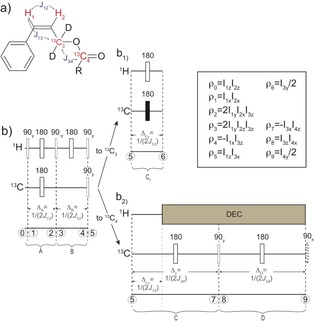
NMR pulse sequences for heteronuclear spin order transfer. a) The asymmetric J‐connectivity in the para‐hydrogenated sidearm is exploited to transfer spin order from the added para‐H2 to the labeled carbons 13C3 and 13C4. The nuclei that compose the spin system of interest are highlighted in red. b) Pulse sequences for spin‐order transfer to 13C magnetization: b1) polarization of 13C3 and b2) polarization of 13C4. The thin and large rectangles represent 90° and 180° pulses, and the subscripts indicate the phases. The black filled rectangle in the sequence block (b1) represent a selective pulse on 13C3. The vertical dashed lines separate the logical blocks of the pulse sequence. The circled numbers highlight specific time points used to follow the evolution of the spin density operator through the sequence as reported in the list beside the drawing. The dashed 90° pulse after timepoint 9 is used to convert S 4y into S 4z for magnetization storage before the cleavage step.
The standard PH‐INEPT sequences used for SOT at high magnetic fields assume that the two nuclei are equally affected by a non‐selective pulse. With a nutation angle of the first pulse on the 1H channel being set to 45°, one can achieve a maximum 50 % polarization transfer.18 Indeed, the effect of a pulse β with phase y on the para‐hydrogen spin order is given by Equation (1):19
| (1) |
so that the nutation angle of 45° converts only half of the para‐hydrogen spin order into anti‐phase proton magnetization for further polarization transfer. The sequence proposed here is adapted to the possibility of distinguishing the two polarized protons, owing to their position on the designed sidearm, because, after para‐hydrogenation, the 3 J 1,3 coupling is large compared to 2 J 2,3 (less than 0.5 Hz is estimated). This allows us to disregard the evolution under 2 J 2,3 and to optimize the first delay ΔA with respect to 3 J 1,3 only. As the sequence can be applied to target heteronuclei other than 13C, the acronym ESOTHERIC (efficient spin order transfer to heteronuclei via relayed INEPT chains) is proposed.
To assess the efficiency of sequential spin‐order transfers after para‐hydrogenation of the precursors, polarization levels at 7 T were detected in a set of three different experiments on 1H2 and on the methylene 13C3 and on the carboxylic 13C4 in cis‐cinnamyl acetate and cis‐cinnamyl pyruvate. Figure 3 a shows that the carboxylate 13C NMR signals of cinnamyl acetate and cinnamyl pyruvate after para‐hydrogenation and application of the ESOTHERIC sequence are enhanced more than 20 000 times for the acetate derivative and 15 000 times for the pyruvate derivative compared to that of a thermally polarized higher concentrated solution of the acetate precursor, 3‐phenyl(1–13C,2H2)prop‐2‐yn‐1‐yl(1–13C)acetate. The enhanced 1H and 13C NMR spectra of the labeled methylene carbon (13C3) of the cinnamyl derivatives are shown in the Supporting Information. The signal enhancements were calculated by taking the ratio of the integrated peaks in the para‐hydrogenated systems over the integrated reference signal (aromatic protons for 1H2, labeled methylene carbon for 13C3, and labeled carboxylic carbon for 13C4) in the thermally polarized acetate precursor, scaled by the respective number of spins, number of scans, and concentrations (we would like to note that utilizing much larger concentrations than 1.5 mm of metabolites resulted in radiation damping, which complicates the proper quantification of the experiments). The proton signal of the products after para‐hydrogenation has anti‐phase character and the signal enhancements were calculated by taking the total of the absolute value of the integral for each H2 proton peak. The polarization values were calculated as the products of the enhancements by the thermal polarizations (22.4 ppm for 1H and 5.66 ppm for 13C) at the temperature of the reaction (320 K) in a 7 T magnetic field. The mean values with standard deviations are summarized in Table 1. As the enhancement values of the products were calculated assuming their complete hydrogenation before the measurement sequence, the reported 1H polarization values are conservative estimates of the achievable polarization levels. As the proton signal is detected after a 45° pulse, the reported polarization values correspond to 50 % of the total para‐hydrogen two‐spin order, which can be utilized for the polarization transfer to the 13C nuclei, according to Equation (1). In our experiments, we obtained P(1H2)=14.4±1.1 % and P(1H2)=10.7±0.6 % for the hydrogenated cinnamyl acetate and cinnamyl pyruvate, respectively. Hence, we estimated that the maximum two‐spin order that can be transferred into 13C polarization is 28.8 % in the acetate derivative and 21.4 % in the pyruvate derivative. By using the ESOTHERIC pulse sequence described above, we achieved P(13C4)=13±0.9 % for the acetate and P(13C4)=8.9±0.2 % for the pyruvate derivative. This gives an experimental transfer efficiency of 45 % for acetate and 42 % for pyruvate. The transfer efficiencies are calculated for the molecules after para‐hydrogen has been added and do not take into account how efficient the hydrogenation is. Theoretically, our approach has a transfer efficiency close to unity. In practice, J‐coupled protons from the acetate and the phenyl group that are not part of the coupling network utilized during the transfer reduce the efficiency by about 5 % (see the Supporting Information for a detailed discussion of the transfer efficiency; other 1H couplings are less than 1 Hz) to 95 %. The main source of loss in the experiments is attributed to spin relaxation, especially to 13C3, which has a transverse relaxation time T 2 (13C3)=1 s (other relaxation times: T 1 (13C3)=50 s, T 1 (13C4)=52 s, T 2 (13C4)=10 s) in the acetate derivative in the presence of the catalyst.
Figure 3.
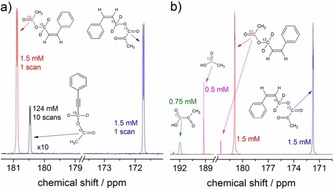
Hyperpolarization of acetate and pyruvate. a) 13C {1H} NMR spectra of hyperpolarized cinnamyl acetate (red) and hyperpolarized cinnamyl pyruvate (blue) in comparison to the thermally polarized acetate precursor (in black). Each spectrum was obtained in a separated experiment at 320 K in a 7 T magnetic field. The delays were ΔA=41.6 ms, ΔB=44.6 ms, ΔC=ΔD= 206 ms for cinnamyl acetate and ΔA=43.4 ms, ΔB=41.6 ms, ΔC=ΔD= 188.6 ms for cinnamyl pyruvate. b) 13C{1H} NMR hyperpolarized spectra of cinnamyl acetate (red) and cinnamyl pyruvate (blue) and the cleaved metabolites: acetate (magenta) and pyruvate (green).
Table 1.
Polarization levels and J‐couplings in acetate and pyruvate precursors. The atom subscripts follow the numbering of Figure 2.
| Product |
P (1H2) [%] |
P (13C3) [%] arm |
P (13C4) [%] metabolite |
3
J
1,2
(J HH) [Hz] |
3
J
1,3
(J HC) [Hz] |
2
J
3,4
(J CC) [Hz] |
|---|---|---|---|---|---|---|
| cinnamyl acetate |
14.4±1.1 | 21.1±0.4 | 13±0.9 | 11.9 | 11.4 | 2.35 |
| cinnamyl pyruvate |
10.7±0.6 | 12.4±0.8 | 8.9±0.2 | 12.0 | 11.5 | 2.65 |
To estimate the polarization losses during the para‐hydrogen bubbling period, we measured the 1H spin‐lattice relaxation time T 1 (1H)=12 s and the 1H singlet lifetime36 T S=17 s of the added protons in the cis‐cinnamyl ester acetate. Assuming an exponential 1H‐polarization decay during the bubbling of para‐hydrogen (20 s), according to T S, we expect an immediate polarization improvement of a factor of 2–3 if the polarization is carried out within two or three seconds inside a more sophisticated polarizer as, for example, presented in Refs. 11, 12, 13, 14.
Lastly, we assessed the polarization levels transferred to the carboxylic 13C in acetic acid and in pyruvate after cleaving off the sidearm, see Figure 3 b. To ensure complete operation of B1 on the whole spin system before and after injection of the cleaving solutions, the volume of the initial [D6]acetone solutions, containing the catalyst and the precursor, was reduced to 0.2 mL, corresponding to half of the height of the RF coil. After the cleavage solution, the total volume corresponded to 0.4 mL, on which the whole B1 acts and allows for quantification of the polarization level. Following para‐hydrogenation, the carboxylic 13C4 nucleus in the cinnamyl derivatives was hyperpolarized by using the ESOTHERIC sequence. An additional 90° pulse on the 13C channel was applied in order to store the hyperpolarization as longitudinal magnetization. A 0.2 mL solution of 0.5 m NaOD in D2O/[D4]MeOH (50 %/50 % in v/v) was injected through a plastic hose to the hyperpolarized derivatives inside the cryomagnet. After injection of the NaOD solution, nitrogen gas was bubbled at atmospheric pressure through the solution to speed up the mixing process. After a fixed reaction time, the remaining polarization was detected by using a 90° pulse. The 13C spectrum (Figure 3 b) thus obtained shows the release of free acetate at 189 ppm and pyruvate at 192 ppm as well as the signal of the residual cinnamyl acetate (182 ppm). That the peaks can be assigned to acetate and pyruvate has been confirmed by adding additional 13C‐labeled metabolite (cleaved metabolite) to the NMR tube to detect the 13C signal in one scan. Taking into account the final concentration of the sample, it was estimated that about a third of the polarization, P(13C)=3.6 %, is preserved in both acetic acid and cinnamyl acetate after 40 s of cleavage reaction. A similar polarization level, P(13C)=3.4 %, is preserved in pyruvate and after 10 s of cleavage reaction.
In summary, we have introduced a pulsed approach that allows for generating high levels of polarization in metabolites within seconds by utilizing para‐hydrogen in the high field of the probing magnet. High fields may not just be generated by superconducting magnets, but, for example, by a portable Halbach array based on permanent magnets with magnetic field strengths larger than 0.5 T. Our success in polarizing metabolites was accomplished by introducing a selective 13C and 2H labeling scheme that allowed us to design an optimized metabolite precursor and combine it with a pulse sequence specifically designed for spin‐order transfer of para‐hydrogen in that molecule. In the proposed molecular systems, the theoretical efficiency for polarization transfer from para‐hydrogen to the 13C nucleus in the metabolite of interest approaches unity when relaxation effects are disregarded, and we obtain more than 42 % experimentally. The transfer efficiency translates into average polarization levels of 13 and 8.9 % for acetate and pyruvate precursors, and we demonstrated that hyperpolarized acetate and pyruvate can be obtained upon cleavage of the sidearm. Although we demonstrate the polarization of metabolites in organic solvents mainly for quantification reasons, we expect that, in the near future, polarization levels >30 % and signal enhancements larger than 50 000 in cleaved metabolites can be obtained, utilizing a more sophisticated para‐hydrogenation reactor in combination with a more efficient automated precursor cleavage protocol (and phase extraction). Improving the metabolite precursors relaxation times may result in a further increase of polarization. The results obtained so far demonstrate that PHIP is a feasible method to rapidly generate hyperpolarized metabolites and the applicability for portable permanent magnet setups is at hand.
Experimental Section
[D6]Acetone samples with a 1.5 mm concentration of the precursor and 5 mm of commercially available catalyst ([1,4‐Bis(diphenylphosphino)butane] (1,5‐cyclooctadiene)rhodium(I) tetrafluoroborate) were placed in a 5 mm NMR tube. para‐Enriched hydrogen gas was obtained by using a Bruker para‐hydrogen generator (BPHG 90). The nominal conversion temperature in the generator was set to 36 K, providing 92 % para‐enriched hydrogen gas. Gaseous para‐hydrogen was delivered through a capillary and bubbled through the solution by using a home‐built, automated setup at 320 K to speed up the reaction. The gas delivery setup follows the idea of Ref. 33. The para‐hydrogen gas was kept at 6 bar to achieve a higher concentration of the dissolved gas and increase the rate of the hydrogenation reaction. The NMR measurements were done in a standard double resonance inverse probehead in a 7 T cryomagnet coupled to a Bruker spectrometer system (Avance III HD, 300 MHz). Following a bubbling period of 20 s and a settling time of 2 s, an NMR pulse sequence was initiated to transfer the para‐hydrogen spin order to observable magnetization, and the FID of the enhanced 1H or 13C signal was recorded. A 45° pulse was used to probe 1H polarization in a single‐scan acquisition. The hyperpolarized 13C spectra were recorded in a single scan by applying the ESOTHERIC sequence for spin order transfer.
More experimental details and synthetic procedures can be found in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank the Max Planck Society for generous funding. We would like to thank Prof. Christian Griesinger for access to his spectrometers and facilities and discussions.
S. Korchak, S. Yang, S. Mamone, S. Glöggler, ChemistryOpen 2018, 7, 344.
References
- 1. Golman K., in ′t Zandt R., Thaning M., Proc. Natl. Acad. Sci. USA 2006, 103, 11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Day S. E., Kettunen M. I., Gallagher F. A., Hu D. E., Lerche M., Wolber J., Golman K., Ardenkjaer-Larsen J. H., Brindle K. M., Nat. Med. 2007, 13, 1382. [DOI] [PubMed] [Google Scholar]
- 3. Gallagher F. A., Kettunen M. I., Day S. E., Hu D. E., Ardenkjaer-Larsen J. H., in ′t Zandt R., Jensen P. R., Karlsson M., Golman K., Lerche M. H., Brindle K. M., Nature 2008, 453, 940. [DOI] [PubMed] [Google Scholar]
- 4. Nelson S. J., Kurhanewicz J., Vigneron D. B., Larson P. E. Z., Harzstark A. L., Ferrone M., van Criekinge M., Chang J. W., Bok R., Park I., Reed G., Carvajal L., Small E. J., Munster P., Weinberg V. K., Ardenkjaer-Larsen J. H., Chen A. P., Hurd R. E., Odegardstuen L.-I., Robb F. J., Tropp J., Murray J. A., Sci. Transl. Med. 2013, 198, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kurhanewicz J., Vigneron D. B., Brindle K., Chekmenev E. Y., Comment A., Cunningham C. H., DeBerardinis R. J., Green G. G., Leach M. O., Rajan S. S., Rizi R. R., Ross B. D., Warren W. S., Malloy C. R., Neoplasia 2011, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ardenkjaer-Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thanning M., Golman K., Proc. Natl. Acad. Sci. USA 2003, 100, 10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walker T. G., Happer W., Rev. Mod. Phys. 1997, 69, 629. [Google Scholar]
- 8. Appelt S., Baranga A. B.-A., Erickson C. J., Romalis M. V., Young A. R., Happer W., Phys. Rev. A 1998, 58, 1412. [Google Scholar]
- 9. Möller H. E., Chen X. J., Saam B., Hagspiel K. D., Johnson G. A., Altes T. A., de Lange E. E., Kauczor H.-U., Magn. Reson. Med. 2002, 47, 1029. [DOI] [PubMed] [Google Scholar]
- 10. Schröder L., Lowery T. J., Hilty C., Wemmer D. E., Pines A., Science 2006, 314, 446. [DOI] [PubMed] [Google Scholar]
- 11. Schmidt A. B., Berner S., Schimpf W., Müller C., Lickert T., Schwaderlapp N., Knecht S., Skinner J. G., Dost A., Rovedo P., Hennig J., von Elverfeldt D., Hövener J.-B., Nat. Commun. 2017, 8, 14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhattacharya P., Chekmenev E. Y., Perman W. H., Harris K. C., Lin A. P., Norton V. A., Tan C. T., Ross B. D., Weitekamp D. P., J. Magn. Reson. 2007, 186, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhattacharya P., Chekmenev E. Y., Reynolds W. F., Wagner S., Zacharias N., Chan H. R., Bünger R., Ross B. D., NMR Biomed. 2011, 24, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goldman M., Johannesson H., Axelsson O., Karlsson M., C. R. Chim. 2006, 9, 357. [Google Scholar]
- 15. Reineri F., Boi T., Aime S., Nat. Commun. 2015, 6, 5858. [DOI] [PubMed] [Google Scholar]
- 16. Cavallari E., Carrera C., Boi T., Aime S., Reineri F., J. Phys. Chem. B 2015, 119, 10035. [DOI] [PubMed] [Google Scholar]
- 17. Cavallari E., Carrera C., Aime S., Reineri F., J. Magn. Reson. 2018, 289, 12. [DOI] [PubMed] [Google Scholar]
- 18. Haake M., Natterer J., Bargon J., J. Am. Chem. Soc. 1996, 118, 8688. [Google Scholar]
- 19. Natterer M. J., Bargon J., Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293. [Google Scholar]
- 20. Bowers C. R., Weitekamp D. P., Phys. Rev. Lett. 1986, 57, 2645. [DOI] [PubMed] [Google Scholar]
- 21. Adams R. W., Aguilar J. A., Atkinson K. D., Cowley M. J., Elliott P. I. P., Duckett S. B., Green G. G. R., Khazal I. G., Lopez-Serrano J., Williamson D. C., Science 2009, 323, 1708. [DOI] [PubMed] [Google Scholar]
- 22. Rayner P. J., Burns M. J., Olaru A. M., Norcott P., Fekete M., Green G. G. R., Highton L. A. R., Mewis R. E., Duckett S. B., Proc. Natl. Acad. Sci. USA 2017, 114, E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barskiy D. A., Kovtunov K. V., Koptyug I. V., He P., Groome K. A., Best Q. A., Shi F., Goodson B. M., Shchepin R. V., Coffey A. M., Waddell K. W., Chekmenev E. Y., J. Am. Chem. Soc. 2014, 136, 3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suefke M., Lehmkuhl S., Liebisch S. A., Blümich B., Appelt S., Nat. Phys. 2017, 13, 568. [Google Scholar]
- 25. Eshuis N., Hermkens N., van Weerdenburg B. J. A., Feiters M. C., Rutjes F. P. J., Wijmenga S. S., Tessari M., J. Am. Chem. Soc. 2014, 136, 2695. [DOI] [PubMed] [Google Scholar]
- 26. Theis T., Truong M. L., Coffey A. M., Shchepin R. V., Waddell K. W., Shi F., Goodson B. M., Warren W. S., Chekmenev E. Y., J. Am. Chem. Soc. 2015, 137, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spannring P., Reile I., Emondts M., Schleker P. P. M., Hermkens N. K. J., van der Zwaluw N. G. J., van Weerdenburg B. J. A., Tinnemans P., Tessari M., Blümich B., Rutjes F. P. J. T., Feiters M. C., Chem. Eur. J. 2016, 22, 9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Truong M. L., Shi F., He P., Yuan B., Plunkett K. N., Coffey A. M., Shchepin R. V., Barskiy D. A., Kovtunov K. V., Koptyug I. V., Waddell K. W., Goodson B. M., Chekmenev E. Y., J. Phys. Chem. B 2014, 118, 13882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iali W., Olaru A., Green G. G. R., Duckett S. B., Chem. Eur. J. 2017, 23, 10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colell J. F. P., Emondts M., Logan A. W. J., Shen K., Bae J., Shchepin R. V., G. X. Ortiz, Jr. , Spannring P., Wange Q., Malcolmson S. J., Chekmenev E. Y., Feiters M. C., Rutjes F. P. J. T., Blümich B., Theis T., Warren W. S., J. Am. Chem. Soc. 2017, 139, 7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trantzschel T., Bernarding J., Plaumann M., Lego D., Gutmann T., Ratajczyk T., Dillenberger S., Buntkowsky G., Bargon J., Bommerich U., Phys. Chem. Chem. Phys. 2012, 14, 5601. [DOI] [PubMed] [Google Scholar]
- 32. Salnikov O. G., Kovtunov K. V., Koptyug I. V., Sci. Rep. 2015, 5, 13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiryutin A. S., Sauer G., Hadjiali S., Yurkovskaya A. V., Breitzke H., Buntkowsky G., J. Mag. Reson. 2017, 285, 26. [DOI] [PubMed] [Google Scholar]
- 34. Schrock R. R., Osborn J. A., J. Am. Chem. Soc. 1976, 98, 2143. [Google Scholar]
- 35. Danieli E., Perlo J., Blümich B., Casanova F., Angew. Chem. Int. Ed. 2010, 49, 4133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4227. [Google Scholar]
- 36. Levitt M. H., Annu. Rev. Phys. Chem. 2012, 63, 89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary