

Abstract
Nosiheptide, a member of the e series of macrocyclic thiopeptide natural products, contains a side-ring system composed of a 3,4-dimethylindolic acid (DMIA) moiety connected to Glu6 and Cys8 of the thiopeptide back-bone via ester and thioester linkages, respectively. Herein, we show that NosN, a predicted class C radical S-adenosylmethionine (SAM) methylase, catalyzes both the transfer of a C1 unit from SAM to 3-methylindolic acid linked to Cys8 of a synthetic substrate surrogate as well as the formation of the ester linkage between Glu6 and the nascent C4 methylene moiety of DMIA. In contrast to previous studies that indicated that 5′-methylthioadenosine is the immediate methyl donor in the reaction, in our studies SAM itself plays this role, giving rise to S-adenosylhomocysteine as a co-product of the reaction.
Graphical Abstract
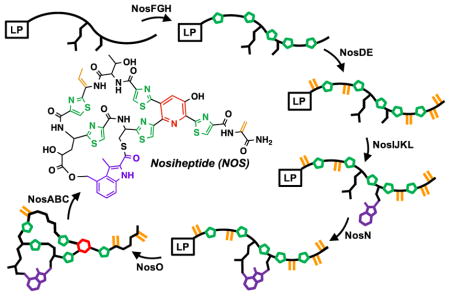
Nosiheptide (NOS) is a thiopeptide natural product that displays potent in vitro activity against a number of bacterial pathogens of urgent clinical relevance, including methicillin-resistant Staphylococcus aureus, penicillin-resistant Streptococcus pneumoniae, and vancomycin-resistant-enterococci.1–3 It is composed of a core peptide macrocycle that contains multiple thiazole rings, dehydrated serine and dehydrated threonine amino acids, and a central tetra-substituted 3-hydroxypyridine ring. In addition to several other post-translational modifications, NOS contains a side-ring system formed via a 3,4-dimethyl-2-indolic acid (DMIA) bridge that connects the 4-methyl group of DMIA to Glu6 of the peptide framework and the carboxylic acid group of DMIA to Cys8.3 Despite its potent in vitro activity, NOS and other similar thiopeptide natural products suffer from poor solubility and gastrointestinal absorption, limiting their use in the clinic.3
In pioneering studies by Wen Liu and coworkers, the gene cluster for the biosynthesis of NOS was identified (nosA-nosP) and the encoded gene products were assigned preliminary functions based on amino acid similarity with proteins of known functions and knock-out studies in Streptomyces actuosus, one of the producers of NOS.1 The thiopeptide is derived from a ribosomally synthesized precursor peptide of 50 amino acids (aa), which is the product of the nosM gene. The first 37 aa of the precursor peptide compose a leader sequence, which is used primarily as a recognition motif for some of the enzymes involved in NOS biosynthesis, while the last 13 aa compose the structural peptide and are incorporated into the final product. A model was put forward, wherein NosD, E, F, G, H, M, and O are responsible for generating the core thiopeptide, while NosA, B, and C are responsible for additional post-translational modifications to the core thiopeptide.1 NosI, K, L, and N were assigned functions related to generating the DMIA moiety and attaching it to the structural peptide, while NosP was suggested to be a regulator of the biosynthetic gene cluster. NosJ, however, was not assigned a function. NosL, a member of the radical S-adenosylmethionine (SAM) superfamily, has been shown to catalyze the first step in the formation of DMIA, which is the conversion of L-tryptophan into 3-methyl-2-indolic acid (MIA), while NosN, annotated as a class C radical SAM (RS) methylase, was predicted to append a methyl group to C4 of MIA or an acylated derivative of it to form DMIA.4 NosK was predicted to transfer MIA or DMIA to Cys8 of the structural peptide, and the 4-methyl group was proposed to be hydroxylated by one of the two annotated cytochrome P450-like enzymes. Lastly, NosI was proposed to generate the Glu6 ester linkage of the side-ring system by adenylating Glu6 and catalyzing the attack of the C4-hydroxyl group of 3-methyl-4-hydroxymethyl-2-indolic acid onto the resulting Glu6 acyl-AMP (Figure 1).1
Figure 1.

Proposed pathway for NOS side-ring formation.
Our recent studies,5 and subsequent studies by Ding et al.,6 have led to a modification of the above work-ing hypothesis. We showed that NosJ is an acyl carrier protein (ACP), and that NosI catalyzes the ATP-dependent activation of MIA to MIA-AMP and the transfer of MIA to NosJ. NosK then transfers MIA from MIA-NosJ to a conserved seryl residue (S102) on itself, presumably for transfer to NosM during some stage in the maturation of NOS.5 When NosK catalyzes this step has not been determined; however, the structure of the protein, which we solved to 2.3 Å resolution, reveals that Ser102 is part of a Ser-Glu-His catalytic triad located at the bottom of an open cleft that is large enough to accommodate the structural peptide.5 Our redetermination of the role of NosI in NOS biosynthesis and the recent findings that both annotated P450-like proteins in the NOS biosynthetic gene cluster have been assigned specific functions7, suggest that the in vivo function of NosN is not that of a methylase, but that it catalyzes both the transfer of a C1 unit from SAM and the subsequent formation of the ester linkage of the side-ring system by nucleophilic addition (Figure 1).
Recent studies from the Zhang laboratory detailed the first in vitro characterization of the NosN reaction.8 As a member of the RS superfamily of enzymes, NosN is believed to use a 5′-deoxyadenosyl 5′-radical (5′-dA•), derived from a reductive cleavage of SAM, to initiate radical-based catalysis.9–10 However, exactly when NosN performs its reaction during NOS maturation is unknown, which has limited mechanistic studies of the enzyme. Although nosN knockout mutants of S. actuosus produce what appears to be a near-fully elaborated NOS that lacks the C4 methyl group of DMIA, and therefore the ester linkage of the side-ring system, this molecule is not a substrate for NosN.8 Moreover, reports from the Liu laboratory have suggested that many of the post-translational modifications to NOS do not take place after the core thiopeptide has been synthesized, but are installed at various steps during the biosynthesis of the core thiopeptide.11
In this work, we provide experimental evidence that NosN catalyzes both carbon transfer from SAM and formation of the ester linkage between Glu6 and the appended C4 carbon unit of DMIA. When a small synthetic substrate containing both Glu6 and Cys8 linked to MIA is used, we find that NosN catalyzes the capture of an intermediate containing a reactive C4 exocyclic methylene group by Glu6, which forms the ester linkage and completes the biosynthesis of the side-ring system of NOS.
RESULTS
Over-production and purification of NosN
NosN containing an N-terminal hexahistidine tag was purified to ≥95% homogeneity (Figure S1, panels A and B) using methods that have already been described (See SI for details).12 Purified NosN exhibits features that are consistent with the presence of a [4Fe–4S] cluster, with a maximum abs orbance at 280 nm and a broad feature at 410 nm (Figure S2, dashed spectrum). The as-isolated enzyme contains 4.6 ± 0.2 iron ions and 3.2 ± 0.7 sulfide ions per poly-peptide. Upon reconstitution and subsequent purification by gel-filtration chromatography, NosN contains 3.7 ± 0.1 iron ions and 5.4 ± 0.4 sulfide ions per protein (Figure S2, solid spectrum).
NosN Recognizes MIA-SNAC as a Substrate
The identity and availability of the substrate for NosN has hindered detailed mechanistic studies of its reaction. Recent reports, however, indicate that NosN can convert the N-acetylcysteamine (SNAC) derivative of MIA to DMIA-SNAC.8 SNAC analogs have been employed to imitate the structure of the 4′-phosphopanthetheine prosthetic group of acyl carrier proteins during polyketide biosynthesis.13 Given its ability to recognize MIA-SNAC, it seemed likely that the true substrate for NosN might be MIA attached to NosJ or MIA attached to Cys8 of the structural peptide at some stage of nosiheptide maturation.6 As shown in Figure S3, NosN is unable to convert MIA-NosJ to the DMIA derivative in our hands, although this ability has recently been reported by Ding et al.6 Although 5′-deoxyadenosine (5′-dA) is produced, neither MTA nor SAH is produced as a co-product, indicating that the transfer of a C1 unit has not taken place. However, as shown previously, NosN is able to convert MIA-SNAC to DMIA-SNAC when incubated under turnover conditions (SAM plus dithionite), as evidenced by the production of a species exhibiting a mass-to-charge ratio (m/z) of 291, and which can be fragmented in the MS to daughter ions exhibiting m/z values of 144 and 172 (Figure S4, panel A, black trace) (Figure S5, panel A, black trace).8 Interestingly, however, in our NosN assays, a product that elutes at 3.5 min by HPLC exhibits an m/z of 289 and gives a daughter ion of 170 m/z during MS/MS fragmentation (Figure 2, trace vi) (Figure S5, panel A, red trace). If either SAM, dithionite, NosN, or MIA-SNAC is omitted from the reaction mixture, these products are not observed (Figure 2, traces i–iv). Unlike the substrate, which exhibits a UV-vis spectrum with an absorbance maximum at 320 nm, the UV-visible spectrum of the 289 m/z species has an absorbance maximum around 300 nm (Figure S5, panel B). The mass, UV-vis spectrum, and elution profile suggests that the observed species may be 4-methylene-3-methyl-indolic acid-SNAC (MMIA-SNAC) that is protonated at N1. This conclusion is consistent with this molecule’s altered conjugation and reduced retention time on a C18 UPLC column, which, in turn is likely due to the molecule’s inherent positive charge, giving rise to its [M+] charge state. The peak that elutes at 4 min (Figure 2, trace vi) is an artifact of the chromatography conditions. When the gradient is shallower, this peak is not present.
Figure 2.

MS MRM chromatogram of the 170 m/z fragment from the 289 m/z parent ion produced in the NosN reaction with 50 μM enzyme, 1 mM SAM, 1 mM MIA-SNAC, 1 mM sodium dithionite, and quenched with H2SO4. Traces are of assays incubated for 90 min without MIA-SNAC (i), without dithionite (ii), without SAM (iii), without NosN (iv), everything present after 1 min (v) and after 90 min (vi). The peak in trace vi marked by the asterisk is an artifact that arises under these chromatography conditions.
Derivatization of MMIA-SNAC to 4-hydroxymethyl-3-methyl-2-indolic acid
To provide further support for formation of a NosN-catalyzed MMIA intermediate when using MIA-SNAC as a substrate in the reaction, reactions were quenched in a final concentration of 2 M NaOH to promote the nucleophilic addition of hydroxide ion onto the electrophilic exocyclic methylene carbon, affording 4-(hydroxymethyl)-3-methyl-2-indolic acid (4OHMMIA). After neutralizing the reaction and separating the small molecules on a C18 column with MRM monitoring, formation of the hydroxymethyl adduct of MMIA (204 m/z) is observed (Figure 3, Panel iii). An additional peak is also observed at 4.2 min at a 1:1 ratio, based on the absorption at 300 nm, and has similar mass and fragment transitions. We suggest that this peak may represent C1 addition at another carbon of MIA-SNAC as a result of the smaller substrate analog hampering specificity for C1 transfer by NosN.
Figure 3.

Derivatization of MMIA-SNAC to 4-hydroxymethyl-3-methyl-2-indolic acid (4OHMMIA). Samples were moni-tored by UV-absorption at 300 nm (blue traces) and the MRM transition of 204→160 m/z for 4OHMMIA (black dotted traces). (Panel i) Standards of 4OHMMIA and MIA were separated by UPLC. (Panels ii and iii) NaOH-quenched NosN assays with 200 μM enzyme and using 1 mM MIA-SNAC as the substrate were analyzed by the developed method and compared at t=1 min and 120 min, respectively. (Panel iv) NaOH-quenched NosN assay at 120 min using 1 mM MIA-t3NosM as a substrate. The shift in elution times between the solid blue traces and the dotted black traces are due to different distances between the end of the column and the respective detector. Peaks under the asterisk represent an unknown product formed during the assay.
Use of [methyl-2H3]-SAM results in deuterium loss during NosN turnover
Reactions were conducted with [methyl-2H3]-SAM to confirm that the added C1 unit in MMIA-SNAC ultimately derives from SAM. When the mass spectrum of MMIA-SNAC produced in this reaction is compared to that of the reaction containing SAM at natural abundance, a mass increase of two is observed in the parent ion, which is consistent with the loss of one deuterium atom from SAM during the transfer of the C1 unit (Figure 4, panel A). Correspondingly, when 5′-dA (m/z 252.1) produced by the reaction containing [methyl-2H3]-SAM is monitored by MS, enrichment of the A+1 isotope (m/z 253.1) is observed, indicating incorporation of deuterium from the methyl group of SAM (Figure 4, panel B). The concentrations of 5′-dA and S-adenosylhomocysteine (SAH) were also quantified as a function of time, and their progress curves at varying concentrations of NosN were compared (Figure 5, panels A and B). The progress curves for SAH and 5′-dA were fitted to Frieden’s equation (Eq. 1) to assess the pre-steady state burst, which we attribute to a slow release of MMIA-SNAC.14 DMIA-SNAC is also observed at a low intensity (Figure S4), which we believe is an off-pathway product, as will be detailed below.
Figure 4.

(Panels A and B) Mass spectrum of MMIA-SNAC and 5′-dA, respectively, produced when 1 mM SAM is used in the NosN reaction (black traces) and when 1 mM [methyl-2H3]-SAM is used in the reaction (red traces) with 50 μM NosN.
Figure 5.

(Panels A and B) Progress curves for formation of 5′-dA and SAH in assays with 10, 20, 30, 40, and 50 μM NosN (black, magenta, blue, green, and orange traces, respectively). All assays contain 1 mM MIA-SNAC, 1 mM SAM, and 2 mM sodium dithionite.
MTA is not the direct methyl donor for the NosN reaction
Intriguingly, previous studies by Zhang and coworkers indicated that MTA is the immediate methyl donor in the NosN reaction.8 We probed this possibility further by performing reactions with [methyl-13C]-SAM in the presence of MTA at natural abundance and monitoring MMIA-SNAC formation. Even upon pre-incubating MTA with NosN and initiating the reactions by adding [methyl-13C]-SAM, only ~4% of the MMIA-SNAC produced is at natural abundance, while MTA appears to act as a modest inhibitor of the reaction (Figure 6). Moreover, in time courses of the NosN reaction, no MTA is observed that is above its background levels in our preparations of SAM.
Figure 6.

Progress curves for C1 transfer to MIA-SNAC using either [methyl-13C]-SAM or MTA as the C1 donor. [methyl-13C]-SAM was added to a 500 μM final concentration in the presence of 0, 250, and 500 μM MTA (black, red, and blue traces, respectively). Formation of 13C-labeled MMIA (closed circles) was compared to that of unlabeled MMIA (open circles).
Use of a peptide mimic to assess the catalytic mechanism of NosN
The observation that NosN catalyzes only one turnover at most with the MIA-SNAC substrate, coupled with the production of MMIA-SNAC that is slowly converted to DMIA-SNAC, suggests the slow release of an intermediate species. We posited that with the natural substrate, NosN might direct Glu6 to react with the C4 exocyclic methylene group of MMIA-SNAC, thereby generating the ester linkage and completing the formation of the side-ring system. Therefore, we synthesized a more natural mimic of the NosN substrate, termed MIA-t3NosM. MIA-t3NosM derives from residues 42–46 of NosM, and contains the three thiazole rings that surround Glu6 and Cys8, as well as the thioester linkage between Cys8 and MIA. Our choice of this substrate was dictated by ease of synthesis—not having to incorporate dehydrated amino acids—while maximizing the peptide length. MIA-t3NosM exhibits an m/z of 684 and elutes at 5 min by UPLC using a C18 column under the described conditions (Figure S6, panel B, trace i). When MIA-t3NosM is incubated with SAM, dithionite and NosN, two peaks, each exhibiting m/z 696, are observed to increase with time (Figure 7, panel A, black trace) (Figure S6, panel B, trace iv) (Figure S6, panel C, black trace). MS/MS analysis of the 696 m/z ion produces prominent daughter ions with m/z values of 170 and 188, the latter of which is consistent with the addition of a hydroxyl group to the exocyclic methylene on the MMIA moiety (Figure 7, panel B, black trace) (Figure S6, panel D, black trace). Correspondingly, when [methyl-13C]-SAM is used in reactions, a dominant ion of m/z 697 is observed, indicating 13C incorporation into the molecule (Figure S6, panel C, red trace). The observation of an increase in mass of one indicates that the carbon unit from SAM is associated with the fragment, which we assign as an MIA derivative (Figure S6, panel D, red trace). Although NosN catalyzes considerable abortive cleavage of SAM in the presence of MIA-t3NosM (Figure S7), the formation of MIA-t3NosM containing the side-ring (NosMSR) system was monitored by MRM (m/z 696), and the progress curve was compared to that for the formation of SAH (Figure 7, panel A, dotted and solid lines, respectively). MTA is also observed; however, its normalized intensity does not change with time (Figure S7, panel C).
Figure 7.

(Panel A) Chromatogram displaying the SIM of the dominant product formed when 1 mM MIA-t3NosM (black), MIA-t3NosMglu→ala (blue), and [γ-carboxy-18O]-MIA-t3NosM (red) are used in the NosN assay with 50 μM enzyme. (Panel B) MS/MS spectrum of the corresponding parent ion from panel A.
Glu6 of NOS traps the exocyclic methylene intermediate
To test the possibility that Glu6 attacks the exocyclic methylene group of MMIA during formation of NosMSR, a variant of the MIA-t3-NosM substrate containing a Glu6→Ala substitution (MIA-t3NosMglu→ala) was synthesized (Figure S8). When this substrate is incubated with NosN under turnover conditions, a dominant signal exhibiting m/z of 640 is observed, which is consistent with incorporation of one methyl group into the MIA-t3NosMglu→ala peptide (Figure 7, panel A, blue trace). The fragmentation pattern of this product (640 m/z) contains a major species at m/z 172, which is consistent with the fragmentation patterns of the methylated products DMIA-SNAC and DMIA-t3NosM (Figure 7, panel B, blue trace). Furthermore, the 188 m/z product ion—produced when fragmenting the NosMSR—is not observed from this product, suggesting that the 188 m/z ion is a result of having Glu6 in the peptide (Figure S8). To further confirm the origin of the hydroxyl group in the 188 m/z product ion, MIA-t3NosM containing a [γ-carboxy-18O]-glutamyl residue was prepared (Figure S9) for use in NosN reactions. The resulting compound is a mixture of two species: one (~37%) contains two incorporated 18O atoms, while the second (~43%) contains only one incorporated 18O atom. When the 18O-containing MIA-t3-NosM is used in a NosN reaction, a mass shift of either 2 or 4 is observed in the NosMSR, indicating retention of the isotopic label in the glutamyl residue during turnover (Figure 7, panel A, red trace). More importantly, during MS/MS analysis of the 700 m/z parent ion, a new peak of 190 m/z is observed, which is consistent with incorporation of one 18O atom into the 188 m/z peak from NosMSR, (Figure 7, panel B, red trace), unambiguously assigning Glu6 as the source of the hydroxyl substituent, and confirming the presence of an ester linkage to the C4 methyl group of DMIA. After base hydrolysis of NosMSR, and analysis by LC-MS, we observe production of 4-(hydroxymethyl)-3-methyl-2-indolic acid at a 9:1 ratio compared to the alternative product made during NosN assays with MIA-SNAC (Figure 3, panel iii, blue trace), indicating that with a more physiological substrate there is better site specificity to the C4 position of the MIA moiety.
DISCUSSION
The formation of the side-ring system of NOS requires the action of five dedicated proteins: NosI, NosJ, NosK, NosL, and NosN. Two of these proteins, NosL and NosN, belong to the RS superfamily of enzymes. NosL catalyzes a complex rearrangement of tryptophan to give MIA, while NosN is annotated as a class C RS methylase. Class C RS methylases belong to the HemN family of RS enzymes, which are believed to use two simultaneously bound molecules of SAM to methylate sp2-hybridized carbon centers. The in vitro characterization of class C methylases has been sluggish, however, because of the complex substrates on which they operate. Our recent studies, which led to a re-evaluation of the proposed NOS biosynthetic pathway, suggest that the substrate for NosN contains the core thiopeptide at some stage during its maturation, but after incorporation of the thioester linkage to MIA and before NosO-catalyzed formation of the central heterocycle.5
Recently, the Zhang laboratory described the first in vitro characterization of NosN and reported some intriguing obser-vations.8 First, they showed that NosN can use MIA-SNAC as a substrate, which it converts to DMIA-SNAC. Second, they showed that NosN first converts one molecule of SAM to MTA, which then becomes the direct methyl donor in the NosN reaction. They propose a mechanism whereby one molecule of SAM is reductively cleaved to generate a 5′-dA•, which then abstracts a hydrogen atom from MTA. The resulting methylene radical then adds to C4 of MIA to give a substrate radical intermediate. Upon loss of the C4 proton, the resulting radical anion species eliminates 5′-thioadenosine (5′-tA) to afford a substrate radical intermediate containing a C4 exocyclic methylene group. The addition of an electron and a proton to this species affords the methylated product. More recently, the Zhang laboratory also showed that NosJ-MIA also serves as a substrate for NosN, which it converts to NosJ-DMIA.6
In our studies herein, we also show that MIA-SNAC is a substrate for NosN; however, in contrast to the observations reported by the Zhang laboratory, we observe SAH as a coproduct rather than 5′-tA. Moreover, when we include MTA in reactions containing [methyl-13C]-SAM, less than 4% of the DMIA product contains an added C1 unit at natural abundance. The C1 unit that is transferred derives almost exclusively from [methyl-13C]-SAM. Moreover, product curves show an initial burst of SAH production followed by a slower steady-state phase of SAH production. Importantly, we observe the production of a species that we believe is an intermediate in the reaction (MMIA-SNAC), and which we believe contains an electrophilic C4 methylene group. This determination is based on the shift in its UV-vis spectrum from that of MIA and DMIA, its mass, its elution profile, and the observation that it is readily converted to 4-hydroxymethyl-3-MIA-SNAC upon treatment with base. MMIA-SNAC is slowly converted to DMIA-SNAC in a reaction that we believe is off pathway. Also, in contrast to observations by the Zhang laboratory, NosJ-MIA is not a substrate for NosN in our hands. Although we observe the production of 5′-dA, we do not observe production of SAH, MTA or NosJ-DMIA. Our observation of SAH rather than 5′-tA formation is consistent with recent studies on ChuW, which also resides in the HemN RS subfamily. This enzyme is proposed to use a similar radical addition mechanism during the anaerobic degradation of heme, and also produces SAH rather than 5′-tA as a coproduct.15
Our working hypothesis for the NosN reaction is depicted in Figure 8. A 5′-dA•, obtained from the reductive cleavage of SAM, is used to abstract a hydrogen atom from the methyl moiety of another simultaneously bound molecule of SAM to give a methylene radical. The methylene radical adds to C4 of MIA that is connected in a thioester linkage to Cys8 of the core peptide that has already been matured to contain thiazole groups. The resulting delocalized substrate radical intermediate loses its C4 proton, which actuates the release of SAH to afford a substrate radical intermediate containing a C4 exocyclic methylene group. This species loses an electron back to the Fe-S cluster to give the electrophilic MMIA species, which subsequently traps Glu6 to complete formation of the side-ring system of NOS.
Figure 8.

Proposed mechanism for the NosN reaction.
To provide evidence for the mechanism in Figure 8, we synthesized an artificial substrate comprised of residues 42–46 of the core peptide, which includes Glu6 and Cys8 in a thioester linkage to MIA as well as the three thiazole rings that surround these two key amino acids. When we use this substrate in NosN reactions, we observe formation of a species by MS that fragments to give a C4 hydroxymethyl group on MIA. If we use a similar substrate that differs only by an alanine substitution for Glu6, the C4 hydroxymethyl-MIA species is not observed. Moreover, if we use a substrate containing a [γ-carboxy-18O]-glutamyl residue, we observe a C4 hydroxymethyl-MIA species that contains one atom of 18O.
In summary, NosN catalyzes the radical-mediated transfer of a C1 unit from SAM to C4 of MIA to generate SAH and a reactive exocyclic intermediate that is captured by Glu6 of the structural peptide to generate the ester linkage in the side-ring system of NOS. Its ability to act as a methylase derives from a slower, non-enzymatic conversion to DMIA in the absence of a good substrate. If a methyl moiety were to be appended to C4 of MIA to give DMIA, there are no remaining unannotated enzymes within the nos operon that could activate it for formation of the Glu6-DMIA ester linkage, implying that NosJ-MIA cannot be the true substrate for NosN.
EXPERIMENTAL PROCEDURES
Materials
All commercial materials were used as received unless otherwise noted. All DNA-modifying enzymes and reagents used were purchased from New England Biolabs. DNA isolation kits were purchased from Machery-Nagel. PfuTurbo DNA polymerase AD was purchased from Agilent Technologies Inc. Sequencing grade trypsin was purchased from Promega Corporation. Primers were purchased from Integrated DNA Technologies. Deoxynucleotides were purchased from Denville Scientific Corporation. The expression vectors pET-26b and pET-28a were purchased from EMD Millipore. N-(2-hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid) (HEPES) was purchased from Fisher Scientific. Imidazole was purchased from J. T. Baker Chemical Co. Potassium chloride and glycerol were purchased from EMD Chemicals. 2-Mercaptoethanol and phenylmethanesulfonyl fluoride (PMSF) were purchased from Sigma-Aldrich. Isopropyl β-D-1-thiogalactopyranoside (IPTG) and dithiothreitol (DTT) were purchased from Gold Biotechnology. ATP (disodium salt) and coenzyme A (trilithium salt) were purchased from Calbiochem. 3-Methyl-2-indolic acid (MIA) was purchased from Matrix Scientific. Talon metal affinity resin was acquired from Clontech Laboratories Inc. Claritas PPT single element Fe (1000 mg/L in 2% HNO3), used to prepare iron standards for quantitative iron analysis, was obtained from SPEX CertiPrep (Metuchen, NJ). Tetrahydrofuran and dichloromethane were obtained from a JC Meyer solvent dispensing system and used without further purification. Silia Flash 60® silica gel (230–400 mesh) for flash chromatography was obtained from Silicycle Inc. All commercially available amino acid building blocks were purchased from Chem-Impex International, Inc. All other chemicals and materials were of the highest grade available and were from Sigma-Aldrich.
General Methods
Size-exclusion chromatography was conducted using a HiPrep 16/60 Sephadex S-200 column connected to an ÅKTA fast pro-tein liquid chromatography (FPLC) system in a Coy anaerobic chamber (Grass Lake, MI). UV-visible spectra were recorded on a Cary 50 spectrometer from Varian (now Agilent Technologies) using the WinUV software package to control the instrument. High performance liquid chromatography (HPLC) with detection by tandem mass spectrometry (LC-MS/MS) was conducted on an Agilent Technologies (Santa Clara, CA) 1200 system coupled to an Agilent Technologies 6410 QQQ mass spectrometer. The system was operated with the associated MassHunter software package, which was also used for data collection and analysis. High-resolution ESI mass spectra were acquired in on a Waters LCT Premier instrument at the Penn State Huck Institute of Life Sciences proteomics and mass spectrometry core facility. DNA sequencing was conducted at the Huck Institutes of the Life Sciences genomics core facility. NMR spectra were recorded on a Bruker AV-3-HD-500 instrument and calibrated using residual solvent peaks as an internal reference. Multiplicities are recorded as: s = singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet, q = quartet, dt = doublet of triplets.
Overproduction and purification of NosN
The gene encoding NosN was codon-optimized for expression in E. coli (sequence below) and synthesized by GeneArt (Thermofisher Scientific). It was supplied in plasmid pMA flanked between an NdeI site on its 5′-terminus and an EcoRI site on its 3′-terminus. The nosN gene was excised from the pMA plasmid by digestion with NdeI and EcoRI and then ligated into pET-28a that had been digested with the same restriction enzymes and subsequently purified by gel-electrophoresis. This cloning strategy affords a NosN protein that contains an N-terminal hexahistidine (His6)-tag that can be exploited for protein purification by immobilized metal affinity chromatography. After transformation of the ligation mixture into E. coli DH5α, DNA was purified from several of the colonies and subjected to sequencing. A plasmid containing the correct sequence (pET28a-nosN) was used to transform E. coli BL21(DE3) for expression of the nosN gene under an inducible T7 promoter.
E. coli Bl-21 (DE3) containing plasmid pDB1282 was transformed with the pET28a-nosN. An LB media starter culture was inoculated with a single colony and incubated overnight at 37 °C with shaking at 250 rpm. Several shake flasks of LB media were then inoculated with the overnight starter culture to initiate growth (180 rpm at 37 °C). Expression of the genes encoded on plasmid pDB1282 was induced at an OD600 of ~0.3 with 0.2% arabinose. Expression of the nosN gene was subsequently induced with 0.5 mM IPTG at an OD600 of ~0.6, and the flasks were immediately placed in an ice bath for 1 h. Once cooled, the cultures were incubated overnight for 18 h at 18 °C with shaking at 180 rpm. The cells were harvested (~30 g), flash-frozen in liquid nitrogen, and stored at −80 °C.
For purification, 30 g of frozen cell paste was resuspended in 150 mL of lysis buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 10% (v/v) glycerol, and 10 mM BME) containing PMSF (1 mM), DNase1 (100 μg mL−1), and lysozyme (1 mg mL−1). Resuspended cells were incubated on ice and subjected to six sonic bursts (40% output) on a QSonica instrument (housed in a Coy anaerobic chamber) for 45 s each with 8-min intermittent pauses. The lysate was then centrifuged for 1 h at 50,000 × g at 4 °C. The resulting supernatant was loaded onto Talon Co2+ resin equilibrated in the lysis buffer. The resin was washed twice with 100 mL of the lysis buffer prior to elution of His-tagged NosN with 50 mL of elution buffer (50 mM HEPES, pH 7.5, 30 mMKCl, 250 mM imidazole, 10% (v/v) glycerol, and 10 mM BME). The pooled eluate was concentrated by ultracentrifugation using an Amicon Ultra centrifugal filter unit with a 10-kDa molecular weight cutoff membrane.
Amino acid analysis of NosN
A correction factor for the Bradford protein assay of 0.55 was obtained for NosN by amino acid analysis as previously de-scribed.12 As-isolated NosN was reconstituted with iron and sulfide according to previously established procedures.12 The stoichiometry of iron and sulfide per as-isolated and reconstituted NosN polypeptide was determined by the methods of Beinert—as described previously12—in concert with the correction factor for the Bradford protein assay.
NosN enzyme assays and quantification of products by LC-MS/MS
All assays were conducted in a Coy anaerobic chamber maintained under 95% N2 and 5% H2. The oxygen concentration was maintained below 1 ppm via the use of palladium catalysts. Typical assays were conducted in a volume of 200 μL, and contained 50 mM HEPES (pH 7.5), 200 mMKCl, 5% glycerol, 2 mM sodium dithionite, 50 μM NosN, 1 mM MIA-SNAC or peptide mimic, and 100 μM L-tryptophan as an internal standard. Reactions were initiated with 1 mM SAM, and at various times, 15 μL volumes were removed and added to 15 μL of either 100 mM H2SO4 or 100% MeOH (for MIA-SNAC-or peptide mimic-containing assays, respectively) to quench the reaction. Quenched samples were centrifuged at 16,000 × g for 15 min before pipetting the supernatant into a sample vial. Components of each quenched reaction were separated on a Zorbax extend-C18 RRHT column (4.6 mm × 5 mm, 1.8 μm particle size) equilibrated in 98% solvent A (0.1% formic acid, pH 2.4) and 2% solvent B (100% acetonitrile). For separation of reaction components in assays containing MIA-SNAC, a linear gradient of solvent B from 2% to 10% was applied from 0 to 0.5 min, which was followed by a linear gradient to 50% from 0.5 to 2.5 min. Solvent B was then increased linearly to 100% from 2.5 to 3.5 min and held constant from 3.5 to 4.5 min. Solvent B was then decreased linearly to 2% from 4.5 to 5 min and held constant for 2.5 minutes to re-equilibrate the column before subsequent sample injections. Products were detected using electrospray ionization in positive mode (ESI+) and quantified with a multiple reaction monitoring (MRM) method for L-tryptophan, SAH, 5′-dA, MTA, thioadenosine, MIA-SNAC, and MMIA-SNAC. For separation and quantification of molecules from the assay containing the peptide mimic, a linear gradient of solvent B from 2% to 10% was applied from 0 to 0.5 min, which was followed by a linear gradient to 40% from 0.5 to 2.5 min. Solvent B was increased linearly to 100% from 2.5 to 4.5 min and then held constant from 4.5 to 5.5 min. Solvent B was then decreased linearly to 2% from 5.5 to 6.0 min and held constant for 2.5 minutes to re-equilibrate the column before subsequent sample injections. Products were detected using ESI+ and quantified with an MRM method for L-tryptophan, SAH, 5′-dA, MTA, thioadenosine, MIA-t3NosM, and NosMSR, MIA-t3NosMglu→ala, and 18O-glutamate-NosMSR. Parameters for the MRM method(s) are in Table S1.
Derivatization of MMIA was performed by quenching assay timepoints with 2 M NaOH and incubating at 37 ºC overnight. The base-hydrolyzed mixture was neutralized with a molar equivalent of HCl and a volume equivalent of 40 mM ammonium acetate. Components of the quenched reaction were separated on a Zorbax extend-C18 RRHT column (4.6 mm × 5 mm, 1.8 μm particle size) equilibrated in 80% solvent A (40 mM ammonium acetate, pH 6.0 and 5 % MeOH) and 20% solvent B (100% MeOH). For separation of reaction components, a linear gradient of solvent B from 20% to 50% was applied from 1 to 4.5 min, which was followed by a linear gradient to 100% from 4.5 to 6.5 min. Solvent B was held constant from 6.5 to 7.5 min before being decreased linearly to 20% from 7.5 to 8.0 min and held constant for 2.5 minutes to re-equilibrate the column before subsequent sample injections. Products were detected using electrospray ionization in negative mode (ESI−) using an MRM method detecting unlabeled and 13C-labeled 4-hydroxymethyl-3-methyl-2-indolic acid, respectively.
Progress curves for SAH and 5′-dA formation from MIA-SNAC experiments were fit to Frieden’s equation using residual analysis (Eq. 1):
| (Eq. 1) |
where t is time, vi is the steady initial velocity, vss is the steady state velocity, and kobs is the rate of change between vi and vss.
Progress curves resulting from the assay with MIA-t3NosM were fit to a single exponential (Eq. 2):
| (Eq. 2) |
where A is the amplitude, k is the rate constant, t is time, and yo is the initial concentration of 5′-dA or SAH
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH GM-122595, AI-133318 and AI-111419 (S.J.B.). SJB is an investigator of the Howard Hughes Medical Institute.
The authors thank the Huck Institute for Life Sciences Genomics Core for DNA sequencing and the Proteomics and Mass Spectrometry Core for small-molecule mass analysis.
ABBREVIATIONS
- 4OHMMIA
hydroxymethyl-3-methyl-2-indolic acid
- 5′-dA
5′-deoxyadenosine
- aa
amino acids
- DMIA
3,4-dimethyl-2-indolic acid
- MIA
3-methyl-2-indolic acid
- MRM
multiple reaction monitoring
- MS
mass spectrometry or mass spectrum
- MTA
methyl-thioadenosine
- NOS
nosiheptide
- RS
radical SAM
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SNAC
N-acetylcysteamine
- SIM
single-ion monitoring
Footnotes
Author Contributions
This manuscript was written through contributions of all authors, each of whom has given approval to the final version.
The Supporting Information is available free of charge on the ACS Publications website.
Optimized gene sequence for nosN, synthetic procedures, Table S1, and Figures S1–S9 (PDF).
References
- 1.Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol. 2009;4(10):855–64. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haste NM, Thienphrapa W, Tran DN, Loesgen S, Sun P, Nam SJ, Jensen PR, Fenical W, Sakoulas G, Nizet V, Hensler ME. Activity of the thiopeptide antibiotic nosiheptide against contemporary strains of methicillin-resistant Staphylococcus aureus. J Antibiot (Tokyo) 2012;65(12):593–8. doi: 10.1038/ja.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benazet F, Cartier M, Florent J, Godard C, Jung G, Lunel J, Mancy D, Pascal C, Renaut J, Tarridec P, Theilleux J, Tissier R, Dubost M, Ninet L. Nosiheptide, a sulfur-containing peptide antibiotic isolated from Streptomyces actuosus 40037. Experientia. 1980;36(4):414–6. doi: 10.1007/BF01975121. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Li Y, Chen D, Yu Y, Duan L, Shen B, Liu W. Radical-mediated enzymatic carbon chain fragmentation-recombination. Nat Chem Biol. 2011;7(3):154–60. doi: 10.1038/nchembio.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badding ED, Grove TL, Gadsby LK, LaMattina JW, Boal AK, Booker SJ. Rerouting the Pathway for the Biosynthesis of the Side Ring System of Nosiheptide: The Roles of NosI, NosJ, and NosK. J Am Chem Soc. 2017;139(16):5896–5905. doi: 10.1021/jacs.7b01497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding W, Ji W, Wu Y, Wu R, Liu WQ, Mo T, Zhao J, Ma X, Zhang W, Xu P, Deng Z, Tang B, Yu Y, Zhang Q. Biosynthesis of the nosiheptide indole side ring centers on a cryptic carrier protein NosJ. Nat Commun. 2017;8(1):437. doi: 10.1038/s41467-017-00439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Xue Y, Ma M, Wang S, Liu N, Chen Y. Multiple oxidative routes towards the maturation of nosiheptide. Chembiochem. 2013;14(13):1544–7. doi: 10.1002/cbic.201300427. [DOI] [PubMed] [Google Scholar]
- 8.Ding W, Li Y, Zhao J, Ji X, Mo T, Qianzhu H, Tu T, Deng Z, Yu Y, Chen F, Zhang Q. The Catalytic Mechanism of the Class C Radical S-Adenosylmethionine Methyltransferase NosN. Angew Chem Int Ed Engl. 2017;56(14):3857–3861. doi: 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- 9.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29(5):1097–106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Booker SJ, Grove TL. Mechanistic and functional versatility of radical SAM enzymes. F1000 Biol Rep. 2010;2:52. doi: 10.3410/B2-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo H, Wang J, Li YM, Yu Y, Zheng QF, Wu JQ, Liu W. Insight into bicyclic thiopeptide biosynthesis benefited from development of a uniform approach for molecular engineering and production improvement. Chem Sci. 2014;5(1):240–246. [Google Scholar]
- 12.Lanz ND, Grove TL, Gogonea CB, Lee KH, Krebs C, Booker SJ. RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol. 2012;516:125–52. doi: 10.1016/B978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
- 13.Franke J, Hertweck C. Biomimetic Thioesters as Probes for Enzymatic Assembly Lines: Synthesis, Applications, and Challenges. Cell Chem Biol. 2016;23(10):1179–1192. doi: 10.1016/j.chembiol.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Frieden C. Kinetic aspects of regulation of metabolic processes. The hysteretic enzyme concept. J Biol Chem. 1970;245(21):5788–99. [PubMed] [Google Scholar]
- 15.LaMattina JW, Nix DB, Lanzilotta WN. Radical new paradigm for heme degradation in Escherichia coli O157:H7. Proc Natl Acad Sci U S A. 2016;113(43):12138–12143. doi: 10.1073/pnas.1603209113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.