

SUMMARY
Piezo proteins form mechanically activated ion channels that are responsible for our sense of light touch, proprioception, and vascular blood flow. Upon activation by mechanical stimuli, Piezo channels rapidly inactivate in a voltage-dependent manner through an unknown mechanism. Inactivation of Piezo channels is physiologically important, as it modulates overall mechanical sensitivity, gives rise to frequency filtering of repetitive mechanical stimuli, and is itself the target of numerous human disease-related channelopathies that are not well understood mechanistically. Here, we identify the globular C-terminal extracellular domain as a structure that is sufficient to confer the time course of inactivation and a single positively charged lysine residue at the adjacent inner pore helix as being required for its voltage dependence. Our results are consistent with a mechanism for inactivation that is mediated through voltage-dependent conformations of the inner pore helix and allosteric coupling with the C-terminal extracellular domain.
In Brief
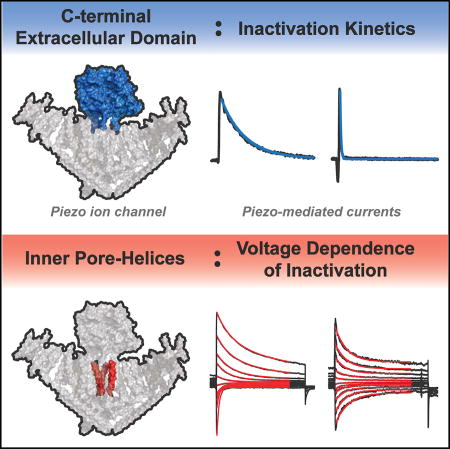
Wu et al. examine how the properties of inactivation are influenced in mechanically activated Piezo ion channels. They identify two structural domains, the C-terminal extracellular domain and the inner pore helix, that correspond with the kinetics and voltage dependence of inactivation, respectively.
INTRODUCTION
Piezo1 and Piezo2 are mechanically activated non-selective cation channels that serve a wide variety of physiological functions (Coste et al., 2010, 2012; Wu et al., 2017). Piezo1 plays a crucial role in several non-neuronal tissues, such as the cardiovascular endothelium and arterial smooth muscle cells, where it senses shear stress, whereas Piezo2 is expressed in dorsal root ganglia (DRG) neurons and Merkel cells, where it functions as a sensor of light touch and proprioception (Maksimovic et al., 2014; Ranade et al., 2014a, 2014b; Retailleau et al., 2015; Woo et al., 2014, 2015).
Upon mechanical stimulation, Piezo-mediated currents rise instantaneously and then decay while the stimulus is still present. In principle, this decay could be due to adaptation of the channel to the stimulus or due to an intrinsic transition toward pore closure known as inactivation (Honoré et al., 2006). While both processes are not mutually exclusive, it has been shown in at least one stimulation paradigm that adaptation has only a minor contribution in Piezo1 and that the predominant mechanism for current decay is, indeed, inactivation, which implies that the molecular mechanism for inactivation resides within the protein itself (Lewis et al., 2017).
Upon their initial discovery in 2010, Piezo proteins were already characterized by their inactivation kinetics, which were correctly described as fast at negative membrane potentials, slow at positive membrane potentials, and distinct between Piezo1 (slower) and Piezo2 (faster) (Coste et al., 2010). Since then, inactivation has emerged as an important mechanism in Piezo function. By decreasing the fraction of channels available for opening, the overall current amplitude and the apparent stimulus sensitivity are changed, and temporal frequency filtering of repetitive stimuli such as mechanical vibration is generated (Lewis et al., 2017; Lewis and Grandl, 2015). More importantly, several point mutations that alter inactivation kinetics in Piezo1 and Piezo2 were identified from human patients with various diseases, such as red blood cell dehydration (xerocytosis) and Gordon syndrome (distal arthrogryposis type 3) (Albuisson et al., 2013; Andolfo et al., 2013; Bae et al., 2013; Coste et al., 2013; Lukacs et al., 2015; McMillin et al., 2014; Okubo et al., 2015; Zarychanski et al., 2012). In addition, endogenous factors such as bradykinin, divalent ion concentration, and extracellular pH affect inactivation, opening the possibility that Piezo function is physiologically regulated through this mechanism (Bae et al., 2015; Dubin et al., 2012; Gottlieb et al., 2012). Given its demonstrated importance for mechanotransduction and its direct link to disease, a molecular understanding of Piezo inactivation is critical for developing treaments for Piezo malfunction-related defects. However, the uncommonly large size of ~2,500 amino acids per Piezo monomer and their lack of homology with other known transmembrane proteins have been obstacles in understanding the mechanism for inactivation.
In the past, a successful strategy for understanding the mechanisms of inactivation in other ion channels has been the identification of structures (residues/domains) that are specifically implicated in inactivation (Goldin, 2003; Hoshi et al., 1991). Here, we use mutagenesis paired with electrophysiology to study the mechanism underlying the inactivation of Piezo channels. We identify two distinct structures that mediate the kinetics and voltage dependence of inactivation.
RESULTS
Voltage Modulates Piezo1 and Piezo2 Inactivation Kinetics in a Continuous Manner
To investigate the readily apparent voltage dependence of Piezo inactivation in greater detail, we performed whole-cell electrophysiology recordings on HEK293T-P1KOcells, which were engineered by CRISPR/Cas9 to lack endogenous Piezo1, and are thus devoid of mechanically activated currents (Dubin et al., 2017).
We mechanically stimulated cells transiently transfected with DNA encoding wild-type mouse Piezo1 by indentation with a blunt glass pipette while holding the cells at potentials ranging from −100 mV to +100 mV (Figure 1A). Mean currents elicited by this protocol were large (1.37 nA ± 0.22 nA at −100 mV and 1.5 nA ± 0.34 nA at +100 mV) and exhibited the previously reported rapid activation and subsequent inactivation, the latter of which was well fit by a single exponential function (Figures 1A and 1B).
Figure 1. Voltage Modulates Piezo1 and Piezo2 Inactivation.

(A) Force probe indentation (arrow) stimulation protocol and representative currents of whole-cell recordings from HEK293T-P1KO cells transiently transfected with wild-type mouse Piezo1 and Piezo2. Red lines indicate single exponential fits to the decaying currents. Insets show currents normalized to their peak amplitudes.
(B) Mean inactivation time constants obtained from exponential current fits as a function of membrane potential for Piezo1 (n = 13 cells) and Piezo2 (n = 14 cells).
(C and D) Shown here: (C) mean peak currents for Piezo1 during force probe stimulation and (D) mean inactivation time constants from single exponential fits, recorded with standard bath and pipette buffers (n = 13 cells), NMDG+ in the bath (n = 10 cells), and NMDG+ in the recording pipette (n = 9 cells) as a function of membrane potential. See Experimental Procedures for buffer compositions.
(E) Mean time constants of inactivation binned as a function of membrane tension within the patch dome during cell-attached pressure-clamp stimulation from HEK293T transiently transfected with Piezo1 (n = 15 cells).
All data are means ± SEM.
Importantly, we observed that the previously reported binary difference in inactivation kinetics between negative and positive potentials is more accurately described as a gradual change; i.e., over a wide range of membrane potentials, inactivation kinetics change exponentially as a function of voltage. Based on this exponential relationship, we applied a two-state mechanism of channel inactivation (open ↔ inactivated) and its associated inactivation rate constant α =A•exp((Einact − zFV)/RT) (see Experimental Procedures for details) to calculate the effective charge (z = 0.31 ± 0.02 e [elementary charge is denoted as e]) associated with voltage-dependent inactivation. Identical experiments on wild-type mouse Piezo2 revealed a similar, albeit shifted, voltage dependence of inactivation kinetics, with a calculated effective charge of z = 0.24 ± 0.02 e (Figure 1B). From these data, we concluded that voltage dependence of inactivation might be generated by one single charged residue within the numerous predicted transmembrane domains within each monomer of the trimeric Piezo channels. The results also suggested that, although mouse Piezo1 and mouse Piezo2 share only ~53% sequence homology, the mechanism and, perhaps, also the structures giving rise to inactivation and its voltage dependence are identical in both proteins.
To determine whether the voltage dependence of inactivation was an effect of ion permeation, we recorded Piezo1 activity while substituting permeant cations on the intracellular (Cs+) or extracellular (Na+ and K+) side with the large cation NMDG+. This resulted in a shift of −25 mV ± 2 mV in reversal potential toward more negative values (NMDG+ extracellular) versus a shift of +22 mV ± 1 mV toward more positive values (NMDG+ intracellular), respectively, compared to the standard recording buffer (3.7 mV ± 2 mV) (Figure 1C). Importantly, we found that inactivation kinetics remained identical across all voltages (p ≥ 0.05, one-way ANOVA followed by post hoc Tukey test), regardless of substitution with NMDG+, demonstrating that the voltage dependence of inactivation is autonomous of the driving force of ion permeation (Figure 1D). These results further suggest that Piezo ion channels inactivate in a voltage-dependent manner that is likely mediated by intrinsic mechanisms within the Piezo protein itself.
Three previous studies concluded that Piezo1 is activated by lateral membrane tension (Cox et al., 2016; Lewis and Grandl, 2015; Syeda et al., 2016). Therefore, we tested whether tension also influences inactivation by performing cell-attached patches on HEK293T cells expressing Piezo1 and applying negative pressure through the patch pipette to induce current and simultaneously visualize membrane geometry to calculate tension. By analyzing these data with respect to current inactivation, we found that inactivation exhibited no substantial dependence on membrane tension (Figure 1E). This result is in stark contrast to the strong modulation by voltage, which ranges over more than one order of magnitude, and thus shows that the mechanism of inactivation is sensitive to modulation by voltage and, to a lesser extent, by tension in the membrane bilayer.
The C-Terminal Extracellular Domain Is Sufficient to Confer Inactivation Kinetics
A prominent domain in Piezo1 called the C-terminal extracellular domain (CED) has previously been implicated in inactivation, as mechanical pulling on this domain can specifically inhibit inactivation (Wu et al., 2016). In the primary sequence, the CED is situated between the two pore helices and connected to them by linker regions that are highly conserved among Piezo orthologs. A Piezo1 cryoelectron microscopy (cryo-EM) structure revealed that the CED is located directly on top of the permeation pathway, which is formed by the inner and outer pore helices (Ge et al., 2015). We therefore hypothesized that the CED might be a structural determinant of inactivation kinetics.
To test this hypothesis, we pursued a chimeric strategy where we engineered a wild-type Piezo1 channel with the CED of Piezo2 (P1−P2CED), and a wild-type Piezo2 channel with the CED of Piezo1 (P2−P1CED) and measured channel activity induced by stimulating transfected cells with a blunt glass pipette in the whole-cell configuration (Figure 2A). When stimulated at a holding potential of +100 mV, both chimeric constructs yielded peak current amplitudes that were statistically identical to those of wild-type channels (wild-type Piezo1: Ipeak = 1.5 nA ± 0.3 nA; wild-type Piezo2: Ipeak = 0.8 nA ± 0.2 nA; P1-P2CED: Ipeak = 1.4 nA ± 0.2 nA; P2−P1CED: Ipeak = 1.2 nA ± 0.3 nA), and no significant difference was measured across all tested voltages (p ≥ 0.05, one-way ANOVA followed by post hoc Tukey test) (Figure 2B). Strikingly, a first qualitative examination of representative currents recorded at −60 mV and at +60 mV suggested that chimera P1-P2CED inactivates with kinetics similar to that of wild-type Piezo2, while chimera P2−P1CED adopts the inactivation kinetics of Piezo1 (Figures 2C and 2D). In order to obtain a detailed quantitative description, we again fit inactivating currents with single exponential functions and plotted the time constants as a function of voltage (Figure 2E). Indeed, chimera P2−P1CED was statistically identical in its inactivation kinetics to wild-type Piezo1 over the entire range of measured voltages from −100 mV to +100 mV (p ≥ 0.05 at each voltage, unpaired Student’s t test). Inactivation kinetics for chimera P1−P2 CED were identical to those for wild-type Piezo2 throughout negative and moderate positive potentials, while for more positive potentials (+40 mV to +100 mV), kinetics were intermediate between wild-type Piezo1 and wild-type Piezo2 (Figure 2E). These results show that the CED is, in principle, sufficient for conferring the distinct inactivation time courses between Piezo1 and Piezo2, although structures/residues outside the CED must exist, at least in Piezo2, that also influence inactivation kinetics at positive potentials.
Figure 2. The CED Confers the Time Course of Inactivation.

(A) Illustrations of chimeric constructs: the CEDs of wild-type Piezo1 (blue) and Piezo2 (red) are swapped to generate constructs P1-P2CED (Piezo1 with the CED of Piezo2) and P2−P1CED (Piezo2 with the CED of Piezo1).
(B) Mean peak currents elicited by force probe stimulation.
(C and D) Representative currents of whole-cell patches from wild-type Piezo1, Piezo2, P1−P2CED, and P2-P1CED elicited by a fore probe stimulus at negative (−60 mV) and positive (+60 mV) holding potentials. Red lines are single exponential fits to the decaying currents.
(E) Mean inactivation time constants (τ) obtained with exponential fits from whole-cell recordings of HEK293T-P1KO cells transiently transfected with wild-type Piezo1 (n = 13 cells), Piezo2 (n = 14 cells), P1−P2CED (n = 14 cells), and P2-P1CED (n = 14 cells) as a function of membrane potential.
n.s., no significance between all constructs, p ≥ 0.05; between Piezo2 and P1−P2CED, *p ≤ 0.05; **p ≤ 0.005, one-way ANOVA followed by post hoc Tukey’s test.
All data are means ± SEM.
The structure of the CED, first resolved in isolation by X-ray crystallography and later again by cryo-EM within the trimeric channel complex, reveals a cavity along the central axis of the CED and multiple large side fenestrations, raising the possibility that the CED mediates inactivation directly by acting as part of the permeation pathway (Ge et al., 2015; Kamajaya et al., 2014). However, we did not find any evidence for this function, as nine residues located within the central cavity, when individually mutated to cysteines and incubated with the cysteine-modifying reagent MTSET, did not show any statistically significant changes in peak current amplitude or the extent of inactivation (Figures S1A–S1D). In addition, several deletion constructs that we engineered to lack the CED had no mechanically activated currents (data not shown; see Experimental Procedures for details). We examined one of these constructs (P2223-S2450delinsTG) further and found that it failed to locate to the plasma membrane but had normal oligomerization as visualized by native gel electrophoresis (Figures S1E and S1F).
These results suggest that the CED may be required for protein trafficking, but not assembly or stability, in addition to determining inactivation kinetics. Similarly, insertion of PreScission Protease sites into the highly conserved adjacent CED linker domains, with the goal of enzymatically cleaving the CED after initial membrane expression, also resulted in non-functional channels (data not shown; Experimental Procedures), altogether precluding a direct test for whether inactivation occurs in the absence of the CED.
A Single Lysine Residue in the Inner Pore Helix Confers Voltage Dependence of Inactivation to Piezo1
The amino acids giving rise to the effective charge that we calculated to be associated with voltage-dependent inactivation could be located within any of the 14–30 transmembrane domains, as predicted from a structural model and hydropathy profiles (Alper, 2017; Ge et al., 2015). However, due to our finding that the CED confers the distinct inactivation kinetics between Piezo1 and Piezo2, and the previous finding that mechanical pulling on the CED directly slows inactivation, we focused our attention on the two transmembrane domains immediately connected to the CED known as the outer pore helix and the inner pore helix (Wu et al., 2016). Specifically, we hypothesized that, similarly to mechanical force transduced by magnetic nanoparticles, Coulomb forces generated by an electric potential on charged residues within these two helices may also influence inactivation kinetics (Figure 3A).
Figure 3. Mutation of Charged Residue Eliminates Voltage Dependence of Inactivation.

(A) Schematic of open-state modulation of inactivation. (i) Piezo in closed-state highlighting CED (green) and inner pore helices (red). (ii) Piezo in tension-induced open state with CED under pulling force by magnetic bead. Orange arrows represent force toward inactivation through pore helices. Movement of the CED away from the channel alters inactivation through interaction with the pore helices. Shaded yellow region depicts permeation pathways. (iii) Piezo in tension-induced open state at a negative holding potential. Larger orange arrows represent stronger force toward inactivation through pore helices. The white plus symbols represent positive charges within the pore helices. Coulombic force holds pore helices and adjacent CED in a fast inactivating state. (iv) Piezo in tension-induced open state held at a positive holding potential. Small orange arrows represent weaker force toward inactivation. Coulombic force induces displaced pore helices and CED similarly to magnetic pulling to induce a slowly inactivating state.
(B) Illustrations of Piezo structure highlighting the CED (green), the inner pore helix (red), and the outer pore helix (blue) and four positively charged residues mutated to charge-neutral glutamines.
(C) Mean inactivation time constants obtained from single exponential fits of force-probe-induced current from whole-cell recordings of HEK293T-P1KO cells transiently transfected with wild-type Piezo1 (n = 13 cells) and single-point mutants K2188Q (n = 8 cells), R2214Q (n = 8 cells), K2479Q (n = 10 cells), and R2482Q (n = 9 cells) as a function of membrane potential.
(D) Force-probe stimulus protocol and representative current traces of single-point mutants K2479Q and K2479E. Red lines indicate single exponential fits to the decaying currents. Insets show currents normalized to their peak amplitudes.
(E) Mean inactivation time constants from single exponential fits of current from whole-cell recordings in HEK293T-P1KO cells transiently transfected with wild-type Piezo1 (n = 13 cells) and single-point mutants K2479Q (n = 10 cells) and K2479E (n = 8 cells) as a function of membrane potential.
All data are means ± SEM.
To test this idea experimentally, we individually neutralized each of the four positively charged residues within these two helices by mutating them to glutamine (outer pore helix: K2188Q, R2214Q; inner pore helix: K2479Q, R2482Q) and measured how this affected the voltage dependence of inactivation (Figure 3B). Strikingly, we found that neutralization of a single lysine (K2479Q) in the inner pore helix resulted in consistent inactivation kinetics at all potentials between −100 mV and +100 mV (τ = 5–9 ms), and calculation of the effective gating charge revealed that it was virtually eliminated (z = 0.06 ± 0.06 e) (Figures 3C and 3D). In contrast, the nearby mutation K2482Q caused a modest decrease in voltage dependence of the inactivation gating charge (z = 0.20 ± 0.02 e). Channels with mutations K2188Q and R2214Q in the outer pore helix exhibited inactivation kinetics and gating charges similar to that of wild-type Piezo1 (K2188Q: z = 0.26 ± 0.03 e; R2214Q: z = 0.26 ± 0.03 e) (Figure 3C). This result shows that two charged residues in the lower part of the inner pore helix of Piezo1 are important for the voltage dependence of inactivation and, in particular, that the single lysine residue K2479 is specifically required for it.
We noticed that the charge neutralization of K2487Q resulted in inactivation kinetics similar to kinetics observed in wild-type Piezo1 at very negative voltages, i.e., −100 mV. We next tested the effect of a charge reversal mutation on that same residue, K2479E. Interestingly, we found not only that this mutation ablates voltage dependence of inactivation similarly to K2479Q but also that it sets inactivation kinetics to values similar to those observed in wild-type Piezo1 at strongly positive potentials (τ = 31–70 ms between −100 and +100 mV, gating charge z = 0.06 ± 0.06 e) (Figures 3D and 3E). This result supports the notion that voltage controls inactivation kinetics over a wide range, specifically through Coulomb forces acting on the inner pore helix.
MTSET Accessibility to Inner Pore Helix Residue V2467 Is Voltage Dependent
The aforementioned results suggested to us that a conformational change within the inner pore helix, whether induced by mechanical force transduced by magnetic nanoparticles or by Coulomb force, might be a mechanism for determining inactivation kinetics in Piezo1. Therefore, we aimed next to test directly for such a voltage-dependent conformational change. For this, we selected five residues in the upper part of the inner pore helix based on their predicted orientation with respect to the permeation pathway, mutated them individually to cysteines (F2460C, G2463C, Y2464C, V2467C, and G2468C), and probed for their accessibility to the extracellular solvent with the cysteine-modifying reagent MTSET in cell-attached recordings using pressure-clamp stimulation (Figure 4A).
Figure 4. MTSET Has Voltage-Dependent Accessibility to the Upper Pore.

(A) Illustration of Piezo1 pore structure highlighting the outer pore helices (blue), inner pore helices (red), and five residues mutated individually to cysteines for MTSET accessibility experiments.
(B) Pressure-clamp stimulation and MTSET labeling protocol. Cells were held at a constant potential of −80 mV while allowing for 2-min incubation with standard buffer or 2 mM MTSET before testing mechanical activation by a pressure-step protocol (5 s, +5 mmHg prepulse, followed by 200-ms pressure steps from 0 to −70 mmHg). Representative current traces from cell-attached patches of HEK293T-P1KO cells transiently transfected with cysteine point mutant V2467C at the −60-mmHg stimulus step with standard buffer (blue) or MTSET (red). Arrow indicates measured time point of inactivation.
(C and D) Shown here: (C) mean peak currents and (D) mean degree of inactivation of currents from cells stimulated using the protocol in (B) upon application of 2 mM MTSET or buffer alone (n = 8–27 cells; *p ≤ 0.05, unpaired Student’s t test; NA, omitted from analysis).
(E) Degree of inactivation for currents recorded from HEK293T-P1KO cells transiently transfected with cysteine point mutant V2467C stimulated using the protocol in (B) in the presence of standard buffer (n = 20 cells), 200 µM MTSET (n = 6 cells), or 2 mM MTSET (n = 8 cells) (n.s., no significance, p ≥ 0.05; *p ≤ 0.05, unpaired Student’s t test).
(F) Pressure-clamp stimulation and MTSET labeling protocol. Cells were recorded in either standard buffer or 2 mM MTSET by first stimulating with a −60-mmHg pressure stimulus, allowing for MTSET labeling for 5 min at a holding potential of either −80 mV or +60 mV, and immediately following with a second −60-mmHg pressure stimulus. Representative current traces from cell-attached patches of HEK293T-P1KO cells transiently transfected with cysteine point mutant V2467C depicting the first pulse (green) and second pulse (magenta) under indicated MTSET labeling conditions. Arrow indicates measured time point of inactivation.
(G) Individual peak current amplitudes from recordings with the stimulation protocol used in (F) of mutant V2467C with indicated MTSET labeling conditions (n = 13–20 cells; *p ≤ 0.05, paired Student’s t test).
(H) Individual measures of degree of inactivation (top) and mean change in inactivation (below) for recordings of mutant V2467C from (G) (n.s., no significance, p ≥ 0.05; *p ≤ 0.05, unpaired Student’s t test; ***p ≤ 0.0005, paired Student’s t test).
All data are means ± SEM.
We designed a first protocol, where we clamped cells at a constant potential of −80 mV for 120 s to allow for cysteine modification by 2 mM MTSET at negative potentials, followed by testing channel function with a standard pressure-step protocol (a 5-s prepulse of +5 mmHg, followed by 200-ms steps from 0 mmHg to −70 mmHg) (Figure 4B). Identical experiments without MTSET in the buffer served as control. Mechanically activated peak current amplitudes at −60 mmHg varied among constructs, and for G2468C, they were too small (<30 pA) to reliably quantify the degree of inactivation, so that it was excluded from further analysis (Figure 4C). Interestingly, the degree of inactivation for construct V2467C was markedly affected by MTSET application (40.4% ± 8.4% with 2 mM MTSET versus 68.6% ± 3.3% with buffer alone), whereas inactivation was not significantly changed for constructs F2460C, G2463C, and Y2464C, nor for wild-type Piezo1 (Figure 4D). Importantly, when we repeated this experiment at a 10-fold lower concentration of MTSET, the extent of inactivation for construct V2467C was reversed to levels observed with buffer alone (69.6% ± 4.7% with 200 µM MTSET) (Figure 4E).
We concluded that residue V2467 can be modified by high concentration and long exposure of MTSET and asked next whether its accessibility was dependent on voltage, which could be driven by a voltage-induced conformational change of the inner pore helix. To test this, we designed a second protocol, where two test pulses (5-s prepulse at +5 mmHg, followed by 200-ms steps to −60 mmHg held at −80 mV) framed a 5-min period of incubation with 2mMMTSET or buffer alone as control, with the membrane potential during the incubation period held either at −80 mV or +60 mV (Figure 4F). Peak current amplitudes of construct V2467 were not affected by voltage during labeling with MTSET (Figures 4G). Consistent with our previous finding, when applied during a negative holding potential, MTSET reduced the degree of inactivation (56.5% ± 3.6% after MTSET labeling, and 69.3% ± 3.7% before MTSET labeling). Strikingly, MTSET failed to induce such an effect on construct V2467 at positive holding potential during the incubation time, as the degree of inactivation was unchanged under this condition (65.7% ± 2.7% after MTSET labeling, and 66.7% ± 2.6% before MTSET labeling) (Figures 4F and 4H). Altogether, these data suggest that residue V2467 in the upper part of the inner pore helix is accessible to MTSET, specifically at negative potentials but not at positive potentials.
DISCUSSION
The mechanisms for inactivation and desensitization are, in principle, understood for virtually all major ion channel families, which has provided a deep understanding and a handle to manipulate countless biological processes. Our long-term goal is to achieve the same depth of understanding for Piezo ion channels, and the first step toward it must be the identification of structures important for inactivation.
We combined previous observations and detailed analysis of Piezo1 function to narrow down and, ultimately, identify structural correlates of inactivation and its voltage dependence. The two pertinent structures that we identified are located proximal to the ion permeation pathway and within the pore itself, bringing to mind known mechanisms for inactivation in other ion channels where inactivation is mediated by direct interactions with or conformational changes of the pore domain.
For example, N-type inactivation, otherwise known as the “ball-and-chain” mechanism, has been most prominently studied in Kv and Nav channels and is described as a mechanism whereby an inactivation domain obstructs the pore and, thus, blocks ion permeation (Goldin, 2003; Hoshi et al., 1990). The facts that the CED domain is a globular structure, is located at the entry to the ion permeation pathway, and confers the characteristic time course of inactivation between Piezo1 and Piezo2 certainly resonate with an N-type mechanism. However, in our hands, Piezo1 was not amenable to the most stringent test of an N-type mechanism, which is the removal and restoration of the inactivation particle leading to a complete loss and rescue of inactivation, respectively. The CED likely provides other functionalities to Piezo1 as well, and our results that CED deletion constructs are non-functional and that at least one of them fails to target to the membrane support this possibility. In addition, the cryo-EM structural analysis of Piezo1 did not capture the CED in any obviously pore-blocking positions, calling into question whether this distinct and unusual structural fold can act as a blocking particle. Certainly, our chimeric constructs suggest that the CED can, at least allosterically, modulate inactivation kinetics.
Another prominent mechanism for channel inactivation is C-type inactivation, which involves structural changes within and around the selectivity filter and the pore entrance (Kurata and Fedida, 2006). The residues we identify to be important for voltage dependence of inactivation are also located within the pore and in tertiary structure, proximal to a residue that influences unitary conductance and reversal potential (E2133) (Coste et al., 2015). By neutralizing or reversing the charge of the residue K2479 in the inner pore helix, we were able to abolish voltage sensitivity of inactivation and set inactivation times at different values, suggesting that we have biased the channel toward distinct conformational states that transition to the inactivated state with different lifetimes. In addition, we find that MTSET has restricted access to residue V2467C, which is located in the upper pore domain, specifically at positive holding potentials. This specific result could be a consequence of electrostatic repulsion of MTSET from the membrane potential or, alternatively, from structural changes within the pore, the latter of which would be characteristic of a C-type mechanism. On the contrary, we do not find a strong dependence of inactivation on ion occupancy, another hallmark of C-type inactivation. Clearly, the exact conformational changes that occur in the pore helix remain to be studied in greater detail.
The mechanism of inactivation in Piezos may involve aspects from both N-type and C-type as well as novel forms of inactivation. Previously, we have shown that Piezo1 can adopt two inactivated states that are kinetically distinct, raising the intriguing possibility that Piezos possess two inactivation mechanisms, perhaps mediated through distinct structures (Lewis et al., 2017). Such a dual mechanism would not be unprecedented, as Shaker channels are known to undergo both N-type and C-type inactivation (Hoshi et al., 1991). It is also possible that inactivation in Piezo channels is mediated through other stimuli, including membrane tension, through mechanisms that we have not identified with our assays.
Alternatively, it also seems possible that the CED and inner pore helix may act in conjunction to mediate one common mechanism of inactivation. While we provide some evidence that the CED may not form part of the permeation pathway itself, we can speculate that highly conserved linkers may provide allosteric coupling to the pore helices, which, in turn, promote structural restriction of the permeation pathway.
Overall, our experiments reveal a mechanism by which the membrane potential of a cell directly determines its mechanical sensitivity. This modulation may be especially important for excitable cells, in which membrane potential varies quickly and over a wide range. The structures we implicated in this mechanism, the CED and the pore domain, are particularly enriched in human disease-related point mutations associated with Piezos, suggesting that they may directly affect this mechanism. Similarly, endogenous modulators of inactivation, such as pH, may act on either the CED or pore helices. Our results provide a rationale for future studies to test whether the CED can be efficiently targeted by small molecules to change overall channel function and mechanotransduction, ultimately having clear implications in addressing disease-related mutations of Piezo.
EXPERIMENTAL PROCEDURES
Construct Subcloning and Characterization
Mouse Piezo1-pIRES-EGFP in pcDNA3.1(+) was obtained from Ardem Patapoutian and previously described (Coste et al., 2012). Mouse Piezo2 was synthesized to be codon optimized for expression in human cells by GENEWIZ and ligated into pcDNA3.1(+) between restriction sites Kpn1 and Not1.
CED chimeric constructs were generated by first inserting AgeI restriction sites into regions flanking the CED of Piezo1 (after amino-acid positions S2211 and G2459) and regions flanking the CED of Piezo2 (after positions S2614 and G2733), using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies). All primers were synthesized and desalted by Sigma-Aldrich. The constructs were then digested with AgeI-HF (New England Biolabs), purified after gel electrophoresis, and re-ligated with T4 DNA ligase (NEB) to insert the Piezo2 CED into the Piezo1 backbone and to insert the Piezo1 CED into the Piezo2 backbone. Lastly, the AgeI restriction sites were removed with the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies).
Piezo1 charge neutralization and reversal constructs, as well as all cysteine mutant constructs, were generated by site-directed mutagenesis using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies).
CED deletion constructs were generated either by digesting, purifying after gel electrophoresis, and self-ligating Piezo DNA with inserted AgeI restriction sites as described for chimeras; or by using the Q5 Site-Directed Mutagenesis Kit (NEB). CED deletion constructs for Piezo1 are as listed: P2223-S2450delinsTG, P2223-P2456delinsTG, L2212-L2461delinsTG, V2226-F2449delinsTG, R2295-K2422del, R2295-K2438del, P2253-K2422del, and P2253-K2438del, where “insTG” indicates when an AgeI restriction site remained in the sequence. P2223-S2450delinsTG was cloned into the background of the previously reported α-bungarotoxin-tagged Piezo1, Piezo1-BTX-86 (Wu et al., 2016) for fluorescence imaging, or the background of the previously reported Piezo1-FLAG construct for NativePAGE electrophoresis (Syeda et al., 2016). All other deletion constructs were in wild-type Piezo1. CED deletion constructs for Piezo2 were cloned into wild-type Piezo2 and are as listed: L2495-L2735delinsTG and V2509-F2723delinsTG.
PreScission Protease sites were inserted into Piezo1 by synthesizing the Piezo1 CED flanked with AgeI restriction sites and the sequence 5′-CTGGAAGTTCTGTTCCAGGGGCCC-3′ at amino-acid positions Q2222 and P2456. AgeI sites were inserted into the Piezo1 vector as well at the described sites. Piezo1 vector and CED fragments were digested and religated with AgeI-HF restriction enzyme (NEB) as described earlier. All constructs were sequence verified by Sanger sequencing (GENEWIZ).
Cell Culture
HEK293T-P1KO cells (Piezo1 knockout human embryonic kidney cells) were obtained from Ardem Patapoutian and previously described (Dubin et al., 2017). Cell culture and transfection were handled as previously described and detailed in Supplemental Experimental Procedures (Wu et al., 2016).
Electrophysiology
Patch-clamp recordings were performed using an EPC10 amplifier and PATCHMASTER software (HEKA Elektronik). Data were sampled at 5 kHz (cel attached) or 10 kHz (whole cell) and filtered at 2.9 kHz. Buffers for whole-cell and cell-attached experiments for Piezo recordings were previously described and are detailed in Supplemental Experimental Procedures (Lewis et al., 2017).
Mechanical stimulation was applied as previously described (Coste et al., 2010; Wu et al., 2016): For force probe stimulation, cells were indented with a fire-polished glass pipette (tip diameter, ~3–5 µm) controlled by an amplifier-controlled piezo-electric driver (E625 LVPZT Controller/Amplifier; Physik Instrumente). The probe was initially positioned ~2–4 µm from the cell and advanced at 0.5 mm/ms in 1-mm increments at an 80° angle, while the cell was held at −80 mV, and the protocol was stopped and the step increment was recorded after eliciting a current of greater than 100 pA. Cells were then stimulated with a voltage-step protocol from −100 mV to +100 mV, with a single indentation stimulus of the previously recorded increment at each voltage step. For pressure-clamp stimulation, negative pressure was applied through the patch pipette with an amplifier-controlled high-speed pressure-clamp system (HSPC-1; ALA Scientific Instruments). Membrane curvature measurements were obtained as previously described (Lewis and Grandl, 2015).
For MTSET experiments, MTSET (Toronto Research Chemicals) was reconstituted in water from powder stock to a 400-mM stock solution and diluted in the cell-attached pipette buffer to a final concentration of 2 mM or 200 µM. Pipette buffer was kept on ice for a maximum for 1.5 hr during recording to minimize hydrolysis. TRPA1 experiments were performed in the whole-cell configuration with a bath buffer consisting of (in millimolar) 150 NaCl, 1 MgCl2, 1 EGTA, 10 HEPES, and 10 glucose (pH = 7.3) with NaOH and an internal buffer consisting of (in millimolar) 150 CsCl, 1 MgCl2, 10 EGTA, and 10 HEPES (pH = 7.3) with CsOH.
Data Analysis
Analysis of current was performed with Igor Pro 6.22A (WaveMetrics) and R + RStudio. Whole-cell electrophysiology recordings were analyzed for cells with a seal resistance of >500 MΩ and a series resistance of <10 MΩ. Cells with maximum currents less than 100 pA in whole-cell recordings were excluded from analysis. Cell-attached recordings were analyzed for patches with a seal resistance of >1 GΩ, and cells with maximum currents less than 30 pA were excluded from analysis for percent inactivation.
The time constant of inactivation t was obtained by fitting a single exponential curve between the peak current and the stimulus offset:
The degree of inactivation for cell-attached recordings was analyzed as previously described (Wu et al., 2016).
Gating charges, z, were calculated with a Faraday constant, F, of 96,485 s•A/mol and a gas constant, R, of 8.31 J/mol•K for a temperature, T, of 298 K. Exponential functions were fit to inactivation time constants in the entire range of reported voltages.
Statistical Analysis
Statistical analyses were performed with paired or unpaired Student’s t tests, or one-way ANOVA when comparing three or more conditions. All data are reported as mean ± SEM. Significance thresholds were set as p < 0.05, as described earlier in the text.
Supplementary Material
Highlights.
Inactivation kinetics of Piezo ion channels are continuously modulated by voltage
The C-terminal extracellular domain confers kinetics of inactivation
A single charged residue in the pore helix confers voltage sensitivity
Substituted cysteine modification in the upper pore helix is voltage dependent
Acknowledgments
This study was supported by NIH grants 1F31NS093777 (to J.W.) and F32NS094088 (to A.H.L.). We thank Jason O. Sosa-Pagan, Son C. Le, and Malcolm F. McDonald for performing preliminary experiments and Lindsey Glickfeld for providing feedback on the manuscript.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and one figure and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.10.120.
AUTHOR CONTRIBUTIONS
J.W. designed the study, performed experiments, analyzed data, and wrote the manuscript. M.Y. and A.H.L. performed electrophysiology experiments and analyzed data. A.N.M. performed protein purification and gel electrophoresis. B.K. performed imaging experiments. J.G. designed the study and wrote the manuscript.
References
- Albuisson J, Murthy SE, Bandell M, Coste B, Louis-dit-Picard H, Mathur J, Fénéant-Thibault M, Tertian G, de Jaureguiberry JP, Syfuss PY, et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 2013;4:1884. doi: 10.1038/ncomms2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper SL. Genetic diseases of PIEZO1 and PIEZO2 dysfunction. Curr. Top. Membr. 2017;79:97–134. doi: 10.1016/bs.ctm.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Andolfo I, Alper SL, De Franceschi L, Auriemma C, Russo R, De Falco L, Vallefuoco F, Esposito MR, Vandorpe DH, Shmukler BE, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121:3925–3935. S1–S12. doi: 10.1182/blood-2013-02-482489. [DOI] [PubMed] [Google Scholar]
- Bae C, Gnanasambandam R, Nicolai C, Sachs F, Gottlieb PA. Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc. Natl. Acad. Sci. USA. 2013;110:E1162–E1168. doi: 10.1073/pnas.1219777110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae C, Sachs F, Gottlieb PA. Protonation of the human PIEZO1 ion channel stabilizes inactivation. J. Biol. Chem. 2015;290:5167–5173. doi: 10.1074/jbc.M114.604033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012;483:176–181. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Houge G, Murray MF, Stitziel N, Bandell M, Giovanni MA, Philippakis A, Hoischen A, Riemer G, Steen U, et al. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc. Natl. Acad. Sci. USA. 2013;110:4667–4672. doi: 10.1073/pnas.1221400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Murthy SE, Mathur J, Schmidt M, Mechioukhi Y, Delmas P, Patapoutian A. Piezo1 ion channel pore properties are dictated by C-terminal region. Nat. Commun. 2015;6:7223. doi: 10.1038/ncomms8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CD, Bae C, Ziegler L, Hartley S, Nikolova-Krstevski V, Rohde PR, Ng CA, Sachs F, Gottlieb PA, Martinac B. Removal of the mechanoprotective influence of the cytoskeleton reveals PIEZO1 is gated by bilayer tension. Nat. Commun. 2016;7:10366. doi: 10.1038/ncomms10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Schmidt M, Mathur J, Petrus MJ, Xiao B, Coste B, Patapoutian A. Inflammatory signals enhance piezo2-mediated mechanosensitive currents. Cell Rep. 2012;2:511–517. doi: 10.1016/j.celrep.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Murthy S, Lewis AH, Brosse L, Cahalan SM, Grandl J, Coste B, Patapoutian A. Endogenous Piezo1 can confound mechanically activated channel identification and characterization. Neuron. 2017;94:266–270. e3. doi: 10.1016/j.neuron.2017.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Li W, Zhao Q, Li N, Chen M, Zhi P, Li R, Gao N, Xiao B, Yang M. Architecture of the mammalian mechanosensitive Piezo1 channel. Nature. 2015;527:64–69. doi: 10.1038/nature15247. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol. 2003;13:284–290. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Gottlieb PA, Bae C, Sachs F. Gating the mechanical channel Piezo1: a comparison between whole-cell and patch recording. Channels (Austin) 2012;6:282–289. doi: 10.4161/chan.21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honoré E, Patel AJ, Chemin J, Suchyna T, Sachs F. Desensitization of mechano-gated K2P channels. Proc. Natl. Acad. Sci. USA. 2006;103:6859–6864. doi: 10.1073/pnas.0600463103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Kamajaya A, Kaiser JT, Lee J, Reid M, Rees DC. The structure of a conserved piezo channel domain reveals a topologically distinct β sandwich fold. Structure. 2014;22:1520–1527. doi: 10.1016/j.str.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata HT, Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Lewis AH, Grandl J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. eLife. 2015;4:e12088. doi: 10.7554/eLife.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Cui AF, McDonald MF, Grandl J. Transduction of repetitive mechanical stimuli by Piezo1 and Piezo2 ion channels. Cell Rep. 2017;19:2572–2585. doi: 10.1016/j.celrep.2017.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs V, Mathur J, Mao R, Bayrak-Toydemir P, Procter M, Cahalan SM, Kim HJ, Bandell M, Longo N, Day RW, et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat. Commun. 2015;6:8329. doi: 10.1038/ncomms9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic S, Nakatani M, Baba Y, Nelson AM, Marshall KL, Wellnitz SA, Firozi P, Woo SH, Ranade S, Patapoutian A, Lumpkin EA. Epidermal Merkel cells are mechanosensory cells that tune mammalian touch receptors. Nature. 2014;509:617–621. doi: 10.1038/nature13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin MJ, Beck AE, Chong JX, Shively KM, Buckingham KJ, Gildersleeve HI, Aracena MI, Aylsworth AS, Bitoun P, Carey JC, et al. University of Washington Center for Mendelian Genomics. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 2014;94:734–744. doi: 10.1016/j.ajhg.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo M, Fujita A, Saito Y, Komaki H, Ishiyama A, Takeshita E, Kojima E, Koichihara R, Saito T, Nakagawa E, et al. A family of distal arthrogryposis type 5 due to a novel PIEZO2 mutation. Am. J. Med. Genet. A. 2015;167A:1100–1106. doi: 10.1002/ajmg.a.36881. [DOI] [PubMed] [Google Scholar]
- Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. USA. 2014a;111:10347–10352. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, Mathur J, Bégay V, Coste B, Mainquist J, et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature. 2014b;516:121–125. doi: 10.1038/nature13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, Martins JR, Jodar M, Moro C, Offermanns S, Feng Y, et al. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep. 2015;13:1161–1171. doi: 10.1016/j.celrep.2015.09.072. [DOI] [PubMed] [Google Scholar]
- Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, Patapoutian A. Piezo1 channels are inherently mechanosensitive. Cell Rep. 2016;17:1739–1746. doi: 10.1016/j.celrep.2016.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SH, Ranade S, Weyer AD, Dubin AE, Baba Y, Qiu Z, Petrus M, Miyamoto T, Reddy K, Lumpkin EA, et al. Piezo2 is required for Merkel-cell mechanotransduction. Nature. 2014;509:622–626. doi: 10.1038/nature13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SH, Lukacs V, de Nooij JC, Zaytseva D, Criddle CR, Francisco A, Jessell TM, Wilkinson KA, Patapoutian A. Piezo2 is the principal mechanotransduction channel for proprioception. Nat. Neurosci. 2015;18:1756–1762. doi: 10.1038/nn.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Goyal R, Grandl J. Localized force application reveals mechanically sensitive domains of Piezo1. Nat. Commun. 2016;7:12939. doi: 10.1038/ncomms12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lewis AH, Grandl J. Touch, tension, and transduction – the function and regulation of Piezo ion channels. Trends Biochem. Sci. 2017;42:57–71. doi: 10.1016/j.tibs.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarychanski R, Schulz VP, Houston BL, Maksimova Y, Houston DS, Smith B, Rinehart J, Gallagher PG. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood. 2012;120:1908–1915. doi: 10.1182/blood-2012-04-422253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.