

Abstract
In animals, the iron storage and detoxification protein, ferritin, is composed of two functionally and genetically distinct subunit types, H (heavy) and L (light), which co-assemble in various ratios with tissue specific distributions to form shell-like protein structures of 24 subunits within which a mineralized iron core is stored. The H-subunit possesses a ferroxidase center (FC) that catalyzes Fe(II) oxidation, whereas the L-subunit does not. To assess the role of the L-subunit in iron oxidation and core formation, two human recombinant heteropolymeric ferritins, designated H-rich and L-rich with ratios of ~20H:4L and ~22L:2H, respectively, were employed and compared to the human homopolymeric H-subunit ferritin (HuHF). These heteropolymeric ferritins have a composition similar to the composition of those found in hearts and brains (i.e., H-rich) and in livers and spleens (i.e., L-rich). As for HuHF, iron oxidation in H-rich ferritin was found to proceed with a 2:1 Fe(II):O2 stoichiometry at an iron level of 2 Fe(II) atoms/H-subunit with the generation of H2O2. The H2O2 reacted with additional Fe(II) in a 2:1 Fe(II):H2O2 ratio, thus avoiding the production of hydroxyl radical. A μ-1,2-peroxo-diFe(III) intermediate was observed at the FC of H-rich ferritin as for HuHF. Importantly, the H-rich protein regenerated full ferroxidase activity more rapidly than HuHF did and additionally formed larger iron cores, indicating dual roles for the L-subunit in facilitating iron turnover at the FC and in mineralization of the core. The L-rich ferritin, while also facilitating iron oxidation at the FC, additionally promoted oxidation at the mineral surface once the iron binding capacity of the FC was exceeded.
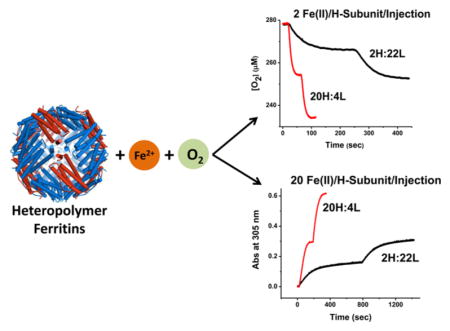
Graphical Abstract
Ferritins belong to a family of highly conserved supra-molecular protein nanostructures found in virtually all life forms. Canonical ferritins are composed of 24 subunits assembled in a shell-like structure designed to sequester thousands of iron atoms in their interior in the form of a biologically available ferric–oxy–hydroxide mineral core.1,2 By storing iron safely and reversibly, ferritins overcome the dual problem of poor iron bioavailability and toxicity and provide a source of iron for the synthesis of heme and iron-containing proteins within the cell. Thus, ferritins play an important role in the biological management of iron and maintenance of cellular iron homeostasis.
The ferritins of most organisms contain polypeptide subunits that can vary, selectively, in amino acid sequence and can be either enzymatically active (H-subunits or H-like subunits) or enzymatically inactive (L-subunits). The active H-subunit contains a ferroxidase center that catalyzes the oxidation of Fe(II) to Fe(III) by molecular oxygen followed by Fe(III) hydrolysis and ultimately formation of the mineral core.1–5 Amphibian ferritins are of two types: homopolymers of M-subunits and of H-subunits both with ferroxidase centers.6 Among prokaryotic ferritins are the homopolymeric bacterioferrritins (BFR’s), which have a heme cofactor, and the bacterial ferritins (Ftn’s), which do not; however, both do possess ferroxidase centers.7 Some ferritins are heteropolymeric proteins consisting of two types of subunits, one with a ferroxidase center and one without. Examples are the phytoferritins of plants,8 insect ferritins,9 and mammalian ferritins.2–5 Mammalian cytosolic ferritins are composed of two functionally and genetically distinct subunit types, H (heavy, ~21000 Da) and L (light, ~19000 Da).2,5 In human ferritins, the subject of the study presented here, the H-subunit has a dinuclear ferroxidase center in which the rapid pairwise conversion of Fe2+ to Fe3+ by molecular oxygen occurs. In contrast, the human L-subunit lacks such a center but, because of the high density of carboxyl groups on the inner surface of the protein cavity, appears to provide efficient sites for iron nucleation and mineralization.1,2 Ferritins with high L-content have fewer ferroxidase centers and thus have iron oxidation activity much slower than that of H-rich ferritins but form larger and more crystalline iron cores.5
The two types of subunits in human ferritins co-assemble in various ratios with a tissue specific distribution to form heteropolymeric 24-mer proteins. Ferritins containing up to 90% H-subunits have a low average iron content (≤1000 Fe atoms/protein shell) and are found in tissues exhibiting high ferroxidation activity (i.e., heart and brain), and those containing up to 90% L-subunits store relatively large amounts of iron (≥1500 Fe atoms/protein shell) and are found in tissues having mainly a storage function and much less iron oxidation activity (i.e., spleen, liver, and placenta).1–3,5–15 Exceptions are human mitochondrial ferritin, a homopolymer composed of 100% Mt-subunits similar to H-subunits,16 and human serum ferritin, a homopolymer composed of ~100% L-subunits.17 The H-subunit:L-subunit ratio in malignant tissues, including breast and pancreatic cancers, hepatocellular carcinoma, and Hodgkin’s lymphoma, changes during differentiation, and the high levels of ferritins detected in the sera of cancer patients have been shown to correlate with aggressive disease and poor clinical outcome.17,18
While the mechanism by which different heteropolymeric human isoferritins assemble in various tissues is unknown, it is important to note that such an assembly must be highly selective because the distribution of species (i.e., H24L0, …, H12L12, … H0L24) covering a significant proportion of the isoferritin spectrum is not observed in tissues. Interestingly, in vitro ferritin reconstitution shows a clear preference for the formation of heteropolymers over homopolymers with a remarkably narrow distribution, suggesting the presence of preferential interactions between H- and L-subunits.19 Arosio et al. recently developed a FRET-based approach to characterize human ferritin self-assembly and found that the self-assembly kinetics of H/L heteropolymers are faster than for H/H homopolymers and that heterodimeric H/L association is preferred during H/L heteropolymer formation.19 The specific recognition between H- and L-subunits is consistent with the fact that H- and L-homopolymeric ferritins are poorly populated in mammalian tissues.
The mechanisms of iron uptake, oxidation, and core formation in ferritins have been extensively studied over the past three decades.1–5 In most ferritins, two widely accepted models have been proposed, the protein catalysis model and the crystal growth model or a combination thereof.2,4 In mammalian ferritins, the overall and generally accepted multistep iron oxidation mechanism involves (a) the binding of ferrous ions at specific dinuclear catalytic sites and (b) the formation of a μ-1,2-peroxo-diFe(III) intermediate at these sites followed by (c) the appearance of a μ-oxo(hydroxo)-bridged diiron(III) complex and finally (d) the formation of small clusters that ultimately lead to large polynuclear aggregates and the mineral core itself. The growing mineral core then provides additional nucleation sites onto which incoming Fe(II) can be further oxidized and deposited.
Mechanistic studies of iron deposition in different types of ferritins have been extensively studied, but in human ferritins, they have been performed almost exclusively using recombinant human homopolymeric H-subunit (HuHF) or homopolymeric L-subunit (HuLF) proteins. The iron oxidation, deposition, and mobilization reactions as well as the detoxification properties of recombinant heteropolymer ferritins of various H-subunit:L-subunit ratios, which mimic naturally occurring ferritins in vivo, have been studied little. This lack of investigation seems particularly surprising given that, as already noted, most mammalian ferritins are heteropolymers. One of the impediments to progress in this area has been an inherent difficulty in cloning, expressing, and producing heteropolymer H/L ferritins. Additionally, earlier attempts to produce heteropolymer ferritins through the denaturation/renaturation procedures were tedious and yielded very small amounts of functional heteropolymer proteins not representative of those occurring naturally.5,10–13 Nonetheless, a few studies have successfully constructed heteropolymer ferritins having a specific H:L ratio of subunits using different cell expression systems.20–22 Native horse spleen ferritin (HoSF), which averages approximately 3–4 H-subunits and 20–21 L-subunits per ferritin molecule, is the most investigated natural isoferritin.23,24 Because of their widespread occurrence, heteropolymeric ferritins are presumed to store and release iron more efficiently than homopolymer ferritins, but additional studies are needed to understand more fully the complementary roles of H- and L-subunits in the biological processing of iron by mammalian ferritins.10,13,21
In this work, we have examined the iron oxidation and mineralization reactions in recombinant heteropolymeric human ferritin samples with compositions of ~20H:4L (termed H-rich ferritin) and ~22L:2H (termed L-rich ferritin) and compared the results with those of human H-subunit homopolymer ferritin (HuHF), human L-subunit homopolymer ferritin (HuLF), and horse spleen ferritin (HoSF). The composition of our H-rich ferritin is reflective of ferritins found in hearts and brains, whereas our L-rich ferritin is similar to those found in livers and spleens. These data on the stoichiometry and kinetics of iron oxidation show a synergism between H- and L-subunits. The presence of the L-subunit in the protein structure accelerates iron turnover at the H-subunit ferroxidase centers and assists with the building of the iron core. Hydrogen peroxide is produced at the ferroxidase site, a fraction of which reacts with the Fe(III)-containing protein and some of which may react with additional Fe(II) in a 2:1 Fe(II):H2O2 stoichiometry, thus avoiding Fenton chemistry. A peroxo-diFe(III) intermediate is formed at the ferroxidase centers of H-rich ferritin and has formation and decay kinetics similar to that reported for the H-subunit homopolymer, HuHF. Some iron oxidation occurs at the mineral surface of all ferritins but especially when the iron binding capacity of the ferroxidase centers is exceeded or when large numbers of L-subunits are present as in L-rich ferritin. At 10-fold higher fluxes of Fe(II) into the protein [i.e., 20 Fe(II) atoms/H-subunit vs 2 Fe(II) atoms/H-subunit], L-rich ferritin favors more iron oxidation at the mineral surface than does H-rich ferritin but displays slower overall kinetics because of its lower H-subunit content. However, the presence of the L-subunit allows the protein to sequester a larger mineral core, in accord with the storage role of human ferritins in tissues with a high iron content. The faster overall iron storage kinetics of human H-rich ferritin is in keeping with a role for this protein in minimizing iron-catalyzed production of reactive oxygen species in tissues of high metabolic activity.
MATERIALS AND METHODS
Plasmid Construction, Expression, and Purification of Recombinant Heteropolymer Ferritins
The human heteropolymers with a high H- or L-subunit content were produced in Escherichia coli by the bicistronic expression vectors as described in ref 25 and purified as expressed, without denaturation and renaturation steps. The polycistronic vector for the H-rich ferritin was constructed by subcloning the human L-ferritin cDNA downstream from the cDNA of human H-ferritin into the pET-Hwt vector. The cDNA for the Lwt was amplified by polymerase chain reaction (PCR) from plasmid pDS-Lwt, by inserting restriction site BamHI at the termini and the Shine-Delgarno sequence (AAGGAG) upstream of the ATG. The fragment was subcloned into pET-Hwt digested with BamHI (pET-H/Lwt). The bicistronic vector for the L-rich ferritin, pDSL/Hwt, was constructed by subcloning the H-ferritin cDNA downstream from the cDNA of L-ferritin into the pDS20pTrp-Lwt vector.26 The cDNA for the Hwt was amplified by PCR from plasmid pUD-Hwt,27 by inserting restriction site BamHI at the termini and the Shine-Delgarno sequence (AAGGAG) upstream of the ATG. The E. coli cells transformed with pET-H and pETH/Lwt were induced with isopropyl β-D-1-thiogalactopyranoside, as described in ref 25, and the E. coli cells transformed with pDSL and pDSL/Hwt in which the ferritins are under the tryptophan promoter were induced as described previously.26 The methods for denaturing and renaturing heteropolymer human ferritins are described elsewhere.19
Proteins and Chemicals
Holoferritins (iron containing ferritin) were rendered iron-free by dialysis against sodium hydrosulfite (dithionite), Na2S2O4, and complexation with 2,2′-bipyridyl at pH 6.0.24 Protein concentrations were determined using the Advanced Protein Assay (http://cytoskeleton.com) or spectrophotometrically using a molar absorptivity of 24000 cm−1 M−1 at 280 nm for the 24-mer apoprotein (iron-free protein, this work). All chemicals were reagent grade and used without further purification. Mops (3-(N-morpholino)-propanesulfonic acid) buffer was purchased from Research Organics (Cleveland, OH) and FeSO4·7H2O from J. T. Baker (Phillipsburg, NJ). Sodium dithionite, Na2S2O4, and 2,2′-bipyridyl were purchased from Sigma-Aldrich (St. Louis, MO). Fe(II) stock solutions were freshly prepared immediately before each experiment in a dilute HCl solution at pH 2.0. Freshly prepared hydrogen peroxide solutions were assayed either by electrode oximetry using catalase (EC 1.11.1.6, 65000 units/mg, Roche Molecular Biochemicals) by following the amount of O2 produced or by ultraviolet–visible (UV–vis) spectroscopy using a molar absorptivity of 43.6 M−1 cm−1 at 240 nm.24
Capillary Electrophoresis
A Beckman PACE model 5510 capillary electrophoresis unit was used to analyze and quantify the subunit composition of the heteropolymer ferritins. The operation of the instrument, the sodium dodecyl sulfate–capillary gel electrophoresis (SDS–CGE) sample preparation and running conditions, and the eCap SDS 14–200 kit used in this study are described in detail elsewhere.28 The PACE 5510 instrument current was set to reverse polarity with protein detection at 214 nm. The voltage setting for the 47 cm capillary was 14.1 kV (300 V/cm), and the separation time for the electropherograms was set to 30 min for the Beckman test mixture of protein and marker standards and the ferritin samples. Because absolute migration times slightly vary from run to run, the data were normalized by dividing the observed migration time of each peak in minutes by the migration time of the Orange G reference to give a relative migration time (RMT). A plot of the logarithm of the molecular weights of the protein standards versus the inverse of the RMT produces a straight line from which the apparent molecular weights of the L- and H-subunits were estimated. The Beckman test mixture with observed migration times consisted of the Orange G reference (12.24 min), α-lactalbumin (15.45 min, 14200 Da), carbonic anhydrase (17.52 min, 29000 Da), ovalbumin (18.97 min, 45000 Da), bovine serum albumin (20.73 min, 66000 Da), phosphorylase B (22.08 min, 97000 Da), β-galactosidase (23.32 min, 116000 Da), and myosin (27.96 min, 205000 Da). Peak areas of the separated ferritin subunits were determined using the Beckman PACE model 5510 CE software. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) slab gels of 15% acrylamide and native PAGE gels of 7.5% acrylamide were stained with Coomassie Blue and spectrophotometrically scanned to determine peak areas.
Oxygen Electrode
The oximetry experiments were performed with an OM-4 oxygen meter (Microelectrodes, Inc., Bedford, NH) equipped with an MI-730 micro-oxygen electrode. The electrode oximetry apparatus and standardization reactions have been described in detail elsewhere.29
UV–Vis Spectroscopy and Stopped-Flow Rapid Kinetics
Conventional UV–vis spectroscopy was performed on a Varian Cary 50 Bio spectrophotometer from Agilent Technologies. Rapid kinetic experiments were conducted with a pneumatic drive Hi-Tech SFA-20 M stopped-flow accessory on a J&M GmbH Tidas diode array spectrophotometer. The absorbance at 670 nm of the peroxo-diFe(III) complex formed after mixing the apoprotein with Fe(II) in the presence of oxygen was monitored in 2.5 ms increments following the first 10 ms of the reaction, the approximate dead time of the stopped-flow/diode array apparatus, including the responses of the hardware and Spectralys 2.00 software plus the 2.5 ms integration time for the first data point. For the stopped-flow experiment, equal 140 μL volumes of a pH 2.0 FeSO4 solution and a buffered apoferritin solution under a 100% O2 atmosphere were rapidly mixed in the quartz stopped-flow cuvette with a path length of 1 cm and a reaction volume of 80 μL. All concentrations given in the figure captions are final concentrations following mixing of the reagents. The spectrophotometer baseline was determined prior to each kinetic run using a cuvette containing equal volumes of apoferritin in buffer and pH 2.0 H2O. Experiments involving the oxidation of Fe(II) by H2O2 under anaerobic conditions were conducted with thoroughly deoxygenated solutions using a flow of moist high-purity grade argon gas (99.995%, <5 ppm O2) over the stirred solutions for a period of ~40–60 min. The spectrophotometric data were further analyzed with OriginLab version 7.5 (OriginLab Corp.). All kinetic experiments were repeated at least three times using two or more independent protein preparations. Kinetic traces represent averages of multiple individual runs.
RESULTS
Analysis of the Subunit Composition of Recombinant Heteropolymer Ferritins by SDS–CGE
SDS–CGE and SDS–PAGE were used to quantify the integrity and the H- and L-subunit composition of recombinant mammalian heteropolymer ferritins. Figure 1A shows the SDS–CGE electropherograms of two heteropolymer ferritin samples plus a mixture of protein standards containing the Orange G reference standard. The top trace shows a well-resolved electropherogram of an H-rich heteropolymer sample with migration times of 16.55 min (RMT of 1.352) and 17.41 min (RMT of 1.423) for the L-subunit and the H-subunit, respectively, from which apparent molecular weights of ~22600 and ~28400 Da, respectively, were estimated. High values of apparent molecular weights of ferritin subunits relative to the accepted values of ~19000 Da for L-subunits and ~21000 Da for H-subunits are typically obtained by SDS–CGE because of interactions of the protein with the detergent and/or with the gel matrix, causing lengthening of migration times.28 From the peak areas of the SDS–CGE trace (Figure 1A), the H-rich sample was found to contain 15% L-subunits and 85% H-subunits, a result in good agreement with the values of 16% L-subunits and 84% H-subunits determined by SDS–PAGE (Figure 1B). The composition of the H-rich ferritin was calculated to be 20.2H:3.8L, which we designate as ~20H:4L.
Figure 1.

Electrophoresis analysis of H-rich and L-rich ferritins. (A) SDS–CGE electropherograms of the Beckman test mixture and the recombinant apo-heteropolymers of human H-rich and L-rich ferritins. The H-rich and L-rich samples were 0.5 mg each and prepared as described elsewhere.25–27 The instrument conditions are specified in Materials and Methods. (B) Electrophoretic analysis of ferritin heteropolymers using 15% SDS–PAGE. The faint bands in the bottom portion of the gel are due to peptide degradation products commonly seen with ferritins. (C) A 7.5% nondenaturing PAGE gel demonstrating the homogeneity of the L-rich and H-rich ferritins and mobilities related to the H- and L-subunit contents. Arrows point to positions of the L- and H-subunit 24-mer homopolymers, designated HuLF and HuHF, respectively. MWM denotes molecular weight markers.
Similarly, the L-rich ferritin showed migration times of 16.72 min (RMT of 1.366) and 17.27 min (RMT of 1.411) for the Land H-subunits, respectively (Figure 1A, middle trace), with a composition of 95% L-subunit and 5% H-subunit comparable to the values of 93% L-subunit and 7% H-subunit from SDS–PAGE (Figure 1B). A composition for the L-rich ferritin of 22.3L:1.7H was obtained, which we designate as ~22L:2H. Nondenaturing PAGE analysis showed that the mobility of the cell-derived ferritins increased with H-subunit content, as expected, and that the bands of the H/L heteropolymers were as sharp as those of the H- and L-homopolymers, indicating a low degree of heterogeneity of these samples (Figure 1C). The slow-moving band present in the four samples of Figure 1C represents ferritin oligomers that are present in all ferritin preparations.
Iron(II) Oxidation and Binding Stoichiometry in the Recombinant H-Rich Heteropolymer
To establish the stoichiometry of binding of Fe3+ to the protein shell, a UV–vis spectrometric titration was performed. Fe(II) was added aerobically in increments of 8 Fe atoms/shell, 2 min apart, to the apo-H-rich ferritin (Figure 2). A discontinuity in absorbance at ~41 Fe(II) atoms/shell was observed corresponding to 2 Fe3+ atoms at each of the ~20 H-subunits, a further verification of the subunit composition of the H-rich protein. The molar absorptivity of the ferroxidase center Fe(III) species was 3900 ± 200 M−1 cm−1 per iron at 295 nm, a value characteristic of oxo/hydroxo-Fe(III) species previously reported for other ferritin types.30–32
Figure 2.

Aerobic spectrophotometric titration of apo H-rich ferritin with Fe(II). Conditions: 2 μM apoprotein in 50 mM Mops and 50 mM NaCl at pH 7.0 and 25 °C, 8 mM stock Fe(II) solution at pH 2.0. Each data point corresponds to the injection of 8 Fe(II) atoms/protein shell [2 μL of Fe(II) added to the 1.0 mL UV cell].
Stoichiometry of Oxygen Consumption in H-Rich Ferritin
The oxygen uptake curves for seven consecutive additions of 40 Fe(II) atoms/protein [i.e., or ~2 Fe(II) atoms/H-subunit] to H-rich ferritin and the corresponding Fe(II):O2 stoichiometric ratios are shown in Figure 3. The first injection of 40 Fe(II) atoms/protein shell [i.e., 2 Fe(II) atoms/H-subunit], produced an Fe(II):O2 stoichiometry of 1.97 ± 0.16, a stoichiometry less than 4 Fe(II):O2, indicating incomplete reduction of dioxygen to water and suggesting the production of hydrogen peroxide. The Fe(II):O2 ratio increased when more Fe(II) was added, reaching a ratio of approximately 2.7 Fe(II) atoms/O2 at a total of 280 Fe(II) atoms added (Figure 3A). When 10-fold larger increments of Fe(II) were added [i.e., 420 Fe(II) atoms/shell per injection or ~20 Fe atoms/H-subunit per injection], an Fe(II):O2 stoichiometry of 2.59 ± 0.21 was observed for the first increment, increasing to ~3.1 for a total of 1680 Fe(II) atoms added (Figure 3B). Similar behavior has been previously reported for recombinant H-subunit homopolymers (HuHF) and other ferritins4,30,33–35 and attributed to two main pathways, the ferroxidase center reaction [i.e., 2 Fe(II) atoms/O2] and the increasing involvement of the mineral surface reaction [i.e., 4 Fe(II) atoms/O2]. Average Fe(II):O2 oxidation stoichiometries increased from ~2 to ~3.1 for H-rich ferritin when Fe(II) was added in increments of 40 and 420 Fe(II) atoms/shell (Figure 3C).
Figure 3.

Oxygen consumption curves vs time for H-rich ferritin following (A) seven injections of 40 Fe(II) atoms/shell and (B) four injections of 420 Fe(II) atoms/shell. (C) Fe(II):O2 stoichiometry vs Fe(II)/shell for 40 Fe(II) atoms/shell per injection (black squares) and 420 Fe(II) atoms/shell per injection (red circles). Conditions: 1.2 μM heteropolymer H-rich ferritin and 48 μM Fe(II) per injection [for the injections of 40 Fe(II) atoms/shell] and 0.12 μM H-rich ferritin and 50.4 μM Fe(II) per injection [for the injections of 420 Fe(II) atoms/shell], 50 mM Mops, 50 mM NaCl, pH 7.05, and 25.0 °C.
The relevant ferroxidase center and mineral surface oxidation reactions are given by the following equations:24,29,31
| (1) |
| (2) |
The H2O2 produced by the ferroxidase reaction can react with further iron via the detoxification reaction.31
| (3) |
The detoxification reaction gains importance when iron is added over the binding capacity of the ferroxidase centers of 2 Fe(II) atoms/H-subunit31 and can contribute to the increase in the measured Fe(II):O2 stoichiometry. We note that the sum of reactions 1 and 3 is equivalent to reaction 2, and therefore, the combination of these two reactions (eqs 1 and 3) taking place cannot be distinguished from the mineral surface reaction itself (eq 2). Nevertheless, the measured Fe(II):O2 stoichiometry provides some insight into the relative importance of the reactions presented above in iron oxidation.
Under the assumption that the ferroxidase and mineral surface reactions are the only reactions taking place, the measured Fe(II):O2 stoichiometry is related to the fraction of iron oxidized at the ferroxidase center, XFe, by the equation Fe(II)/O2 = 4/(XFe + 1) from which XFe can be calculated, namely, XFe = 4/[Fe(II)/O2] – 1.31 Values of XFe for the ferroxidation reaction calculated from experimental Fe(II):O2 stoichiometries should be considered lower limits to the true values because of possible contribution from the detoxification reaction (eq 3), as small as it may be, to the measured stoichiometry. [Note that an Fe(II):O2 stoichiometry of 3, which is midway between the stoichiometries of 2 and 4 for eqs 1 and 2, gives an XFe value of 0.333. This value corresponds to ⅓ of the iron being oxidized by the ferroxidase reaction and ⅔ by the mineral surface reaction, the factor of 2 being due to the difference in the stoichiometric coefficients for Fe in eqs 1 and 2.]
With the information mentioned above in mind, the Fe(II):O2 stoichiometry of 3.1 at 1680 Fe atoms added to H-rich ferritin (Figure 3C) corresponds to an XFe of 0.29 or only 29% of the iron being oxidized through the ferroxidase center reaction, a value down from 100% for the first addition of 2 Fe(II) atoms/H-subunit to the protein in which an Fe(II):O2 stoichiometry of ≈2 was observed (Figure 3C). We note that no precipitation was observed up to 2500 Fe(II) atoms/shell added, a result indicating that all of the added iron is sequestered by the H-rich protein. Precipitation occurs only beyond 3000 Fe(II) atoms added (vide infra).
H2O2 Production in H-Rich and L-Rich Ferritins
Catalase was used in conjunction with electrode oximetry to confirm the production of H2O2 in both H-rich and L-rich ferritin solutions. Addition of catalase to H-rich ferritin following ferroxidation of 40 Fe(II) atoms/shell (2 Fe atoms/H-subunit) caused the evolution of 7.5 ± 1.5 μM oxygen (Figure 4A), a clear indication of the presence of H2O2 in the solution. The observed Fe(II):O2 oxidation stoichiometry of 2.05 ± 0.12 indicates that virtually all the Fe(II) has been processed by the ferroxidase center reaction to produce H2O2. However, the amount of O2 generated was only 62% of the 12 μM expected from the catalase-facilitated disproportionation of the H2O2, namely, H2O2 → H2O + ½O2. Thus, 38% of the hydrogen peroxide produced by the ferroxidase reaction had been consumed by unknown reaction(s) (vide infra). Moreover, because virtually all the iron was oxidized via the ferroxidase reaction, the detoxification reaction (eq 3) could not be significantly involved in the consumption of the H2O2.
Figure 4.

Oxygen consumption curves vs time following four consecutive injections of (A) 40 Fe(II) atoms per H-rich protein shell, (B) 420 Fe(II) atoms per H-rich protein, and (C) 3.6 Fe(II) atoms per L-rich protein. The Fe(II):O2 stoichiometry is indicated next to each O2 consumption curve following Fe(II)/protein additions. Catalase (1 μL) was added at the end of the first oxidation reaction as indicated on the graph. Conditions: (A) 1.2 μM H-rich ferritin and 48 μM Fe(II) per addition [or ~2 Fe(II) atoms/H-subunit], (B) 0.12 μM H-rich protein and 50.4 μM Fe(II) [or ~20.5 Fe(II) atoms/H-subunit], and (C) 3.5 μM L-rich ferritin and 12.6 μM Fe(II) per addition [or ~2 Fe(II) atoms/H-subunit], 50 mM Mops, 50 mM NaCl, pH 7.05, and 25.0 °C.
Upon further addition of 40 Fe(II) atoms/shell to the same H-rich protein sample containing catalase (additions 3 and 4), an Fe(II):O2 oxidation stoichiometry of 3.72 ± 0.18 was obtained (Figure 4A), which is higher than the Fe(II):O2 stoichiometry of ~2.4 in the absence of catalase (Figure 3A), a clear indication that H2O2 is also produced during subsequent additions of Fe(II) to the protein as previously reported for HuHF.31 The fact that an Fe(II):O2 stoichiometry of 4:1 was not achieved in the presence of catalase again suggests that some of the H2O2 produced has been consumed by other reactions. [If all the H2O2 produced at the ferroxidase center had undergone catalyzed disproportionation to O2 and H2O, the apparent Fe(II):O2 stoichiometry would have been 4.0.]
Similar observations were recorded when the experiment was performed with four successive additions of 420 Fe(II) atoms/shell [i.e., 20.5 Fe(II) atoms/H-subunit] to H-rich ferritin (Figure 4B), again showing evolution of O2 upon addition of catalase following the first addition of Fe(II) and demonstrating the production of H2O2 at these higher fluxes of iron into the protein. From an Fe(II):O2 stoichiometry of 2.42 ± 0.18, an XFe value of 0.65 was calculated. However, only 6.0 ± 1.0 μM O2 was produced or 73% of the expected value of 8.2 μM O2 from the catalase-facilitated disproportionation of the H2O2, once again pointing to other H2O2-consuming reaction(s) occurring. In the presence of catalase, the Fe(II):O2 stoichiometry progressively increased from 3.0 to 3.6, still short of the limit of 4.0, a further indication that other reactions consume much of the hydrogen peroxide produced at the ferroxidase center. These reactions largely involve the protein,24 but the detoxification reaction (eq 3) might also contribute.31
A similar experiment was performed with L-rich ferritin containing 1.8 H-subunits per 24-mer to which 2 Fe(II) atoms/H-subunit were added followed by the addition of catalase (Figure 4C). The observed Fe(II):O2 oxidation stoichiometry of 2.95 ± 0.15 (Figure 4C) corresponds to an XFe value of 0.36. The amount of O2 generated (0.62 ± 0.12 μM) with catalase was only ~38% of that expected from the ferroxidation reaction.
Taken together, the results from the experiments with H-rich and L-rich ferritins described above confirm the production of H2O2 in both proteins and indicate that undefined reactions involving H2O2 occur but more H2O2 is consumed in L-rich ferritin than in H-rich ferritin by these reaction(s) (Figure 4). Such phenomena have been previously observed with bullfrog H-ferritin36 and with HuHF and HoSF ferritins24,31 and studied in some detail in these latter proteins (see Discussion).
Fe(II) Oxidation by H2O2 in H-Rich and L-Rich Ferritins
Because hydrogen peroxide is produced in both H-rich or L-rich ferritins, these proteins were examined for their ability to prevent the formation of significant amounts of hydroxyl radicals via Fenton chemistry, i.e., Fe2+ + H2O2 → Fe3+ + HO− + HO•. Anaerobic titrations of H-rich ferritin containing 41 Fe(II) atoms/shell (ferroxidation) and L-rich ferritin containing 200 Fe(II) atoms/shell (mineral surface oxidation) with H2O2 in increments of 0.1 H2O2 molecule/Fe(II) were performed. In both instances, the spectrophotometric titrations showed a discontinuity in absorbance at ~0.5 H2O2 molecule/Fe(II), indicating that one H2O2 molecule oxidizes two Fe(II) atoms and that almost complete reduction of H2O2 to H2O occurs (Figure 5A). These results suggest that both H-rich and L-rich proteins attenuate the production of hydroxyl radicals produced by Fenton chemistry, in line with the previously established detoxification properties of other ferritins.4
Figure 5.

Reaction of hydrogen peroxide with iron(II) in H-rich and L-rich ferritins. (A) Spectrophotometric titration of 1.0 μM apo-H-rich ferritin containing 41 Fe(II) atoms/shell and 0.5 μM apo-L-rich ferritin containing 200 Fe(II) atoms/shell with H2O2 under anaerobic conditions. Each addition corresponds to 0.1 H2O2 molecule/Fe(II). The dashed curve is a fourth-order polynomial fit, and the straight lines are computed tangents. (B) Fe(II) oxidation kinetics in H-rich ferritin by O2 and H2O2 in the presence of 41 or 200 Fe(II) atoms/shell. (C) Fe(II) oxidation kinetics in L- rich ferritin by O2 and H2O2 in the presence of 41 Fe(II) atoms/shell. H2O2 was added in one shot to the Fe(II)-containing protein solution at a ratio of 0.5 H2O2 molecule/Fe(II). All protein solutions were prepared in 0.1 M Mops and 50 mM NaCl, pH 7.0 and 25.0 °C.
The rates of iron oxidation by hydrogen peroxide were measured and compared to those of dioxygen. Upon addition of small amounts of iron to H-rich ferritin [i.e., 2 Fe(II) atoms/H-subunit], the initial rate of Fe(II) oxidation by H2O2 was comparable to that by O2 (Figure 5B, green and blue curves), as also reported for HuHF at the same level of iron loading.31 The final absorbances were also the same (Figure 5B). However, upon addition of large amounts of iron to H-rich ferritin [i.e., 10 Fe(II) atoms/H-subunit or 200 Fe(II) atoms/shell], oxidation by H2O2 was ~3 times faster than by O2 and the final absorbance for the O2 sample was lower than that for the H2O2 sample (Figure 5C), implying some difference in the mineral cores produced.
In contrast to H-rich ferritin, iron oxidation by H2O2 in L-rich ferritin [41 Fe(II) atoms/shell, i.e., ~20 Fe(II) atoms/H-subunit] was ~120 times faster than by O2, i.e., 1.03 × 10−3 s−1 versus 8.6 × 10−4 s−1 (Figure 5C). Similar differences in rates for the two oxidants have been reported for HuLF.31,37 The kinetic differences between the two oxidants in L-rich ferritin were also reflected in a significant difference in the absorbance values of the resultant cores (Figure 5C). The molar absorptivity value of the μ-oxo/hydroxo-Fe(III) core produced by H2O2 was 2200 ± 100 M−1 cm−1 versus 3400 ± 200 M−1 cm−1 for that produced by O2 as the oxidant, indicating significantly different mineral structures.
We conclude that the molecular structure and/or composition of cores formed largely via the mineralization reaction as in L-rich ferritin and HuLF depends significantly on the oxidant, O2 versus H2O2, but less so for H-rich ferritin and HuHF where the ferroxidase center reaction dominates (Figure 5C).31,34 In this connection, Mössbauer spectra of HuHF cores prepared using O2 versus H2O2 as the oxidant are very similar, with the O2 derived cores being slightly more crystalline,38 a finding in keeping with the observations here with H-rich ferritin.
Conventional Kinetics of Fe(II) Oxidation Measured by UV–Vis Spectrophotometry
Fe(II) oxidation in ferritin has been traditionally followed by UV–vis absorption spectroscopy in the 300–330 nm spectral region that is attributed to the formation of μ-oxo/hydroxo-Fe(III) species. Figure 6A shows the increase in absorbance at 305 nm for six successive additions of 41 Fe(II) atoms/shell to the same H-rich protein sample [i.e., 2 Fe(II) atoms/H-subunit]. Except for the initial rate of oxidation following the first injection, i.e., (4.51 ± 0.36) × 10−3 s−1, the initial rates of iron oxidation for injections 2–6 were similar and slower with an average rate of (1.65 ± 0.17) × 10−3 s−1 (Table 1).
Figure 6.

Kinetic curves and initial rates of Fe(II) oxidation following multiple additions of Fe(II) to H-rich apoferritin. (A) Sequential addition of six injections of 41 Fe(II) atoms/shell followed by four additional injections 1 h later to the same protein sample. (B) Addition of 41 Fe(II) atoms/shell to different Fe(III)-preloaded H-rich samples that have sat for various amounts of time. (C) Plot of the initial rates of Fe(II) oxidation in H-rich ferritin following multiple additions of 100, 200, 300, and 400 Fe(II) atoms/shell to the same protein sample. Conditions: 0.2 μM protein, 0.1 M Mops, 50 mM NaCl, pH 7.4, and 25 °C.
Table 1.
Initial Rates (k) of Fe(II) Oxidation in H-Rich Ferritin following Different Additions of Fe(II) to the Same Protein Sample at the Indicated Timesa
| k for injection 1 (×10−3 s−1) | k for injection 2 (×10−3 s−1) | k for injection 3 (×10−3 s−1) | k for injection 4 (×10−3 s−1) | k for injection 5 (×10−3 s−1) | k for injection 6 (×10−3 s−1) | |
|---|---|---|---|---|---|---|
| apo-H-rich ferritin with 41 Fe(II) atoms/shell | 4.51 ± 0.36 | 1.45 ± 0.12 | 1.54 ± 0.12 | 1.60 ± 0.13 | 1.76 ± 0.14 | 1.89 ± 0.15 |
| 1 h-aged holo-H-rich ferritin preloaded with 246 Fe(III) atoms/shell and 41 Fe(II) atoms/shell | 4.42 ± 0.35 | 2.30 ± 0.18 | 1.99 ± 0.15 | 2.06 ± 0.16 | ||
| 5 h-aged holo-H-rich ferritin preloaded with 410 Fe(III) atoms/shell and 41 Fe(II) atoms/shell | 1.59 ± 0.13 | |||||
| 5 h-aged holo-H-rich ferritin preloaded with 2000 Fe(III) atoms/shell and 41 Fe(II) atoms/shell | 1.50 ± 0.12 |
Conditions: 0.2 μM protein, 0.1 M Mops, 50 mM NaCl, pH 7.4, and 25 °C.
Whether successive additions of 41 Fe(II) atoms/shell were made to the same apo-H-rich sample (Table 1, injections 2–6) or to aged H-rich samples preloaded with 410 or 2000 Fe(III) atoms/shell, similar iron oxidation rates were observed (Table 1), suggesting that iron oxidation occurs to a significant extent at the preformed mineral surface, a result in accord with the observed increase in the Fe(II):O2 oxidation stoichiometry (Figure 3). Interestingly, the H-rich protein regains its full ferroxidation activity of (4.42 ± 0.35) × 10−3 s−1 after 1 h for a core size of 246 Fe(III) atoms/shell (Figure 6B, red curve; Table 1), suggesting a complete clearance of iron from the ferroxidase centers within this time frame. A core size of <400 Fe(III) atoms/shell appears to be the cutoff for the H-rich protein to regain full ferroxidase activity upon standing (Figure 6B and Table 1). Notably, it has been reported for HuHF that an incipient core of 200 Fe(III) atoms/shell is the minimal core size beyond which oxidation of Fe(II) appreciably occurs on the surface of the mineral core.31 The result for H-rich ferritin was supported by a series of Fe(II) oxidation kinetics whereby multiple additions of 100, 200, 300, and 400 Fe(II) atoms/shell were made to the same protein sample (Figure 6C). As seen in Figure 6C, a break around 400 Fe(III) atoms/shell for H-rich ferritin was observed regardless of the number of Fe(II) atoms per shell added per injection beyond which a constant rate of oxidation was observed, corresponding to the mineral surface reaction.
Rates of Fe(II) Oxidation by O2 in H-Rich, HuHF, and L-Rich Ferritins
To further assess the role of the L-subunit in iron oxidation and core formation, we compared the initial rates of Fe(II) oxidation by O2 and the Fe(II):O2 oxidation stoichiometries of three ferritin samples with different L-subunit contents, namely, H-rich ferritin, HuHF, and L-rich ferritin. The initial rates of Fe(II) oxidation from the 305 nm absorbance change with Fe(II) added in increments of 2 Fe(II) atoms/H-subunit or 20 Fe(II) atoms/H-subunit to apo-H-rich ferritin and apo-HuHF are plotted in Figure 7 against the total iron added. The presence of only ~4 L-subunits in H-rich ferritin had a significant effect on its kinetic properties relative to those of HuHF. Initial rates of oxidation in H-rich ferritin with added Fe(II) [i.e., 2 Fe(II) atoms/H-subunit per addition] decreased slightly from ~1.1 × 10−2 to ~0.9 × 10−2 s−1 (Figure 7A) with a corresponding increase in Fe(II):O2 stoichiometry from ~2:1 to ~2.4:1 (Figure 8A). For HuHF, in marked contrast to H-rich ferritin, a steady decrease in the initial rate from ~8 × 10−3 to ~4 × 10−3 s−1 was observed when increments of 2 Fe(II) atoms/H-subunit were added (Figure 7A). This loss of 50% activity in HuHF with added iron (Figure 7A) occurs with an increase in Fe(II):O2 stoichiometry from ~2:1 to ~3.4:129 and is reflective of the iron oxidation reaction shifting from the ferroxidase centers largely to the surface of the growing iron core.29 In contrast, the ferroxidase centers of H-rich ferritin remained largely involved in Fe(II) oxidation at all additions of iron albeit at a slightly lower rate. The increase in the Fe(II):O2 stoichiometry from ~2:1 to only ~2.4 (Figure 8A) reflects the onset of some mineral surface reaction (Figure 7A); i.e., the fraction of iron oxidized at the ferroxidase center, XFe, decreased from ~1.0 to ~0.7.
Figure 7.

Comparison of initial rates of Fe(II) oxidation in H-rich ferritin and HuHF for multiple additions of (A) 2 Fe(II) atoms/H-subunit and (B) 420 or 480 Fe(II) atoms/shell to the same protein sample. (C) Maximal iron loading capacity of H-rich ferritin and HuHF. All protein solutions are prepared in 0.1 M Mops and 50 mM NaCl at pH 7.0 and 25 °C. The numbers in parentheses after the accolades are the difference in initial rates between H-rich ferritin and HuHF.
Figure 8.

Comparison of oxygen consumption curves and iron oxidation kinetics for H-rich and L-rich ferritins following (A) three injections of 2 Fe(II) atoms/H-subunit, (B) six injections of 40 Fe(II) atoms/shell, and (C) two injections of 20 Fe(II) atoms/H-subunit. Conditions: (A) 1.2 μM H-rich ferritin and 7.2 μM L-rich ferritin, (B) 1.2 μM H-rich ferritin and 1.2 μM L-rich ferritin, and (C) 0.2 μM H-rich ferritin and 1.0 μM L-rich ferritin, with 50–100 mM Mops, 50 mM NaCl, pH 7.05, and 25.0 °C. The Fe(II):O2 ratios and initial rates of Fe(II) oxidation by O2 are recorded by each curve.
The situation changed when iron was added in 10-fold larger increments of 20 Fe(II) atoms/H-subunit to both H-rich and HuHF proteins (Figure 7B). As reported in Figure 7B, the initial rate for H-rich ferritin initially was 5 times higher than that of HuHF but dropped to 2 times as the core developed and oxidation shifted toward the mineral surface at larger fluxes of iron into both proteins. Taken together, the data shown in panels A and B of Figure 7 imply a role for L-subunits in facilitating iron oxidation at the H-subunit ferroxidase centers by increasing the rate of iron turnover at the centers and its concomitant movement to the mineral core. Notably, the kinetics of iron oxidation are faster in H-rich ferritin than in HuHF, and the protein holds a larger amount of iron; the maximal iron loading capacity of H-rich ferritin is ~3000 Fe(III) atoms/shell compared to ~2000 Fe(III) atoms/shell for HuHF, at which point precipitation occurs (Figure 7C).
Kinetic oxygen uptake experiments were also conducted to compare the properties of L-rich ferritin (~22L:2H) with those of H-rich ferritin (~20H:4L). Repeated aerobic additions of only 2 Fe(II) atoms/H-subunit to L-rich ferritin consistently produced an Fe(II):O2 stoichiometry of ~2:1 (Figure 8A, top curve), indicative of iron oxidation solely at the ferroxidase centers. In contrast, the same experiment performed with H-rich ferritin produced kinetics faster than those of the L-rich protein but an increase in Fe(II):O2 stoichiometry from ~2:1 to ~2.4:1 (Figure 8A, bottom curve), indicative of ~33% of the iron being oxidized at the mineral surface (XFe = 0.67). Upon six additions of 40 Fe(II) atoms/shell to L-rich ferritin [i.e., 10-fold higher, 22 Fe(II) atoms/H-subunit] (Figure 8A), the Fe(II):O2 stoichiometry increased from ~3.0 and ~3.5 (Figure 8B, top curve), whereas in H-rich ferritin at the same 40 Fe(II) atoms/shell per addition [i.e., 2 Fe(II) atoms/H-subunit], it varied from ~2 to ~2.7 (Figure 8B, bottom curve). Thus, at a high iron flux, the few H-subunits present in L-rich ferritin cannot sustain the oxidation of Fe(II) at the ferroxidase sites and the protein favors iron oxidation on the surface of a growing mineral core (Figure 8B, top curve), the Fe(II):O2 stoichiometry of ≈3.5 reflecting mostly mineral surface reaction (XFe = 0.14). These results corroborate our findings in panels A and B of Figure 7 and suggest that at a low iron flux of 2 Fe(II) atoms/H-subunit, iron oxidation occurs at the dinuclear iron sites of the H-subunits in the L-rich heteropolymer and that the few L-subunits present in the H-rich protein shell modulate H-subunit activity and facilitate iron turnover at the ferroxidase centers.
Whether 2 or 22 Fe(II) atoms/H-subunit are added, the oxidation kinetics in L-rich ferritin were 4–6 times slower than in H-rich ferritin as determined by oximetry measurements, (Figure 8A, B). Similarly, when 20 Fe(II) atoms/H-subunit were added at once to either L-rich or H-rich ferritin, the iron oxidation kinetics measured spectrophotometrically at 305 nm were also approximately 4–6 times slower for L-rich than for H-rich ferritin (Figure 8C). A 10-fold difference in oxidation kinetics of H-rich compared to L-rich ferritin is expected solely on the basis of their 10-fold difference in subunit content, 20 H-subunits in H-rich versus 2 H-subunits in L-rich ferritin. The fact that the H-rich ferritin exhibits, on average, kinetics only 5 times faster than those of L-rich ferritin when a stoichiometric amount of Fe(II) is added [i.e., 2 Fe(II) atoms/H-subunit] is further evidence that the presence of L-subunits also enhances the inherent rate of oxidation at the ferroxidase centers as well as turnover of Fe(III) at the centers upon addition of more Fe(II) to the protein.
Formation of a Peroxo-DiFe(III) Complex in H-Rich Ferritin
A μ-peroxo-diFe(III) intermediate is the first detectable iron-protein species (λmax ~ 670 nm) observed in some ferritins.4,23,32,39,40 Stopped-flow experiments were conducted to examine whether such an intermediate is also formed in heteropolymer H-rich ferritins. Figure 9 illustrates the family of stopped-flow UV–vis spectra for a sample with 40 Fe(II) atoms/shell [i.e., 2 Fe(II) atoms/H-subunit]. The top left inset of Figure 9A shows the series of spectra for the first 40 ms of the reaction corresponding to the formation of the peroxo intermediate with a λmax of ~670 nm. The top right inset illustrates the series of spectra for the decay of the intermediate in the time interval of 40 ms to 19 s. Because the decay of the peroxo intermediate does not show a clean isosbestic point, the kinetic trace at 670 nm (Figure 9B) must involve more than one product and was thus fitted to the following sequential kinetic model as previously done with HuHF:32
Figure 9.

Stopped-flow kinetics of H-rich ferritin. (A) Family of stopped-flow UV–vis spectra for a H-rich ferritin sample with 40 Fe(II) atoms/shell where the increase in the absorbance of the peroxo complex is colored red and its decay black. (B) Absorbance–time curve at 670 nm from the data in panel A. Experimental (black) and simulated (red) curves are shown. The rate constants from the simulation are as follows: k1 = 92.38 ± 5.78 s−1, k2 = 3.17 ± 0.55 s−1, and k3 = 0.24 ± 0.02 s−1. Final experimental conditions after mixing in the stopped-flow apparatus: 2.5 μM H-rich apoferritin saturated with 100% O2, 100 μM FeSO4 (pH 2.0), 50 mM Mops, 25 mM NaCl, pH 7.0, and 25.0 °C.
| (4) |
In this reaction scheme, the diFe(II)-protein complex (A) rapidly reacts with O2 at the ferroxidase center of the H-subunit with a rate constant k1 to form a blue μ-peroxo-diFe(III) complex (B) that then decays via a rate constant k2 to form a related species (B′) (i.e., a hydroperoxo complex) before it decays to a μ-oxo/hydroxo diiron(III) species (C). The excellent fit to the data in Figure 9 yielded the following values for the rate constants: k1 = 92.4 ± 5.8 s−1, k2 = 3.17 ± 0.55 s−1, and k3 = 0.24 ± 0.02 s−1. The maximal absorbance of the μ-peroxo-diFe(III) complex (species B) corresponds to a molar absorptivity (ε670) of 400 ± 75 M−1 cm−1/Fe(III) dimer, a value similar to those reported for peroxo species in other ferritins.16,23,32
DISCUSSION
Under proper physiological conditions, the H- and L-subunits of mammalian ferritins assemble into 24-mer heteropolymer cagelike nanostructures of variable subunit composition.13,41–45 Given the tissue-dependent composition of mammalian isoferritins, a preferred association between H- and L-subunits to form heteropolymer ferritins is expected and has been demonstrated here (Figure 1C) and in a recent investigation by Arosio and co-workers.19 We note that, despite a preferred association between H- and L-subunits, 100% homogeneity in subunit composition among ferritin molecules for a specific sample of known H/L content is unlikely. Heteropolymer ferritins probably manifest a narrow distribution of hybrid molecules of various combinations of H- and L-subunits distributed about the protein shell in different positions relative to one another. These subspecies of ferritins cannot be resolved by today’s techniques (Figure 1C). Thus, although self-assembled with a cell, the recombinant human H-rich (~20H:4L) and L-rich (~2H:22L) ferritins used in this study likely represent a collection of molecular subspecies, the average properties of which are reported here. Their integrity and composition have been demonstrated by electrophoresis (Figure 1) and iron binding measurements (Figure 2).
Homopolymeric ferritins, including bacterial ferritins (H-type),46 phytoferritins (H-type),47 amphibian ferritins (H-type and M-type, a close variation of H-type),39,48 and human HuHF (H-type),1–5 all contain ferroxidase centers and deposit iron within their central cavities by processes involving the oxidation and hydrolysis of iron. However, the mechanisms differ in detail among various ferritins, although a common mechanism for all ferritins has been put forth49 but recently challenged.50,51 A ferroxidase center is not a requirement for in vitro core formation because the human L-subunit homopolymer, HuLF, is also capable forming a mineral core albeit at a slower rate.4,33,51 Heteropolymeric human ferritins, being composed of both types of subunits (i.e., H and L), are a special case. Here, we have explored two such heteropolymers, an H-rich ferritin consisting of ~20H:4L and an L-rich ferritin of ~2H:22L, representing opposite ends of the spectrum of subunit composition, and have studied the complementary functions of H- and L-subunits and compared the results with those of homopolymeric HuHF and HuLF ferritins.
The two reaction pathways, ferroxidase and the mineral surface, depend significantly on both the amount of iron presented to the protein and its L-subunit content (Figures 3 and 4). For example, the incremental addition of 2 Fe(II) atoms/H-subunit to the same H-rich ferritin sample up to 280 Fe(II) atoms/shell produces a steady increase in Fe(II):O2 stoichiometry from 1.97 ± 0.16 to 2.67 ± 0.22 (Figures 3A and 8B), corresponding to a decrease in the percentage of iron oxidized by the ferroxidase reaction from ~100 to 50% as the core develops and the mineral surface reaction becomes increasingly important. By comparison, incremental additions of 2 Fe(II) atoms/H-subunit to the L-rich ferritin shifted the Fe(II):O2 stoichiometry from ~2:1 (Figure 8A) to ~3.5:1 when up to 240 Fe(II) atoms/shell have been added, corresponding to a decrease in the percentage of iron oxidized by the ferroxidase reaction from ~100 to 14%, in support of the hypothesis that the higher L-subunit content of L-rich ferritin favors the mineral surface (Figure 8B). When larger increments of Fe(II) [i.e., 20 Fe(II) atoms/H-subunit] are added to the H-rich ferritin up to 1680 Fe(II) atoms/shell, the measured Fe(II):O2 stoichiometry ranges from 2.59 ± 0.21 to 3.10 ± 0.25 (Figure 3B), corresponding to a decrease in the percentage of iron processed by the ferroxidase reaction from 54 to 29%. The relatively large flux of Fe(II) into the protein exceeds the capacity of the ferroxidase center to process all the iron, and a significant portion is oxidized at the mineral surface. As the mineral core grows, more surface sites become available for Fe(II) binding and oxidation, causing the increases in the observed stoichiometry (Figure 3B, C).
The production of H2O2 in both H-rich and L-rich ferritins is expected from their H-subunit contents and eq 2 and confirmed by the catalase experiments (Figure 4). The amount of O2 produced by the addition of catalase is smaller than that expected from the H2O2 produced in both H-rich and L-rich ferritins, but even less O2 than expected is produced for L-rich ferritin (Results and Figure 4), implying that the L-subunit is more reactive toward H2O2. These findings are in keeping with previous studies29 demonstrating that H2O2 added to both HuHF and HoSF reacts with both proteins in the absence of Fe(II) but more so with HoSF and probably accounts for the abundance of peptide fragments previously observed with the latter protein by SDS–CGE.28 Reaction of H2O2 with the Mops buffer to form an N-oxide is unlikely to occur significantly in the experiments presented here as the rate is very slow.52
Importantly, Fenton chemistry does not appear to occur to a significant extent in either L-rich or H-rich ferritin as the observed H2O2:Fe(II) oxidation stoichiometry is ~0.5 for both proteins (Figure 5A) according to the detoxification reaction (eq 3). The detoxification reaction has also been observed in HuLF as well as HuHF, demonstrating that a ferroxidase center is not a requirement for this reaction. Despite the detoxification reaction, sensitive EPR spin-trapping experiments have detected the presence of very small amounts of hydroxyl radical produced during the oxidative deposition of iron in both HuHF53 and HoSF,54 i.e., corresponding to <1% of the Fe(II) oxidized.
The rates of oxidation of Fe(II) by H2O2 compared to the rates by O2 have been studied in several ferritins under different conditions. While the two rates are comparable at a stoichiometric amount [i.e., 2 Fe(II) atoms/H-subunit] in H-rich ferritin (Results and Figure 5B) and in HuHF and Azotobacter vinelandii bacterioferrin,37 the rate of oxidation by H2O2 in HuHF becomes ~100 times faster than that by O2 at an Fe(II):protein ratio of 500, i.e., ~20 Fe(II) atoms/H-subunit.31 In contrast to H-rich ferritin, L-rich ferritin shows a 120-fold difference in rate at ~20 Fe(II) atoms/H-subunit (Results and Figure 5C). The rate is approximately 20–50 times faster in HoSF at an Fe(II)/protein shell ratio of 8–30, i.e., 2–8 Fe(II) atoms/H-subunit,55 and ~10 times faster in homopolymeric E. coli bacterioferritin at an Fe(II)/shell ratio of 48–160, i.e., 2–7 Fe(II) atoms/H-subunit,56 suggesting that H2O2 is a better oxidant for Fe(II) in these proteins. Therefore, the increasing involvement of the detoxification reaction as the mineral core develops could, in part, drive the increase in Fe(II):O2 stoichiometry measured for L-rich ferritin from 2 toward a value of 4 (Figure 8B), a result in keeping with a detailed analysis of stoichiometry changes observed for HuHF31 and that presented here (Results).
In principle, all H2O2 produced at the ferroxidase center through eq 2 could subsequently react with further Fe(II) either at the ferroxidase or mineral surface by the detoxification reaction (eq 4), especially given the fast oxidation rates of H2O2 compared to those of O2 (Figure 5B, C), but that does not happen as evidenced by the Fe(II):O2 stoichiometries of <4. It appears that, under stoichiometric conditions [i.e., 2 Fe(II) atoms/H-subunit] and dissolved oxygen (i.e., ~0.25 mM), where the Fe(II):O2 stoichiometry is 2 (Figures 3A and 4A), the micromolar quantities of H2O2 produced at the ferroxidase centers of either H-rich or L-rich ferritin cannot compete with the higher concentrations of O2 for Fe(II) oxidation or with preformed diFe(II)-O2 ferroxidase center complexes. Detailed studies with HuHF have shown that the detoxification reaction gains importance only at intermediate levels of iron loading [i.e., ~200–400 Fe atoms/shell], but never exceeds the ferroxidase pathway, and declines at higher levels.31
Stopped-flow kinetic measurements of oxidation of Fe(II) by O2 in H-rich human ferritin reveal the formation of a μ-peroxo-diFe(III) complex at the ferroxidase center (Figure 9), an intermediate species first identified in the M-subunit of amphibian ferritin (BfMF),39 and later found in HuHF32,40 and in HoSF.23 Therefore, a ferroxidase-centered peroxo-diFe(III) species is a common intermediate regardless of whether L-subunits are present and is independent of the ratio of subunits, from ~20H:4L as in H-rich ferritin to ~3H:21L as in HoSF.23 The presence of only 4 L-subunits in H-rich ferritin does not significantly modulate the kinetics of formation (k1) and decay (k2) of the peroxo-diFe(III) intermediate (B) (eq 4). The values for the rate constants (k1 = 92.4 ± 5.8 s−1, k2 = 3.17 ± 0.55 s−1, and k3 = 0.24 ± 0.02 s−1) for H-rich ferritin (Figure 9) are very similar to the values (k1 = 81.8 ± 0.3 s−1, k2 = 2.83 ± 0.05 s−1, and k3 = 0.365 ± 0.001 s−1) reported for HuHF.32 In contrast, for HoSF (3.3H:20.7L), the kinetics of formation (k1) and decay (k2) of the peroxo-diFe(III) intermediate (B) to form the hydroperoxo complex (B′) are considerably slower, ~ 5- and ~2-fold, respectively, the rate constants being k1 = 17.7 ± 0.3 s−1 and k2 = 1.51 ± 0.06 s−1 for HoSF.23 However, k3 = 0.34 ± 0.06 s−1 for HoSF is comparable to those of the other two proteins. Thus, k3 for formation of the μ-oxo/hydroxo diFe(III) complex (C) with the concomitant release of H2O2 into solution is ~0.3 s−1, corresponding to the slow step in the reaction sequence (eq 4) for all three proteins. It is the clearance of this species from the ferroxidase center and subsequent slow step(s) measured by conventional kinetics (Figures 5 and 6 and Table 1) that govern the overall rate of core formation via the protein catalysis mechanism. Finally, we note that the kinetics of the equine protein are considerably more complicated than those of the two human proteins mentioned above because of the occurrence of an additional and more dominant ferroxidase center pathway lacking an observable peroxo-diFe(III) intermediate.23 This pathway accounts for two-thirds of the iron oxidized at the ferroxidase center.23 In this regard, peroxo intermediates have not been observed with E. coli bacterioferritin (EcBFR)46 or with the H-subunit bullfrog ferritin BfHF48 despite structurally similar ferroxidase centers for these proteins.
The kinetic and stoichiometry data depicted in Figures 7 and 8 particularly emphasize the influence of the L-subunit on the chemistry of core formation in ferritin. The enhanced rate of iron oxidation due to the four L-subunits in H-rich ferritin compared to homopolymeric HuHF is demonstrated by the data in Figure 7 at both low [i.e., ~2 Fe(II) atoms/H-subunit] (Figure 7A) and high [i.e., ~20 Fe(II) atoms/H-subunit] (Figure 7B) levels of iron. The H-rich protein can also sequester a considerably larger core, 3000 Fe(III) for H-rich ferritin versus 2000 Fe(III) for HuHF (Figure 7C), presumably because of the greater negative charge density on the inner surface of the protein shell due to the presence of the four L-subunits. Comparisons between the H-rich and L-rich ferritins in Figure 8 further illustrate the role of the L-subunit in ferroxidase activity. For example, when iron is delivered to these proteins at a stoichiometric ratio of 2 Fe(II) atoms/H-subunit, all the iron is processed through the ferroxidase center as evidenced by the constant Fe(II):O2 stoichiometry of ~2:1 for L-rich ferritin, although it oxidizes iron more slowly because of its lower H-subunit content (Figure 8A, top curve). On the other hand, the H-rich ferritin, while showing faster oxidation (Figure 8A, bottom curve) and 2-fold slower rates on a per H-subunit basis (Results), displays some shift toward the mineral surface reaction with the Fe(II):O2 stoichiometry increasing from ~2:1 to ~2.4. Pronounced differences are also evident at 10-fold larger additions of iron, i.e., 20 Fe(II) atoms/subunit, in which case both proteins shift toward mineral surface reactions but L-rich ferritin more so as indicated by the higher Fe(II):O2 stoichiometries (Figure 8B, C), a result consistent with its high L-subunit content and again emphasizing its role in mineralization.
Previous studies reported a maximal ferroxidase activity with ferritin samples consisting of ~8 H-subunits and 16 L-subunits,13,19 suggesting that the structural distribution of the appropriate mixture of the two subunits in heteropolymer ferritins plays an important role in the protein’s functionality. The data depicted in Figure 8 corroborate these findings and indicate that the inherent rate of oxidation (rate per H-subunit) at the ferroxidase centers is ~2-fold faster in L-rich than in H-rich ferritins, demonstrating a modulating effect of L-subunits on H-subunit ferroxidase activity.
A recent study57 has proposed a new function for L-subunits in electron transport across the protein shell and concluded that this specific function does not necessarily require the presence of the H-subunit. That L-subunits can facilitate electron transfer suggests that iron oxidation in the H-rich protein should occur faster than in HuHF as the results in Figure 7 demonstrate, consistent with the findings of a several-fold increase in activity for two heteropolymer samples (12H:12L and 22.5H:2.5L) compared to HuHF in the reported study.57 However, we note that diffusion of molecular O2 to the ferroxidase center has been shown to be very fast relative to iron oxidation,33 so a specific mechanism for electron transfer across the protein shell via the L-subunit may not be required. The enhanced rate of iron oxidation seen in our experiments for the H-rich ferritin compared to that for HuHF may simply be due to the modulating effect of the L-subunit on the kinetics of the H-subunit as presented here. Interestingly, this newly discovered rapid electron transfer was shown to be bidirectional, allowing not only rapid oxidation of Fe(II) but also reduction of Fe(III) and demineralization of the iron core of H-rich ferritin.57
In conclusion, this work further illustrates differences in iron oxidation activity between ferritins with different subunit compositions. It demonstrates an important effect of L-subunits on ferritin functionality in that the L-subunit enhances H-subunit activity and iron turnover, and its presence increases the capacity of the protein to store iron, in agreement with the different distributions of isoferritins in tissues of different organs. Specifically, our data indicate that L-rich ferritins found in iron-rich tissues such as liver and spleen are slow in iron incorporation but can hold more iron, whereas H-rich ferritins exhibit fast iron incorporation, hold less iron, and are more suited for tissues with high metabolic activity such as heart, kidney, brain, and developing erythroid cells. The faster regeneration of ferroxidase activity in the H-subunit, due to the presence of the L-subunit within the protein shell, is another property of heteropolymer H/L ferritins, supporting the synergistic involvement of both H- and L-subunits in rapid iron oxidation and core formation.
Acknowledgments
Funding
This work is supported by National Institutes of Health Grants R15GM104879 (F.B.-A.) and R01 GM20194 (N.D.C.). It is also partially supported by Grant PRIN10-11 of MIUR, from the Italian Ministry of Research (P.A.).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Theil EC, Tosha T, Behera RK. Solving biology’s iron chemistry problem with ferritin protein nanocages. Acc Chem Res. 2016;49:784–791. doi: 10.1021/ar500469e. [DOI] [PubMed] [Google Scholar]
- 2.Chasteen ND, Harrison PM. Mineralization in ferritin: an efficient means of iron storage. J Struct Biol. 1999;126:182–194. doi: 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- 3.Arosio P, Ingrassia R, Cavadini P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta, Gen Subj. 2009;1790(7):589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Bou-Abdallah F. The iron redox and hydrolysis chemistry of the ferritins. Biochim Biophys Acta, Gen Subj. 2010;1800:719–731. doi: 10.1016/j.bbagen.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Harrison PM, Arosio P. Ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta, Bioenerg. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 6.Ha Y, Shi D, Small GW, Theil EC, Allewell NM. Crystal structure of bullfrog M ferritin at 2.8 Å resolution: analysis of subunit interactions and the binuclear metal center. JBIC, J Biol Inorg Chem. 1999;4:243–256. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 7.Le Brun NE, Crow A, Murphy MEP, Mauk AG, Moore GR. Iron core mineralisation in prokaryotic ferritins. Biochim Biophys Acta, Gen Subj. 2010;1800:732–744. doi: 10.1016/j.bbagen.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Briat JF, Duc C, Ravet K, Gaymard F. Ferritins and iron storage in plants. Biochim Biophys Acta, Gen Subj. 2010;1800:806–814. doi: 10.1016/j.bbagen.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Pham DQD, Winzerling JJ. Insect ferritins: Typical or atypical? Biochim Biophys Acta, Gen Subj. 2010;1800:824–833. doi: 10.1016/j.bbagen.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim HJ, Kim HM, Kim JH, Ryu KS, Park SM, Jahng KY, Yang MS, Kim DH. Expression of heteropolymeric ferritin improves iron storage in Saccharomyces cerivisiae. Appl Environ Microbiol. 2003;69(4):1999–2005. doi: 10.1128/AEM.69.4.1999-2005.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levi S, Santambrogio P, Cozzi A, Rovida E, Corsi B, Tamborini E, Spada A, Albertini A, Arosio P. The role of the L-chain in ferritin iron incorporation. Studies of homo and heteropolymers. J Mol Biol. 1994;238:649–654. doi: 10.1006/jmbi.1994.1325. [DOI] [PubMed] [Google Scholar]
- 12.Hann HWL, Levy HM, Evans AE. Serum ferritin as a guide to therapy in neuroblastoma. Cancer Res. 1980;40:1411–1413. [PubMed] [Google Scholar]
- 13.Santambrogio P, Levi S, Cozzi A, Rovida E, Albertini A, Arosio P. Production and characterization of recombinant heteropolymers of human ferritin H and L chains. J Biol Chem. 1993;268:12744–12748. [PubMed] [Google Scholar]
- 14.Andrews SC. Iron storage in bacteria. Adv Microb Physiol. 1998;40:281–351. doi: 10.1016/s0065-2911(08)60134-4. [DOI] [PubMed] [Google Scholar]
- 15.Drysdale JW, Adelman TG, Arosio P, Casareale D, Fitzpatrick P, Harzard JT, Yokota M. Human isoferritins in normal and disease states. Semin Hematol. 1977;14:71–88. [PubMed] [Google Scholar]
- 16.Bou-Abdallah F, Santambrogio P, Levi S, Arosio P, Chasteen ND. Unique iron binding and oxidation properties of human mitochondrial ferritin: A comparative analysis with human H-chain ferritin. J Mol Biol. 2006;347:543–554. doi: 10.1016/j.jmb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: Past, present and future. Biochim Biophys Acta, Gen Subj. 2010;1800:760–769. doi: 10.1016/j.bbagen.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alkhateeb AA, Connor JA. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim Biophys Acta, Rev Cancer. 2013;1836:245–254. doi: 10.1016/j.bbcan.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Carmona F, Poli P, Bertuzzi M, Gianoncelli A, Gangemi F, Arosio P. Study of ferritin self-assembly and heteropolymer formation by the use of Fluorescence Resonance Energy Transfer (FRET) technology. Biochim Biophys Acta, Gen Subj. 2017;1861(3):522–532. doi: 10.1016/j.bbagen.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Rucker P, Torti FM, Torti SV. Recombinant ferritin: modulation of subunit stoichiometry in bacterial expression system. Protein Eng, Des Sel. 1997;10(8):967–973. doi: 10.1093/protein/10.8.967. [DOI] [PubMed] [Google Scholar]
- 21.Lee J, Seo HY, Jeon ES, Park OS, Lee KM, Park CU, Kim KS. Cooperative activity of subunits of human ferritin heteropolymers in Escherichia coli. Biochem Mol Biol. 2001;34(4):365–370. [Google Scholar]
- 22.Masuda T, Goto F, Yoshihara T, Ezure T, Suzuki T, Kobayashi S, Shikata M, Utsumi S. Construction of homo- and heteropolymers of plant ferritin subunits using an in-vitro protein expression system. Protein Expression Purif. 2007;56:237–246. doi: 10.1016/j.pep.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Zhao G, Su M, Chasteen ND. μ-1,2-Peroxo diferric complex formation in horse spleen ferritin. A mixed H/L-subunit heteropolymer. J Mol Biol. 2005;352(2):467–477. doi: 10.1016/j.jmb.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 24.Zhao G, Bou-Abdallah F, Yang X, Arosio P, Chasteen ND. Is hydrogen peroxide produced during iron(II) oxidation in mammalian apoferritins? Biochemistry. 2001;40:10832–10838. doi: 10.1021/bi011052j. [DOI] [PubMed] [Google Scholar]
- 25.Luscieti S, Santambrogio P, Langlois d’Estaintot B, Granier T, Cozzi A, Poli M, Gallois B, Finazzi D, Cattaneo A, Levi S, Arosio P. Mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J Biol Chem. 2010;285:11948–11957. doi: 10.1074/jbc.M109.096404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santambrogio P, Cozzi A, Levi S, Rovida E, Magni F, Albertini A, Arosio P. Functional and immunological analysis of recombinant mouse H- and L-ferritins from Escherichia coli. Protein Expression Purif. 2000;19:212–218. doi: 10.1006/prep.2000.1212. [DOI] [PubMed] [Google Scholar]
- 27.Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. Overexpression of wild type and mutated human ferritin H-chain in hela cells. J Biol Chem. 2000;275:25122–2512. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 28.Grady JK, Zang J, Laue TM, Arosio P, Chasteen ND. Characterization of the H- and L-subunit ratios of ferritins by sodium dodecyl sulfate–capillary gel electrophoresis. Anal Biochem. 2002;302:263–268. doi: 10.1006/abio.2001.5561. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Chen-Barrett Y, Arosio P, Chasteen ND. Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry. 1998;37:9743–9750. doi: 10.1021/bi973128a. [DOI] [PubMed] [Google Scholar]
- 30.Bou-Abdallah F, Biasiotto G, Arosio P, Chasteen ND. The putative nucleation site in human H-chain ferritin is not required for mineralization of the iron core. Biochemistry. 2004;43:4332–4337. doi: 10.1021/bi0498813. [DOI] [PubMed] [Google Scholar]
- 31.Zhao G, Bou-Abdallah F, Arosio P, Levi S, Janus-Chandler C, Chasteen ND. Multiple pathways for mineral core formation in mammalian apoferritin. The role of hydrogen peroxide. Biochemistry. 2003;42:3142–3150. doi: 10.1021/bi027357v. [DOI] [PubMed] [Google Scholar]
- 32.Bou-Abdallah F, Zhao G, Mayne HR, Arosio P, Chasteen ND. Origin of the unusual kinetics of iron deposition in human H-chain ferritin. J Am Chem Soc. 2005;127:3885–3893. doi: 10.1021/ja044355k. [DOI] [PubMed] [Google Scholar]
- 33.Bou-Abdallah F, Zhao G, Biasiotto G, Poli M, Arosio P, Chasteen ND. Facilitated Diffusion of Iron(II) and Dioxygen Substrates into Human H-Chain Ferritin. A Fluorescence and Absorbance Study Employing the Ferroxidase Center Substitution Y34W. J Am Chem Soc. 2008;130:17801–17811. doi: 10.1021/ja8054035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Le Brun NE, Thomson AJ, Moore GR, Chasteen ND. The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin. Biochemistry. 2000;39:4915–4923. doi: 10.1021/bi992631f. [DOI] [PubMed] [Google Scholar]
- 35.Jameson GNL, Jin W, Krebs C, Perreira AS, Tavares P, Liu X, Theil EC, Huynh BH. Stoichiometric production of hydrogen peroxide and parallel formation of ferric multimers through decay of the diferric-peroxo complex, the first detectable intermediate in ferritin mineralization. Biochemistry. 2002;41:13435–13443. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 36.Waldo GS, Theil EC. Formation of iron-tyrosinate is the fastest reaction observed in ferritin. Biochemistry. 1993;32:13262–13269. doi: 10.1021/bi00211a039. [DOI] [PubMed] [Google Scholar]
- 37.Bunker J, Lowry T, Davis G, Zhang B, Brosnahan D, Lindsay S, Costen R, Choi S, Arosio P, Watt GD. Kinetics of iron deposition catalyzed by recombinant human liver heavy and light ferritins and Azotobacter vinelandii bacterioferrin using O2 and H2O2 as oxidants. Biophys Chem. 2005;114:235–244. doi: 10.1016/j.bpc.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Bou-Abdallah F, Carney E, Chasteen ND, Arosio P, Viescas AJ, Papaefthymiou GC. A comparative Mössbauer study of the mineral cores of human H-chain ferritin employing dioxygen and hydrogen peroxide as iron oxidants. Biophys Chem. 2007;130:114–121. doi: 10.1016/j.bpc.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fetter J, Cohen J, Danger D, Sanders-Loehr J, Theil EC. The influence of conserved tyrosine-30 and tissue dependent differences in sequence on ferritin function – use of blue and purple Fe(III) species as reporters of ferroxidation. JBIC, J Biol Inorg Chem. 1997;2:652–661. [Google Scholar]
- 40.Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM. Iron(II) oxidation by H chain ferritin: evidence from – site-directed mutagenesis that a transient blue species is formed at the dinuclear iron center. Biochemistry. 1995;34:15204–15213. doi: 10.1021/bi00046a028. [DOI] [PubMed] [Google Scholar]
- 41.Kim M, Rho Y, Jin KS, Ahn B, Jung S, Kim H, Ree M. pH-dependent structures of ferritin and apoferritin in solution: disassembly and reassembly. Biomacromolecules. 2011;12:1629–1640. doi: 10.1021/bm200026v. [DOI] [PubMed] [Google Scholar]
- 42.Santambrogio P, Levi S, Arosio P, Palagi L, Vecchio G, Lawson DM, Yewda S, Artymiuk P, Harrison PM, Jappelli R. Evidence that a salt bridge in the light chain contributes to the physical stability difference between heavy and light human ferritins. J Biol Chem. 1992;267:14077–14083. [PubMed] [Google Scholar]
- 43.Gerl M, Jaenicke R. Mechanism of the self-assembly of apoferritin from horse spleen. Eur Biophys J. 1987;15:103–109. doi: 10.1007/BF00257503. [DOI] [PubMed] [Google Scholar]
- 44.Santambrogio P, Pinto P, Levi S, Cozzi A, Rovida E, Albertini A, Artymiuk P, Harrison PM, Jappelli R, Arosio P. Effects of modifications near the 2-, 3-and 4-fold symmetry axes on human ferritin renaturation. Biochem J. 1997;322:461–468. doi: 10.1042/bj3220461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huard DJ, Kane KM, Tezcan FA. Re-engineering protein interfaces yields copper-inducible ferritin cage assembly. Nat Chem Biol. 2013;9:169–176. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- 46.Le Brun NE, Wilson MT, Andrews SC, Guest JR, Harrison PM, Thomson AJ, Moore GR. Kinetic and structural characterization of an intermediate in the biomineralization of bacterioferritin. FEBS Lett. 1993;333:197–202. doi: 10.1016/0014-5793(93)80404-i. [DOI] [PubMed] [Google Scholar]
- 47.Zhao G. Phytoferritin and its implications for human health and nutrition. Biochim Biophys Acta, Gen Subj. 2010;1800:815–823. doi: 10.1016/j.bbagen.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Pereira AS, Tavares P, Lloyd SG, Danger D, Edmondson DE, Theil EC, Huynh BH. Rapid and parallel formation of Fe3+ multimers, including a trimer, during H-type subunit ferritin mineralization. Biochemistry. 1997;36:7917–7927. doi: 10.1021/bi970348f. [DOI] [PubMed] [Google Scholar]
- 49.Honarmand Ebrahimi K, Hagedoorn PL, Hagen WR. Unity in the Biochemistry of the Iron-Storage Proteins Ferritin and Bacterioferritin. Chem Rev. 2015;115:295–326. doi: 10.1021/cr5004908. [DOI] [PubMed] [Google Scholar]
- 50.Bou-Abdallah F, Yang H, Awomolo A, Cooper B, Woodhall MR, Andrews SC, Chasteen ND. Functionality of the three-site ferroxidase center of Escherichia coli bacterial ferritin (EcFtnA) Biochemistry. 2014;53:483–95. doi: 10.1021/bi401517f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bradley JM, Moore GR, Le Brun NE. Diversity of Fe2+ entry and oxidation in ferritins. Curr Opin Chem Biol. 2017;37:122–128. doi: 10.1016/j.cbpa.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 52.Zhao G, Chasteen ND. Oxidation of Good’s buffers by hydrogen peroxide. Anal Biochem. 2006;349:262–267. doi: 10.1016/j.ab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Zhao G, Arosio P, Chasteen ND. Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry. 2006;45:3429–3436. doi: 10.1021/bi052443r. [DOI] [PubMed] [Google Scholar]
- 54.Grady JK, Chen Y, Chasteen ND, Harris DC. Hydroxyl radical production during oxidative deposition of iron in ferritin. J Biol Chem. 1989;264:20224–20229. [PubMed] [Google Scholar]
- 55.Lowery TJ, Jr, Bunker J, Zhang B, Costen R, Watt GD. Kinetic studies of iron deposition in horse spleen ferritin using H2O2 and O2 as oxidants. Biophys Chem. 2004;111:173–181. doi: 10.1016/j.bpc.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Bou-Abdallah F, Lewin AC, Le Brun NE, Moore GR, Chasteen ND. Iron detoxification properties of Escherichia coli bacterioferritin. J Biol Chem. 2002;277:37064–37069. doi: 10.1074/jbc.M205712200. [DOI] [PubMed] [Google Scholar]
- 57.Carmona U, Li L, Zhang L, Knez M. Ferritin light-chain subunits: key elements for the electron transfer across the protein cage. Chem Commun. 2014;50:15358–15361. doi: 10.1039/c4cc07996e. [DOI] [PubMed] [Google Scholar]