

Abstract
Many microorganisms use flavin-dependent thymidylate synthase (FDTS) to synthesize the essential nucleotide 2′-deoxythymidine-5′-monophosphate (dTMP) from 2′-deoxyuridine-5′-monophosphate (dUMP), 5,10-methylenetetrahydrofolate (CH2THF), and NADPH. FDTSs have a structure that is unrelated to the thymidylate synthase used by humans, and a very different mechanism. Here we report NMR evidence that FDTS ionizes N3 of dUMP using an active-site arginine. The ionized form of dUMP is largely responsible for the changes in the flavin absorbance spectrum of FDTS upon dUMP binding. dUMP analogs also suggest that the phosphate of dUMP acts as the base that removes the proton from C5 of the dUMP-methylene intermediate in the FDTS-catalyzed reaction. These findings establish additional differences between the mechanisms of FDTS and human thymidylate synthase.
TOC image
INTRODUCTION
2′-Deoxythymidine-5′-monophosphate (dTMP) – an essential precursor for the biosynthesis of DNA – is formed by the reductive methylation of 2′-deoxyuridine-5′-monophosphate (dUMP) to form dTMP by thymidylate synthases (TSs). Two classes of TSs are known. Classic TSs are homodimeric enzymes that use 5,10-methylenetetrahydrofolate (CH2THF) as both the carbon-donor and reductant to form dTMP from dUMP without the use of a prosthetic group.1 Flavin-dependent thymidylate synthases (FDTSs), on the other hand, are tetrameric and use a flavin adenine dinucleotide (FAD) prosthetic group to synthesize dTMP from dUMP, CH2THF and NADPH.2 Whereas the folate becomes dihydrofolate (DHF) in the classic TS reaction, the folate remains in the fully-reduced tetrahydrofolate (THF) state in the reaction catalyzed by FDTSs. The electrons required to form dTMP come from NADPH by way of the FAD prosthetic group in FDTSs instead of coming from the folate as in classic TSs.
The mechanism of the well-studied classic TS is worth considering in detail because it too converts dUMP to dTMP using CH2THF and, therefore, faces similar chemical tasks. Classic TS has been studied for decades and its chemical mechanism is well-understood (Figure 1A).1 Classic TS first activates dUMP through a Michael-like addition of an active-site cysteine to C6 of dUMP (step 1). The resulting enolate then attacks the iminium form of CH2THF, forming a bridged intermediate between the cysteine, dUMP, and CH2THF (step 2). Upon deprotonation at C5 of the pyrimidine, the adduct breaks down into THF and an exocyclic methylene intermediate still covalently attached to the active-site cysteine (step 3). Finally, a hydride is transferred from THF to the methylene of the intermediate to eliminate the cysteine, forming dTMP (step 4).
Figure 1.
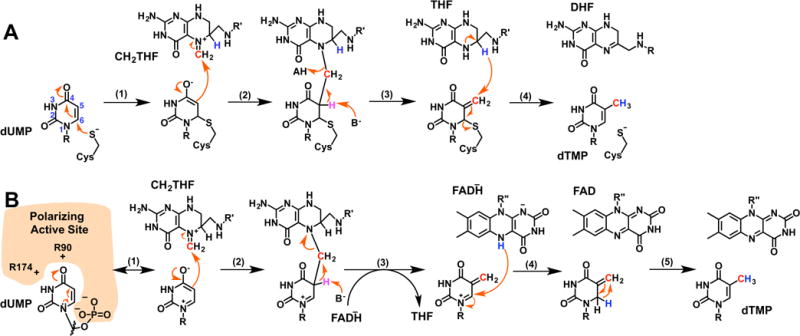
Thymidylate synthase chemical mechanisms. (a) the mechanism for classic TS using an enzymatic cysteine-nucleophile.1 (b) a proposed mechanism for FDTS using an electrostatically polarizing active site to activate dUMP.6
In contrast, the chemical mechanism of FDTS has not been fully determined. A major question is the mechanism of dUMP activation. Unlike classic TS, FDTS does not contain an active-site cysteine to activate dUMP through a Michael-like addition, and mutagenesis has ruled out all other potential enzymatic nucleophiles.3 Hydride transfer from reduced FAD has been proposed to activate dUMP;3,4 however, this is unlikely since 5-fluoro-2′-deoxyuridine-5′-monophosphate (5F-dUMP) cannot oxidize reduced FDTS in the presence of CH2THF,5 and stopped-flow experiments on the oxidative half-reaction indicate multiple intermediates prior to flavin oxidation.6
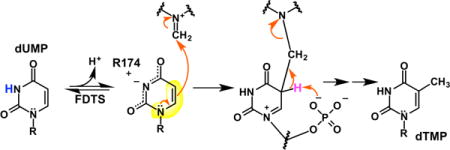
The electrostatically polarizing active site was recently proposed to activate dUMP.6,7 In this mechanism, the arrangement of two arginines (R90 and R174 in Thermotoga maritima FDTS) near O4 of dUMP and the phosphate of dUMP near N1 stabilizes the resonance form of dUMP shown in Figure 1B (step 1). The enolate of this resonance-form was proposed to be nucleophilic at C5, attacking CH2THF, forming a bridged intermediate between dUMP and CH2THF (step 2). Upon deprotonation of C5 by an unidentified base, the bridged intermediate breaks down into THF and an exocyclic methylene intermediate (step 3). A hydride is then transferred from reduced FAD to C6 of the intermediate (step 4).3 The exocyclic methylene tautomer of dTMP then undergoes a 1,3-hydride shift to form dTMP (step 5). Very recently, this mechanism was modified by the detection of a flavinmethylene-deoxynucleotide adduct, suggesting that CH2THF actually transfers the methylene carbon to reduced FAD,8 and this iminium adduct, rather than the one shown in Figure 1B, transfers carbon to the nucleotide. Regardless, the mechanism of uracil activation remains of critical importance.
While the mechanism for dUMP activation presented in Figure 1B is consistent with most published data, it has not been completely verified. Here, 13C-NMR was used to test the hypothesis that dUMP is activated by an electrostatically polarizing active site. Polarization turns out to be more extreme than previously proposed (Figure 1B); the enzyme actually deprotonates N3 of dUMP using an active site arginine. Additional evidence suggests that the phosphate of dUMP acts as the general base that deprotonates C5 of the dUMP-methylene adduct in the reaction catalyzed by FDTS.
RESULTS
A mechanism was proposed where dUMP is activated as a nucleophile by three charged groups in the active site of FDTS oriented around polar ends of the uracil ring – R90, R174, and the phosphate of dUMP (Figure 2).6 These charges were proposed to enhance the contribution of the enolate resonance form of the uracil ring, increasing nucleophilicity at C5 (Figure 1B). Mutagenesis supported this idea6 – the rate constants for flavin oxidation of the R90A variant and the R174A variant were 8-fold and 3000-fold smaller, respectively, than wild-type FDTS in oxidative half-reactions with dUMP and CH2THF.
Figure 2.
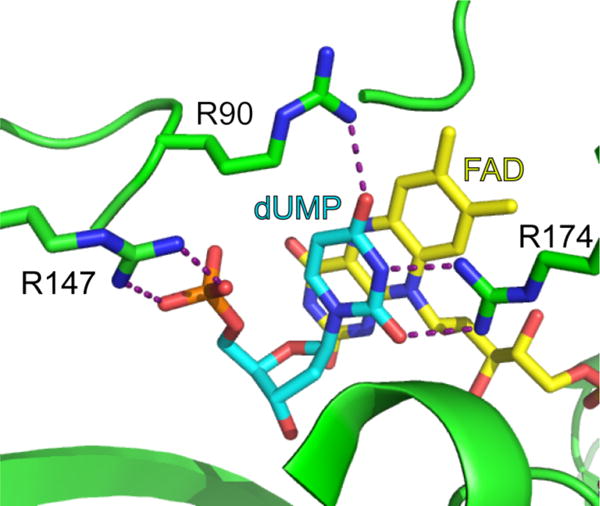
Active site of flavin-dependent thymidylate synthase (PDB code: 1O26). In the mechanism shown in Figure 1B, the arrangement of R90, R174, and the phosphate of dUMP (cyan) are proposed to stabilize a resonance form of dUMP with enhanced nucleophilicity at C5. Also, note the close proximity of the phosphate oxygens to C5 of the uracil.
Binding
A large change in the flavin UV/visible absorbance spectrum of oxidized WT FDTS occurs upon binding of dUMP (Figure 3A) – the extinction coefficients for the peaks at 375 nm and 450 nm increase and decrease, respectively. This spectral change can be used to track dUMP binding in titrations, allowing the dissociation constant of dUMP for FDTS to be determined. dUMP bound stoichiometrically to WT FDTS, providing only an estimate of the Kd (30 nM; Table 1). 2′-Deoxyuridine (dU) – which lacks the 5′ phosphate group – induces a change similar to that by dUMP in the flavin absorbance spectrum of WT FDTS upon binding to the enzyme (SI Figure 1A). The dissociation constant for dU binding to oxidized WT FDTS was 78 μM (Table 1). The R90A variant and the R174A variant bound dUMP with dissociation constants of 15 μM and 109 μM, respectively (Table 1). The flavin spectral change upon binding of dUMP to the R90A variant was similar to dUMP binding to WT FDTS (SI Figure 1B). However, the spectral change in the flavin of the R174A variant was clearly different from the rest of the enzymes tested (Figure 3B) – the 450 nm peak blue-shifted slightly and only the extinction coefficient of the 450 nm peak decreased upon binding dUMP.
Figure 3.
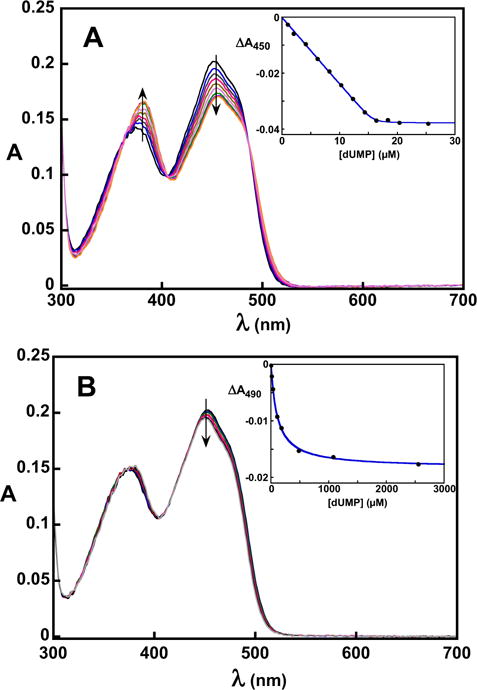
Spectrophotometric titrations of WT (A) and R174A (B) FDTS with dUMP in 0.1 M Tris-HCl, pH 8 at 25 °C. The insets show the change in absorbance as a function of ligand concentration. The spectral change for dUMP binding to R174A FDTS was noticeably different from other FDTS variants. Fitting to Eq. 1 gives a Kd of 30 nM for dUMP binding to WT FDTS. Fitting to a square hyperbola gives a Kd of 109 μM for dUMP binding to R174A FDTS.
Table 1.
Ligand Binding to WT and Variant FDTSs
| enzyme + ligand complex | Kd (μM)a |
|---|---|
| WT + dUMP | 0.03b ± 0.02 |
| WT + dU | 78 ± 5 |
| R90A + dUMP | 14 ± 2 |
| R174A + dUMP | 109 ± 6 |
| WT + dUMS | 0.17b ± 0.08 |
0.1 M Tris-HCl, pH 8 at 25 °C
Estimate from fitting to Equation 1. Errors are standard errors on the fits.
NMR
Polarization of the uracil ring of dUMP was probed by comparing the 13C-NMR spectra of 13C/15N-labeled dUMP in aqueous solution at pH 8 with 13C/15N-labeled dUMP bound to FDTS. Due to the size of the enzyme tetramer (110 kDa), multinuclear NMR experiments on the complex were not possible. Instead, 1-dimensional 13C-NMR spectra were collected of labeled dUMP bound to WT FDTS (Figure 4). Spectra were recorded at 45 °C. An excess of FDTS (1.5 mM) over dUMP (500 μM), well above the Kd of dUMP, ensured that the signals resulted from dUMP bound to the enzyme. The resonances associated with the protein were subtracted from the 13C-NMR spectra of the 13C-labeled dUMP-FDTS complexes by using the spectrum of the unlabeled complex. The peaks associated with dUMP in the ligand-protein complex were substantially broadened, confirming binding.
Figure 4.
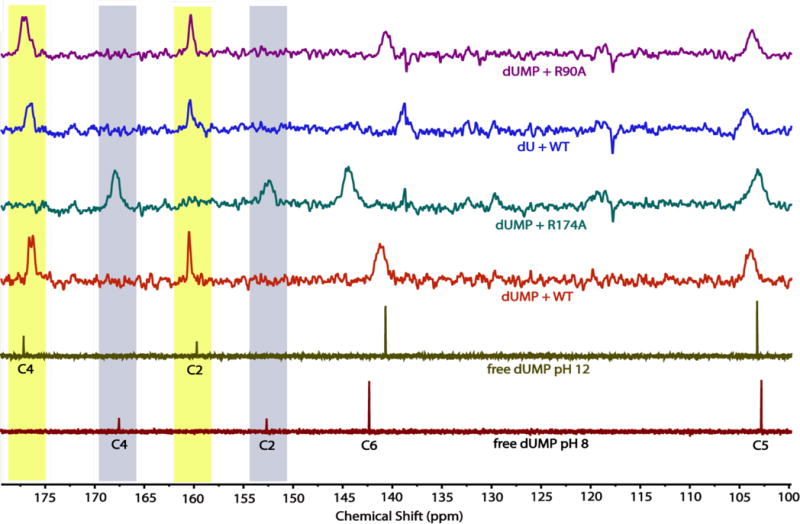
The 13C-NMR spectra of uracil carbons of dUMP/dU free in solution or bound to WT and variant FDTSs at 45 °C. All of the ligand-FDTS complexes were at pH 8. The C4 and C2 signals for dUMP shifted upfield ~8 ppm when bound to WT FDTS, having nearly identical chemical shifts as free dUMP at pH 12. Mutagenesis of R174 – which is 2.8 Å from N3 of dUMP in the dUMP-WT FDTS structure – to alanine eliminated the changes in chemical shifts of the dUMP-FDTS complex. In contrast, mutagenesis of R90 to alanine or removal of the phosphate of dUMP had little effect on the 13C-NMR spectrum of the uracil carbons in the complex. Highlighted in maize and blue, respectively, are the chemical shifts of the carbonyl carbons of N3-ionized and N3-unionized uracil.
Binding of dUMP to WT FDTS produced substantial changes in the 13C chemical shifts of the uracil ring (Figure 4 and Table 2). Most notably, the 13C-NMR signals of the two carbonyl carbons, C2 and C4, shifted downfield by approximately 8 ppm each. dUMP bound to WT FDTS had the same 13C spectrum whether the flavin was in the oxidized form or the hydroquinone form (SI Figure 2), indicating that the redox state of the flavin does not affect the electrons of the uracil of dUMP. It is worth noting that there is no evidence in the NMR spectrum of the reduced enzyme complex for rapid chemical exchange between uracil and dihydrouracil, which would be expected if the uracil moiety were rapidly reduced and re-oxidized by the flavin as invoked recently in a mechanism that proposed hydride transfer to C6 activated uracil as a nucleophile.4
Table 2.
Chemical Shifts for Uracil Carbonsa
| C2 (ppm) | C4 (ppm) | C5 (ppm) | C6 (ppm) | |
|---|---|---|---|---|
| unbound dUMP pH 8 | 152.7 | 167.6 | 102.8 | 142.3 |
| unbound dUMP pH 12 | 159.7 | 177.2 | 103.2 | 140.7 |
| dUMP + WT | 160.5 | 176.4 | 103.9 | 141.2 |
| dUMP + R174A | 152.6 | 167.9 | 103.2 | 144.4 |
| dUMP + R90A | 160.3 | 177.0 | 103.7 | 140.7 |
| dU + WT | 160.4 | 176.6 | 104.3 | 138.8 |
45 °C, pH 8 unless stated otherwise.
The polarization hypothesis was further tested by individually removing the charged groups interacting with the uracil ring and obtaining chemical shifts. To test the importance of the phosphate of dUMP, a 13C-NMR spectrum was also collected of dU bound to WT FDTS. The 13C chemical shifts of the uracil in unbound dU are identical to the chemical shifts of the uracil in unbound dUMP. The 13C spectrum of dU bound to WT FDTS was nearly identical to the 13C spectrum of dUMP bound to WT FDTS (Figure 4); the only notable difference was that the signal for C6 (which is 3.3 Å away from the phosphate of dUMP in the dUMP-FDTS structure) was ~2 ppm lower than the C6 resonance of the dUMP-FDTS complex. Likewise, dUMP bound to R90A FDTS also had 13C chemical shifts that were similar to dUMP bound to WT FDTS. However, dUMP bound to R174A FDTS had chemical shift values that were similar to those of free dUMP at pH 8 (Figure 4). The peaks were still broadened in the complex, indicating that dUMP was bound to the R174A variant.
Ionization of bound uracil explains the observed chemical shifts. The values of the 13C-chemical shifts of dUMP bound to WT FDTS are nearly identical to those of unbound dUMP at pH 12, well above the pKa for N3 of dUMP (9.39), showing that N3 of dUMP is ionized when bound at pH 8 (Figure 4). The 13C-NMR spectra at pH 8 of free dUMP and dUMP bound to WT FDTS and the variants show that R174 is largely responsible for producing the changes in the chemical shifts of the uracil of dUMP bound to FDTS. The crystal structure of the dUMP-WT FDTS complex10 shows that R174 is positioned 2.8 Å away from N3 of dUMP (Figure 2), consistent with the strong hydrogen bond that would be expected between a guanidinium cation and a uracil anion. Thus the enzyme decreases the pKa of N3 from 9.3 in aqueous solutions9 to well below 8 by the interaction of R174.
Linkage of Binding and Deprotonation
NMR spectroscopy showed that the enzyme binds deprotonated uracil moieties of ligands, and this requires R174. Deprotonation of uracil could be accomplished in two ways. R174 could act as a base if it is neutral (deprotonated) in the free enzyme in an internal proton transfer from dUMP. Several instances of arginine acting as a base have been documented in enzymology.11 Alternatively, if the side-chain is in the typical cationic (protonated) state in the free enzyme, then the N3 proton must be expelled into solution upon dUMP binding. To distinguish these possibilities, a solution of enzyme (10 μM in active sites) in unbuffered 50 mM NaCl, pH 8.0, and 20 μM phenol red was titrated with unbuffered dUMP in 50 mM NaCl, pH 8.0. Phenol red, a pH indicator dye, has an absorbance peak of high extinction at 588 nm when deprotonated; protonating the dye causes a loss of this absorbance. Absorbance spectra recorded after each addition of dUMP (Figure 5) showed that binding of dUMP was accompanied by the decrease of the absorbance of deprotonated phenol red, indicating the expulsion of a proton upon binding of dUMP. Thus, R174 does not act as a base and is likely protonated in the free enzyme.
Figure 5.
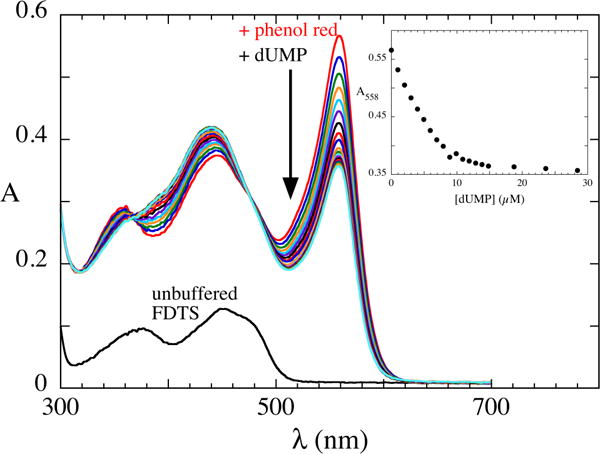
Proton expulsion upon dUMP binding. Phenol red was added to unbuffered FDTS (10 μM in FAD) in 50 mM NaCl, pH 8.0. Aliquots of concentrated dUMP at pH 8.0 was added and spectra were recorded. Proton release upon dUMP binding was evident by the decrease in absorbance of deprotonated phenol red. The inset shows that proton releases ceases at ~1:1 stoichiometry of dUMP:FAD.
Structures
In order to confirm that dUMP and dU bind in a catalytically competent conformation in the active site variants of FDTS, the crystal structures were determined of dUMP-bound complexes of the R90A and R174A FDTS variants and the dU complex of WT FDTS (SI Table 1). In all three cases the nucleotide/nucleoside binds in the same position as in the WT FDTS-dUMP complex (Figure 6). The active sites are identical in all of the structures with one notable exception. In the dU-FDTS structure R147 (which forms a salt bridge with the phosphate of dUMP) is flipped away from the phosphate binding site (SI Figure 3) and a polyethylene glycol molecule fills the space.
Figure 6.
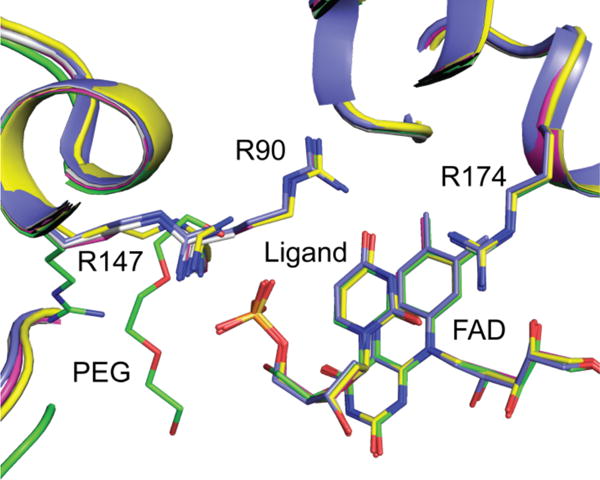
Overlay of WT and variant structures. dUMP/dU/dUMS bind in the same position and orientation in all of the active site variants used in this study. dUMP + WT (PDB code: 1O26), blue; dU + WT, green; dUMP + R90A, gray; dUMP + R174A, magenta; dUMS + WT, maize.
Lack of Reactivity of dUMP Analogs
In addition to the two arginine variants, dU – which still binds to the WT enzyme – failed to oxidize reduced WT FDTS in the presence of CH2THF and could not be converted into 2′-deoxythymidine (dT), indicating that the phosphate of dUMP is needed for the reaction (Figure 7). However, the NMR spectrum shows that the phosphate of dUMP does not play a role in ionizing the uracil ring of dUMP because dU ionizes upon binding (Figure 4). In addition, the crystal structure of the dU-FDTS complex shows that dU binds in a very similar manner to dUMP, with no difference in the position of the uracil moieties (Figure 6). Therefore it appears that the phosphate participates chemically in a step of the oxidative half-reaction that follows the deprotonation of dUMP.
Figure 7.
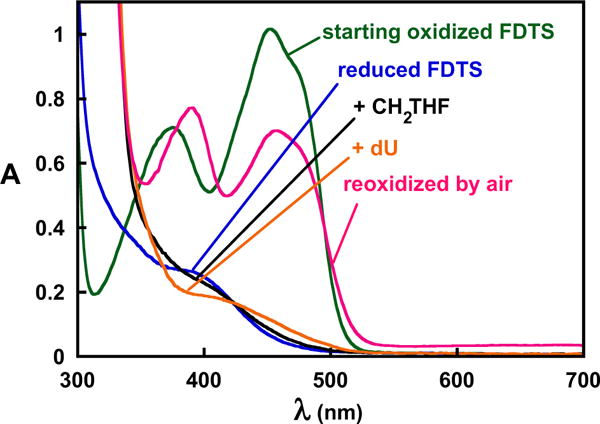
dU failed to oxidize reduced FDTS in the presence of CH2THF. Anaerobic oxidized FDTS (green) in 0.1 M Tris-HCl, pH 8 at 25 °C was titrated stoichiometrically with dithionite to reduce the flavin (blue). Anaerobic addition of CH2THF perturbed the spectrum, indicating that it formed a complex with the enzyme (black). Anaerobic addition of dU to the reduced FDTS-CH2THF complex perturbed the spectrum further but failed to oxidize the flavin (orange), indicating that the phosphate of dUMP plays an essential role in the FDTS-catalyzed reaction. The flavin could then be re-oxidized by air (red).
Any mechanism for FDTS requires a base to remove the proton at C5 of the dUMP-methylene-bridged intermediate. However, the base in the mechanism of FDTS is currently unknown. The base would have to be on the opposite face of the uracil ring from the side that reacts with the methylene. The methylene is proposed to be transferred to the uracil via the isoalloxazine of FAD;8 thus, the base would have to be on the side of the uracil opposite the flavin in Figure 2.12 In the dUMP-WT FDTS structure no side-chains are properly positioned to act as a base. However, dUMP is bound in an unusual curled conformation where one of the phosphate oxygens of dUMP is 3.4 Å away from C5, close enough for the phosphate to act as a general base8.
To test whether the phosphate might be the base, an analog of dUMP was synthesized with the phosphate replaced by sulfate – 2′-deoxyuridine-5′-monosulfate (dUMS). Sulfate is structurally similar to phosphate, but the pKa1 for sulfate is ~10 units lower than the pKa2 for phosphate,13 making it a very poor general base. dUMS binds to oxidized FDTS with an affinity similar to that of dUMP and produced a similar change in the flavin UV/visible absorbance spectrum upon binding to FDTS (Table 1, Figure 8A). The crystal structure of the dUMS-FDTS complex at 1.9 Å resolution showed that dUMS binds in the same position as dUMP (Figure 6 and SI Figure 4), without affecting any protein residues. However, dUMS was unable to oxidize reduced FDTS in the presence of CH2THF (Figure 8B), and no 2′-deoxythymidine-5′-monosulfate was detected afterwards by HPLC (data not shown).
Figure 8.
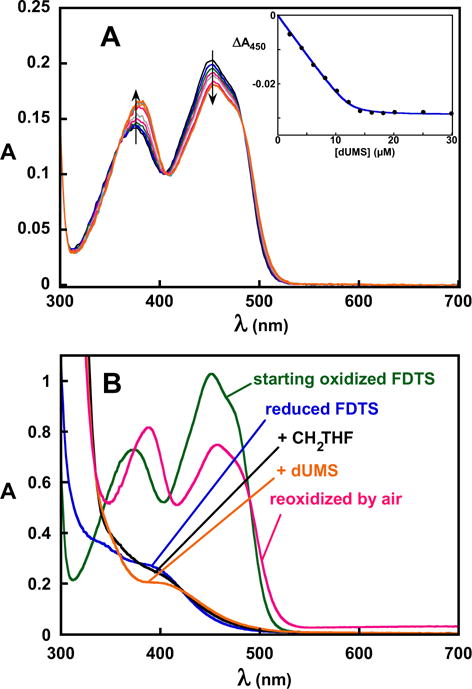
Binding and reactivity of dUMS in 0.1 M Tris-HCl, pH 8 at 25 °C. A, spectrophotometric titration of WT FDTS with dUMS. dUMS produced a flavin spectral change similar to that produced by dUMP. Fitting to Eq. 1 gave a Kd of 170 nM. B, dUMS failed to oxidize reduced FDTS in the presence of CH2THF. Anaerobic oxidized FDTS (green) was titrated stoichiometrically with dithionite to reduce the flavin (blue). Anaerobic addition of CH2THF perturbed the spectrum of the flavin (black), indicating that it formed a complex with the enzyme. Anaerobic addition of dUMS to the reduced FDTS-CH2THF complex also perturbed the spectrum (orange), indicating binding, but it failed to oxidize the enzyme. The flavin was then re-oxidized by exposure to air (red).
DISCUSSION
The 13C chemical shift values for the uracil carbons of dUMP bound to FDTS at pH 8 are nearly identical to those of unbound dUMP at pH 12 (see Figure 4). At pH 12, dUMP in water is deprotonated (pKa for N3 is 9.39). This result suggests that N3 of dUMP is also fully ionized when bound to FDTS at pH8. R174 in T. maritima FDTS appears to be solely responsible for causing this ionization. When dUMP is bound to R174A FDTS, the chemical shifts of the uracil carbons are nearly identical to those of unbound dUMP at pH 8, yet the chemical shifts of dUMP bound to R90A FDTS are nearly identical to dUMP bound to WT FDTS. In order for the uracil moiety to be fully ionized (within the limit of detection) at pH 8, the positively charged R174 must stabilize the anion by at least 2.8 kcal mol−1 in order to lower the pKa of N3 by more than two units.
Two mechanisms may be imagined for generating dUMP deprotonated at N3. Neutral R174 could deprotonated dUMP after binding. Several enzymes use arginine as a base.11 Alternatively, if R174 is positively charged in the free enzyme, the binding of dUMP would be followed by expulsion of a proton into solution from the initial complex, which was, in fact, detected using a pH-indicator dye.
The large change in the flavin absorbance spectrum of WT FDTS upon binding dUMP is likely due to the N3-ionized form of the uracil stacking against the isoalloxazine of the flavin because it was observed in each complex where ionization was verified by NMR: dUMP-WT, dU-WT, and dUMP-R90A. However, the absorbance spectral change upon binding dUMP by R174A FDTS is significantly different, indicating that the environment of the flavin is different.
Charges also play a significant role in the binding of dUMP by FDTS. Removal of the phosphate or substitution of the two arginines with alanine each cause a substantial lowering of the binding affinity of FDTS for dUMP and dU. dU binds to WT FDTS as much as 5300-fold weaker than dUMP, corresponding to a loss of ~5.2 kcal/mol of binding energy. The phosphate of dUMP forms a salt bridge with R147 and hydrogen bonds with S88 and Q75, interactions which are unavailable with dU. dUMP binds to R90A FDTS ~670-fold weaker than WT, corresponding to a loss of ~4.0 kcal/mol of binding energy. R90 would form a hydrogen bond with the partially negatively charged O4. Finally, dUMP binds to R174A FDTS ~7300-fold weaker than WT FDTS, corresponding to a loss of ~5.4 kcal/mol of binding energy. R174 is positioned to form a salt bridge with N3 of the ionized form of dUMP. Substitution of R174 with alanine removes this interaction.
Besides its important role in binding, the phosphate of dUMP appears to act as the base that deprotonates pyrimidine C5 of the dUMP-methylene adduct during carbon transfer, explaining why neither dU nor dUMS cause oxidation of the reduced flavin of FDTS in the presence of CH2THF. The absence of the phosphate in the complexes of these analogs does not alter the position of the uracil moiety, as evidenced by their crystal structures; does not prevent deprotonation of N3, as evidenced by the NMR spectra of the complexes; and the influence of the anionic uracil on the isoalloxazine remains the same, as evidenced by the unchanged perturbations to the isoalloxazine absorbance spectra. Thus, the dU and dUMS complexes differ from the dUMP complex only in the absence of an anionic phosphate oxygen, an oxygen with a pKa of about 7. The pKa of the sulfate of dUMS is many units lower than that of phosphate, making base catalysis inaccessible to dUMS, while dU has no oxygen at all in a position to act as a base. A role as a base also rationalizes the unusual conformation of dUMP bound to FDTS which, to our knowledge, is unique among nucleotide-binding proteins.
A new mechanism for dUMP activation based on ionization of N3 follows from a modification of the mechanism6 shown in Figure 1B. The negative charge of the ionized nitrogen produced when dUMP binds FDTS delocalizes onto O2 and O4 (Figure 9). This enhances nucleophilicity at C6 by partially isolating electronically the enamine substructure within uracil. Enamines are inherently nucleophilic on the β-carbon, but will be less so if substituted with electron-withdrawing groups on the nitrogen or β-carbon, as is the enamine in uracil. Delocalizing the charge from ionized N3 onto the C2 carbonyl lessens the delocalization of the lone-pair of N1 towards this carbonyl. Likewise, delocalization of the charge of N3 onto the C4 carbonyl prevents the delocalization of electron density away from C6. Both of these effects enhance the chemical behavior of the enamine substructure by making it more electronically isolated, increasing nucleophilicity at C5, leading to direct attack on the electrophilic imine on the flavin8 generated from the carbon donated by CH2THF (Figure 9). Ionization of N3 is clearly important for chemistry – the observed rate constant for flavin oxidation for the R174A variant is 3000-fold lower than that of wild-type,6 and the crystal structure shows this is not due to improper positioning of dUMP in the active site. A precedent for activation of dUMP by deprotonation rather than attack by Michael-nucleophiles for reaction with electrophiles is the reaction of dUMP with formaldehyde under basic conditions to make 5-hydroxymethyl dUMP.14,15
Figure 9.
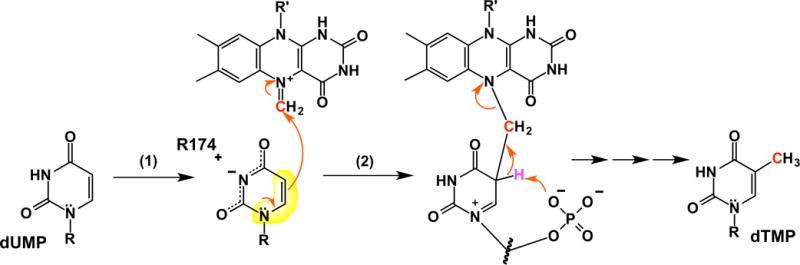
Proposed mechanism for the reaction of FDTS. dUMP is activated by deprotonating N3 (1). The negative charge delocalizes onto O4 and O2, isolating electronically the enamine substructure (highlighted in yellow), enhancing its nucleophilicity for reaction with the methylene carbon of the flavin-iminium adduct proposed.8 Removal of the C5 proton of the bridged adduct by the phosphate of dUMP leads, ultimately, to production of dTMP and oxidized FAD.
The findings presented here establish that FDTS uses a novel mechanism to methylate a pyrimidine that does not use a Michael-like addition to make an adduct. Activation of uracil by ionization of N3 has never been reported, but might be used by other enzymes that transfer an electrophile to uracil. It is noteworthy that the reaction of an anionic pyrimidine intermediate with an electrophile has been observed in an unrelated reaction; uracil DNA glycosylases use a strong hydrogen bond with O2 to stabilize anionic uracil deprotonated at N1, 16–19 suggesting that anionic pyrimidine intermediates might be more common than is currently appreciated. The mechanism for dUMP activation by FDTS is clearly distinct from the mechanism of activation by classic TS, which activates dUMP through a Michael-like addition of an enzymatic cysteine. Thus enzymes have evolved independently that exploit the chemical pliability of the uracil heterocycle. The classic TSs activate C6 by creating an enolic carbon, while FDTS electronically isolates the enamine substructure. In addition, the phosphate of dUMP appears to be important for catalysis by FDTS, which is not the case with classic TS.
CONCLUSIONS
FDTS ionizes N3 of dUMP, which is largely caused by an active site arginine, providing a new mechanism for dUMP activation. The phosphate of dUMP is crucial for flavin oxidation, suggesting that it acts as the base that deprotonates C5 of the dUMP-methylene adduct. Notably, both ionization of N3 and acid-base catalysis by the phosphate of dUMP are not implicated in the mechanism used by classic TS, demonstrating the evolution of two independent chemical routes for synthesizing dTMP.
EXPERIMENTAL SECTION
Enzyme Preparations
Recombinant WT and mutagenized FDTSs from T. maritima were purified as previously reported,6 followed by formation of apo-protein by treatment with concentrated sodium chloride10 and reincorporation of exogenous FAD after desalting.
Synthesis of 13C/15N-labeled dUMP and dU
5 mg commercially obtained 13C/15N-labeled dCMP was deaminated by incubation with 1 M sodium nitrite in 1 mL aqueous 3 M ammonium acetate, pH 4 for 24 hours at 37 °C. Conversion of dCMP to dUMP was confirmed by a shift in the UV/Vis λmax from 271 nm to 261 nm. The 13C/15N-labeled dUMP was purified on a Shimadzu HPLC with a UV/Vis diode array detector at room temperature on a 5 μm, 25 cm × 4.6 mm Supelcosil LC-18-DB column using isocratic aqueous 10 mM ammonium acetate, pH 5 at 1 mL/min. The purified 13C/15N-labeled dUMP was then lyophilized three times to remove the ammonium acetate. When needed, 13C/15N-labeled dUMP was converted to dU using alkaline phosphatase and 13C/15N-labeled dU was purified using the same HPLC method used for dUMP.
Synthesis of dUMS
The protocol for dUMS synthesis was adapted from a procedure for making sulfated guanosine.20 134 mg sulfur trioxide-dimethylformamide complex (0.88 mmol) was added to a solution of 200 mg deoxyuridine (0.88 mmol) in 10 mL dry dimethylformamide under argon at −10 °C with stirring. After two minutes, the reaction was allowed to warm to room temperature where it remained for two hours with stirring under argon. The reaction was then placed on ice and 1 mL saturated aqueous potassium bicarbonate was added with stirring. After removing the solvent by lyophilization, the powder was dissolved in 1 mL water and dUMS was purified from dU-disulfate, dU-3′-monosulfate, and unreacted dU on a preparative 5 μm 250 mm × 21 mm NUCLEODUR®C18 Gravity HPLC column using isocratic 0.2 M ammonium acetate, pH 5 at 5 mL/min. The ammonium acetate was then removed from purified dUMS by five rounds of lyophilization. 1H-NMR (400 MHz, D2O, 298 K): δ 7.67 ppm (d, 1H), 6.12 ppm (t, 1H), 5.71 ppm (d, 1H), 4.37 ppm (m, 1H), 4.06 ppm (m, 3H), 2,19 ppm (m, 2H). 13C-NMR (101 MHz, D2O, 298 K): δ 166.1 ppm, 151.4 ppm, 141.5 ppm, 102.1 ppm, 85.5 ppm, 84.4 ppm, 70.7 ppm, 67.5 ppm, 38.8 ppm. MS (ESI, neg. mode): [M-H]− 307. Product ion scan fragments of 307[1−]: 195, 111, 97. dUMS was also observed in the crystal structure of the FDTS-dUMS complex, further verifying its structure.
Spectrophotometric Titrations
Spectrophotometric titrations were performed on a Shimadzu UV-2501PC scanning spectrophotometer at 25 °C. ~16 μM enzyme in 0.1 M Tris-HCl pH 8 was titrated with ligand in the same buffer. Absorbance spectra were recorded after each addition. The change in flavin absorbance due to ligand binding was plotted against ligand concentration. For weak or moderate binding the data were fit to a square hyperbola in KaleidaGraph to determine the Kd. When binding was tight, data were fit to Eq. 1, where Δεmax is the maximum change in extinction coefficient, E0 is the initial enzyme concentration, and L0 is the total ligand concentration.
| Eq. 1 |
Reactions of dU and dUMS with FDTS
Experiments testing the reactivity of dU and dUMS with FDTS were performed in anaerobic cuvettes21 and monitored in a scanning spectrophotometer. Cuvettes were made anaerobic by repeated cycles of vacuum and equilibration with anaerobic argon. ~80 μM FDTS in 0.1 M Tris-HCl pH 8 at 25 °C was anaerobically titrated with ~30 mM dithionite in the same buffer until the flavin was completely reduced. 200 μM CH2THF (after mixing) was added, followed by addition of either 10 mM dU or 200 μM dUMS (after mixing). No oxidation of the flavin occurred over four hours with either dU or dUMS.
13C-NMR Spectroscopy
All 13C-NMR spectra of enzyme complexes were acquired on a Varian/Agilent 600 MHz NMR spectrometer equipped with a 1H-15N-13C triple resonance cryo-probe and cryogenic 13C preamplifier in 50 mM Tris-HCl, pH 8, 0.2 % sodium azide, 10 % D2O, at 45 °C using 5 mm NMR tubes. Protons were decoupled during data acquisition. Peak assignments were obtained from 13C-1H, 1H-1H 13C-13C and 15N-13C-1H chemical shift correlation spectra of the unbound nucleotide. 500 μM 13C/15N-labeled dUMP or dU and 1.5 mM enzyme were used for ligand complexes of the WT and mutagenized FDTS. Data were processed using MestReNova. The 13C-NMR spectrum of the 12C dUMP-FDTS complex was subtracted from the spectra of the 13C-labeled complexes to remove the resonances associated with the protein. For the reduced FDTS-dUMP complex, the flavin was reduced by addition of 10 mM glucose-6-phosphate and catalytic amounts of glucose-6-phosphate dehydrogenase and NADPH in an anaerobic chamber. The sample was placed in an NMR tube, capped with an anaerobic septum, and removed from the anaerobic chamber for data collection. The sample was pale yellow throughout the data collection, indicating that the flavin remained reduced. Spectra for the uncomplexed ligands were acquired separately. The sample for unbound dUMP at pH 12 was acquired in 50 mM sodium phosphate buffer, 0.2 % sodium azide, 10% D2O.
Detection of Proton Release
The buffer of stock solutions of FDTS was removed by passing through a desalting column (Econo-Pac 10DG, Bio-Rad) equilibrated with a solution of 50 mM NaCl whose pH had been adjusted to 8.0 using HCl and/or NaOH. Enzyme was diluted into the same solvent to a concentration of 10 μM FAD, and phenol red was added (20 μM) from a concentrated stock solution in 50 mM NaCl whose pH had been adjusted to 8.0. The pH of the enzyme-dye mixture was checked at the start of an experiment and adjusted to 8.0 with HCl or NaOH if deviations were detected. Titrations were performed on a Shimadzu UV-2501PC scanning spectrophotometer at 25 °C. Small aliquots were added of a concentrated stock solution of dUMP made in 50 mM NaCl, pH 8.0, and absorbance spectra were recorded.
Crystallization and Structure Determination
Crystals of FDTS were grown under previously reported conditions using the sitting drop vapor diffusion method.12 For the R90A FDTS-dUMP, R174A FDTS-dUMP, and WT FDTS-dU complexes, crystals were grown by mixing protein-ligand stock solution (10 mg/mL FTDS variant, 20 mM dU(MP), 10 mM Tris-HCl pH 7.9) with 40-50% PEG 200, 0-150 mM NaCl, 100 mM Tris-HCl pH 8.0 at a 1:1 ratio. The R174A crystals were grown by streak-seeding WT FDTS-dUMP crystals (using an eyelash) into a pre-equilibrated drop containing R174A FDTS and dUMP in the crystallization solution. Crystals were flash cooled in liquid N2 without additional cryoprotection. Crystals of the FDTS-dUMS complex (stock solution containing 10 mg/mL FDTS, 5 mM dUMS, 10 mM Tris-HCl pH 7.9) were grown in 7% PEG 4K, 100 mM NaCl, 100 mM Na/K phosphate pH 6.6. The crystals were serially transferred to solutions of increasing PEG 4K and glycerol to a final concentration of 24% PEG 4K, 10% glycerol, 200 mM NaCl, 100 mM Na/K phosphate pH 6.6 before being flash frozen in liquid N2.
Diffraction data were collected at GM/CA at APS on beamline 23ID-D. Data were processed in HKL2000,22 XDS23 or MOSFLM24. A single chain of FTDS (PDB ID 4GT9) was used for molecular replacement in MolRep25 and iterative rounds of model building in Coot26 and refinement in Refmac27 were used to generate the final model. TLS groups were identified using the TLSMD server.28 Restraints for dUMS were generated using the GRADE server.29 An additional FAD was observed near the active site in the FTDS-dUMS complex, the adenine portion is disordered and crosses a crystallographic 2-fold axis so riboflavin was built in place of FAD. The final models were validated with MolProbity.30
Supplementary Material
Acknowledgments
CH2THF was a gift from Merck Eprova AG, Switzerland.
Funding Sources
This work was supported by NSF CHE 1213620 (B.A.P.), NIH R01DK042303 (J.L.S.), NIH R01GM081544 (J.L.S.) and an NIH training grant (GM008597) to S.M.B. GM/CA at APS is supported by the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104).
Footnotes
Supporting Information. Binding, structural and crystallographic data collection and refinement statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carreras CW, Santi DV. Annu Rev Biochem. 1995;64:721. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 2.Koehn EM, Kohen A. Arch Biochem Biophys. 2010;493:96. doi: 10.1016/j.abb.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koehn EM, Fleischmann T, Conrad JA, Palfey BA, Lesley SA, Mathews II, Kohen A. Nature. 2009;458:919. doi: 10.1038/nature07973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishanina TV, Corcoran JM, Kohen A. J Am Chem Soc. 2014;136:10597. doi: 10.1021/ja506108b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gattis SG, Palfey BA. J Am Chem Soc. 2005;127:832. doi: 10.1021/ja0432214. [DOI] [PubMed] [Google Scholar]
- 6.Conrad JA, Ortiz-Maldonado M, Hoppe SW, Palfey BA. Biochemistry. 2014;53:5199. doi: 10.1021/bi500648n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishanina TV, Koehn EM, Conrad JA, Palfey BA, Lesley SA, Kohen A. J Am Chem Soc. 2012;134:4442. doi: 10.1021/ja2120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishanina TV, Yu L, Karunaratne K, Mondal D, Corcoran JM, Choi MA, Kohen A. Science. 2016;351:507. doi: 10.1126/science.aad0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aylward NN. J Chem Soc B Phys Org. 1967;401 [Google Scholar]
- 10.Mathews I, Deacon A, Canaves J, Mcmullan D, Lesley S, Agarwalla S, Kuhn P. Structure. 2003;11:677. doi: 10.1016/s0969-2126(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 11.Guillén Schlippe YV, Hedstrom L. Arch Biochem Biophys. 2005;433:266. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Koehn EM, Perissinotti LL, Moghram S, Prabhakar A, Lesley SA, Mathews II. Proc Natl Acad Sci U S A. 2012;109:15722. doi: 10.1073/pnas.1206077109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jencks WP, Regenstein J. In: Handbook of Biochemistry and Molecular Biology. Fourth. Lundblad RL, MacDonald FM, editors. 2010. pp. 595–636. [Google Scholar]
- 14.Alegria AH. Biochim Biophys Acta. 1967;149:317. doi: 10.1016/0005-2787(67)90159-1. [DOI] [PubMed] [Google Scholar]
- 15.Cline RE, Fink RM, Fink K. J Am Chem Soc. 1959;293:2521. [Google Scholar]
- 16.Drohat AC, Stivers JT. J Am Chem Soc. 2000;122:1840. [Google Scholar]
- 17.Drohat AC, Stivers JT. Biochemistry. 2000;39:11865. doi: 10.1021/bi000922e. [DOI] [PubMed] [Google Scholar]
- 18.Dong J, Drohat AC, Stivers JT, Pankiewicz KW, Carey PR. Biochemistry. 2000;39:13241. doi: 10.1021/bi001437m. [DOI] [PubMed] [Google Scholar]
- 19.Werner RM, Stivers JT. Biochemistry. 2000;39:14054. doi: 10.1021/bi0018178. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder FC, Taggi AE, Gronquist M, Malik RU, Grant JB, Eisner T, Meinwald J. Proc Natl Acad Sci U S A. 2008;105:14283. doi: 10.1073/pnas.0806840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams CHJ, Arscott DL, Matthews RG, Thorpe C, Wilkinson KD. Methods Enzymol. 1979;62:185. doi: 10.1016/0076-6879(79)62217-6. [DOI] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Methods Enzym. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Kabsch W. Acta Crystallogr D Biol Crystallogr. 2010;66:125. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leslie AGW, Powell HR. In: Evolving Methods for Macromolecular Crystallography. Read RJ, Sussman JL, editors. 2007. pp. 41–51. [Google Scholar]
- 25.Vagin A, Teplyakov A. J Appl Crystallogr. 1997;30:1022. [Google Scholar]
- 26.Emsley P, Cowtan K. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. Acta Crystallogr D Biol Crystallogr. 2004;60:2184. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 28.Painter J, Merritt EA. J Appl Crystallogr. 2006;39:109. [Google Scholar]
- 29.Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G. Grade, version 1.1.1. Cambridge, United Kingdom: Global Phasing Ltd.; 2011. http://www.globalphasing.com. [Google Scholar]
- 30.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta Crystallogr D Biol Crystallogr. 2010;66:12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.