

Summary
Objective
Juvenile myoclonic epilepsy (JME) is the most common form of idiopathic generalized epilepsies (IGEs) and is genetically heterogeneous. Mutations in EFHC1 cause JME. Because about 2 million people in India are affected by JME alone, we investigated the prevalence of mutations in the EFHC1 gene in the Indian population with JME. We studied 63 patients with JME and 80 healthy controls.
Methods
Clinical identification of JME was evaluated using established criteria. Following clinical evaluation of the patients and confirming presence of JME, blood samples were collected from each patient and healthy individual. Subsequently, genomic DNA was extracted from the blood samples. Eleven exons of the EFHC1 gene were individually amplified by polymerase chain reaction (PCR) for each DNA sample. The PCR products were then purified and sequenced commercially. The identified DNA variants were sequenced at least twice in both the forward and reverse directions and compared with the Exome Aggregation Consortium (ExAC) database.
Results
We found five heterozygous and one homozygous variant. We found three novel coding variants 661C→T, 779 G →A, and 730 C→T, which lead to R221C, R260Q, and R244STOP amino acid substitutions, respectively. The coding variant 475 C→T, resulting in the amino acid substitution R159W, reported earlier as polymorphism, was also identified in both patient and control populations.
Significance
Detection of these three novel variants, excluding R159W, which is considered polymorphism, expands the range of possible mutations in the EFHC1 gene. The novel variants that we are reporting herein have not been mentioned before as occurring in JME patients of other ethnic population. Therefore, these novel coding variants may be confined to the Indian JME population. Further studies on the mutational spectrum of EFHC1 in a larger number of Indian JME patients concurrent with their mode of inheritance and underlying functional assays should establish whether EFHC1 could be a panethnic gene for JME.
Keywords: JME, EFHC1, Indian population, Mutation, Polymorphism
Key Points.
EFHC1 mutation status in Indian population is reported
Novel EFHC1 variants and mutations are identified
The EFHC1 coding polymorphism R159W is very rare in the Indian population
Juvenile myoclonic epilepsy (JME) is the most common form of idiopathic generalized epilepsy (IGE). It accounts for 10–15%1, 2, 3 of all cases of epilepsy. About 2 million people in India are affected by JME. The seizures of JME may begin between late childhood and early adulthood, usually around the time of puberty. JME usually has an onset in adolescence, with the typical age of onset between 12 and 18 years;4 however, children as young as 6 years and adults as old as 36 years can develop JME. It is characterized by adolescent onset, infrequent absence seizures, awakening myoclonic seizures, and generalized tonic‐clonic (GTC) or clonic‐tonic‐clonic (CTC) seizures. It is also more likely to occur in people who have family members with generalized epilepsy. Studies investigating the genetic contribution to JME have mainly focused on familial forms with an autosomal dominant inheritance of IGE syndromes.5, 6
A number of studies have demonstrated that mutations in ion channels as well as in neurotransmitter receptors are associated with JME and include calcium channel subunit CACNB4,5 the γ‐aminobutyric acid (GABA) receptor subunit, GABRA1,7 ligand‐gated chloride channel for GABA subunit GABRD,8 and the chloride channel CLCN2.9 For each of these genes, mutations have been reported mostly in a single family with JME and most often it is de novo, that is, not observed in the parents. Importantly, these mutations were not observed at all in other family‐based JME studies, neither in the same nor in different ethnic populations. Therefore, mutations in ion channel genes can be considered a rare cause of JME.
Analyzing the Mexican cohort samples as described by Bai et al.,10 Suzuki et al.6 isolated a new gene, EFHC1, which has apoptotic activity and encodes a protein of 640 amino acids with a calcium‐binding motif, within the 6p12–11 mapped locus and identified five missense mutations in six independent Mexican JME families out of 31 Mexican JME families. All mutations resulted in single amino acid substitutions. Stogmann et al.11, 12 also sequenced 61 Austrian JME patients and identified three heterozygous missense mutations in the EFHC1 gene. Annesi et al.13 studied 27 Italian JME families with 86 affected individuals and reported two heterozygous mutations in EFHC1 in three unrelated families. Medina et al.14 further identified five novel mutations in transcripts A and B of the EFHC1 gene in 4 (9%) of 44 Hispanic patients from Mexico and Honduras and in 2 (3%) of 67 Japanese patients with juvenile myoclonic epilepsy. The latter three studies, therefore, further support EFHC1 as a JME‐causing gene.
However, Pinto et al.15 reported heterogeneity at the 6p12–11 locus and observed the absence of mutation in the EFHC1 gene in 112 Dutch JME patients who were previously mapped to the 6p12–p11 locus. Therefore, it seems that multiple genes may be involved in JME, and these may vary between and within ethnicities. One large JME family from Belize that mapped to the 6p12–11 locus carried a common polymorphism of EFHC1 that cosegregated with JME with higher frequency than in normal individuals. This polymorphism did not have any effect on normal EFHC1 function, as judged by EFHC1‐mediated cell death analysis, suggesting the possibility that this family may be linked to EFHC1 and that another nearby mutation most likely may be responsible for JME in this family.16
Subaran et al.17 questioned the effect of reportedly pathogenic EFHC1 mutations on JME. The group emphasizes that pathogenicity of EFHC1 coding variants may depend on genetic backgrounds of the population being studied. Further, the study cautions us about the level of evidence necessary to attribute causation.
Therefore, to establish whether EFHC1 mutations contribute to JME in populations with different ethnic backgrounds, we screened the EFHC1 gene for mutations among 63 JME patients originating from 63 independent families in India alongside 80 controls with no history of epilepsy.
Material and Methods
Patient samples
Sixty‐three Indian JME patients were analyzed for the present study according to the criteria of the Commission on Classification and Terminology of the International League Against Epilepsy (1989).
Clinical identification of JME was evaluated using the following inclusion criteria: (1) had to have seizure onsets around the adolescent period, between 10 and 21 years of age. All had experienced early morning events, and precipitation of seizures included sleep deprivation. (2) Electroencephalogram (EEG) of the patient often displayed bilateral, diffuse, symmetrical, and synchronous 4‐ to 4.5‐Hz spikes, including polyspikes and wave complexes, either spontaneously or on photic, hyperventilation and in a sleep deprived state. (3) History of seizures with myoclonus and absence seizures. The exclusion criteria for patients included: (1) symptomatic seizures history, (2) presence of progressive myoclonic epilepsy and/or progressive neurological disease in the family, and (3) evidence of complex or partial seizures.
Following clinical evaluation of the patients and confirming presence of JME, blood samples were collected. All subjects provided written informed consent, as required by the institute's ethics committees. It was very difficult to access the blood samples from family members.
DNA sequencing
Peripheral blood samples were collected from each participant, and genomic DNA was extracted using Flexigene DNA Kit (Qiagen). Eleven exons of the EFHC1 gene were individually amplified by polymerase chain reaction (PCR) using intronic primers. The PCR products were then purified using QIAquick gel extraction kit (Qiagen) and sequenced commercially. The identified variants were sequenced at least twice in both the forward and reverse directions. Screening for mutations in the EFHC1 gene was performed in all index patients and controls.
Results
Clinical characteristics
In our cohort of 63 JME patients, 39 (62%) were male and 24 (38%) were female. The average age at onset of epilepsy was 13 years (range 10–21 years). Most cases (73%) were singletons having no positive family history of epilepsy. All the patients met the criteria for classic JME.
Mutation analysis
In 63 JME patients, we identified four heterozygous coding variants, 661C→ T, 779 G → A, 881 G → A, and 730 C→ T, of EFHC1 that resulted in the amino acid substitutions R221C, R260Q, R294H, and R244STOP, respectively, and one homozygous and one heterozygous coding variant 475 C→T that resulted in the amino acid substitution R159W (Fig. 1). The identified heterozygous coding variant R221C was found in exon 4, and the other three variants R260Q, R294H, R244STOP were found in exon 5. Homozygous and heterozygous coding variants R159W were found in exon 3 (Fig. 2).
Figure 1.

Chromatogram of the five EFHC1 variants. (A) WT and (B) R260Q; (C) WT and (D) R244STOP; (E) WT and (F) R221C; (G) WT and (H) R294H; (I) WT and (J) & (K) R159W (homozygous and heterozygous, respectively)
Figure 2.
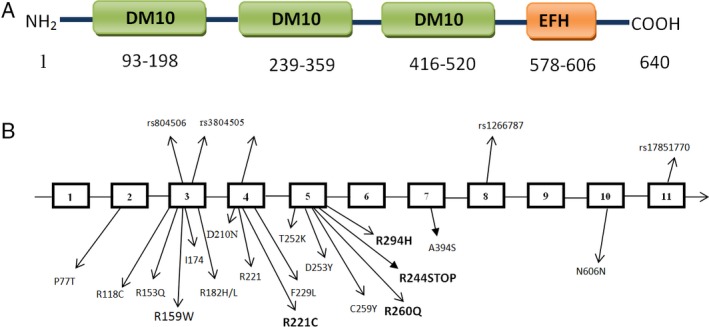
(A) Schematic diagram of EFHC1 protein. (B) Genomic organization of the EFHC1 gene showing the previously reported mutations in different exons, whereas the new mutations observed in the Indian JME patient are shown in bold type.
The coding variant R159W is considered a polymorphism and occurred at a frequency of 16% and 14% in Mexican JME families and in the control population, respectively.6 We detected this variant in 2 JME patients (frequency 3.17%) and in 1 in the control sample (frequency 1.25%). This could be a relatively rare polymorphism in the Indian population.
The coding variants R221C, R260Q, and R244STOP seem to be novel variants because these were never reported in any other ethnic populations. These variants are within the second DM10 domain of the EFHC1 gene, except R221C, which is very close to the second DM10 domain. The frequencies of the coding variants of R294H, including the novel ones, were 2.32% for each in the Indian JME patient population we studied. None of these variants was found in our Indian control samples. In the Exome Aggregation Consortium (ExAC) database, the frequencies of R221C, R260Q, R294H, and R244STOP variants were found to be 0.18%, 0.057%, 1.92%, and 0.0034%, respectively. As mentioned earlier, because of the lack of samples available from family members, we were unable to infer their EFHC1 mutational status, which consequently limits our efforts to determine the mode of inheritance. This has prompted us to treat any EFHC1 variants found in the studied patients as singletons.
R294H mutation was detected in a female patient with JME who had unaffected parents but who had a history of generalized seizures and myoclonus with awakening preponderance in a paternal uncle (Fig. 3A). She had a febrile seizure at the age of 3–4 years, and at the age of 12 years had a myoclonus seizure and proceeded to display generalized seizures at the age of 15 years. She had complaints of headache, frontal throbbing, and phonophobia, but no photophobia, nausea, or vomiting. The R294H variant was reported earlier in two probands of German JME patients who had a subtype of JME developing from childhood absence epilepsy.18
Figure 3.

Pedigrees of the families with JME: (A) R294H and (B) R159W. Filled symbols indicate affected individuals. Arrows indicate the probands of each family.
A male patient carrying the R159W allele had a mother with generalized seizures and myoclonus with awakening preponderance (Fig. 3B). He had abnormal EEG with generalized spike and wave discharges, but no focal deficits. He had a myoclonic seizure at the age of 16 years that proceeded to form generalized seizures at the age of 18 years. Patients carrying R221C, R260Q, and R244STOP had no affected members in their extended family, including their parents. These patients displayed classical JME features. Therefore, it seems that in these patients JME appears as a singleton.
Discussion
Mutation in EFHC1 causes JME. We evaluated 63 Indian JME patients for the presence of EFHC1 mutation. Five heterozygous and one homozygous EFHC1 variants were found in six independent JME families. Because their mode of inheritance was not ascertained and in some cases there were signs of dominant inheritance, these variants were considered as singletons. Detection of these three novel variants, excluding R159W, which is considered a polymorphism, expands the range of possible mutations in the EFHC1 gene. It also demonstrates that because mutations in EFHC1are found more often in JME patients irrespective of their geographical origin or ethnicity, EFHC1 may be considered a panethnic JME gene. Because of the limited number of variants we unearthed in the Indian JME patients, we could not establish any correlation between nature of variants and clinical features. However, all affected individuals displayed classic JME symptoms with typical EEG traits of 4‐ to 4.5‐Hz generalized polyspike waves, further supporting the notion that EFHC1 mutations are associated with classic JME without pyknoleptic absences.6
On the possible effects of variants in the functioning of EFHC1, polymorphism phenotyping (PolyPhen) analysis predicts that R221C and R294H variants may have damaging effects, whereas R260Q may have negligible effects. The three mutations R221C, R294H, and R260Q received scores of 0.776, 0.998, and 0.087, respectively. Another tool, SIFT, that predicts variation effects on protein function showed scores of 0.01, 0.00, and 0.06 for R221C, R294H, and R260Q, respectively. According to these scores, R221C and R294H are deleterious and R260Q is tolerated. One of the identified EFHC1 mutations (R244STOP) results in an abrupt stop codon in the middle of the second DM10 domain. Because both of these tools predicted similar consequences for the variants and because their frequency of occurrence in the general population is much lower, according to the ExAC database unphenotyped for epilepsy, including their absence in our control sample comprising 80 people, we assume that these variants may be mutations. Moreover, because these novel variants, except R221C, lie within the DM10 domain of EFHC1, it is most likely that functions of EFHC1 might be compromised.
Interestingly, in a recent article Bailey et al.19 reanalyzed 54 EFHC1 variants associated with epilepsy from 17 cohorts and classified 9 variants as pathogenic. This study also included JME mutations (H89R, Y355C, R372W, R436C, N519S, V556L, I619S, and Y631C) reported from India. The majority of these EFHC1 coding variants originated from Bangalore (R372W, R436C, N519S, V556L, I619S, and Y631C) and may not be pathogenic.19 Therefore, these are far from the New Delhi EFHC1 variants that are located primarily between DM10 (1) and DM10 (2) domains. It might be a reflection of the differences in “peopling” effect and ancestral origin of the two different populations of New Delhi (North India) and Bangalore (South India). Subaran et al.17 recently reported that the pathogenic EFHC1 P77T‐R221H (rs149055334–rs 79761183) JME haplotype is present in both Hispanic and African American controls, including in the public database of unphenotyped West African ancestry populations. We believe that some EFHC1 mutations may be pathogenic only when introduced into specific genetic backgrounds.
Having said that, however, in the absence of specific biological assays that can monitor the altered function of the protein, the identified novel coding variants of EFHC1 reported herein cannot be termed mutants with certainty. The same notion may be extended to earlier reported coding variants of EFHC1, which are often considered mutants on the basis of prevalence and cosegregation in JME patients, including modeling on PolyPhen tools.6, 11, 13, 14, 20
About 263 missense variants for EFHC1 have been reported in the ExAC database. All the coding variants that have so far been reported to occur in JME patients, including the ones that we have uncovered in Indian JME samples, are found in the ExAC database except N606N, which was reported earlier. However, the novel variants that we are reporting herein have not been mentioned before as occurring in JME patients of other ethnic populations. Therefore, these novel coding variants may be confined to the Indian JME population. Because we identified only four mutations in 63 independent families, it points out that EFHC1 mutations may not be the only cause of JME in the Indian population. Further studies on the mutational spectrum of EFHC1 in Indian JME patients concurrent with their mode of inheritance and underlying functional assays should establish whether EFHC1 could be a panethnic gene for JME. Absence of parental samples in our present study limited our efforts and scientific evaluation of the genetic nature of the variants, that is, whether they are de novo or sporadic. Additionally, we have not analyzed the promoter sequence of EFHC1 that may also harbor polymorphism that probably affects the transcriptional regulation of EFHC1.Therefore, further work is necessary to evaluate and identify the EFHC1 coding variants status in Indian JME patients.
Conclusion
Mutation in EFHC1 causes JME. We are one of the first to report studies on the status of EFHC1 mutation in Indian JME patients. We observed three coding variants (R221C, R260Q, and R244STOP) that were never reported before in any ethnic community. These variants were absent in the Indian control sample. All these novel variants were singletons, and none of their parents have JME. In the absence of a definitive biological process wherein we can assess the mutational effect of EFHC1, whether the identified variants of EFHC1 in Indian JME patients and/or in other ethnic communities are truly mutants remains questionable, and as of now these variants would most likely be risk‐allele for JME.
Disclosure
The authors declare no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
This study was supported by a grant from the Department of Biotechnology (DBT), Govt. of India (Grant No. BT/522NE/TBP/2013).
Biography
Ms. Romita Thounaojam, PhD student in the Department of Biotechnology & Bioinformatics, NEHU Shillong‐793022.

References
- 1. Janz D, Christian W. Impulsive petit mal. Dtsch Z Nervenheilkd 1957;176:346–386. [Google Scholar]
- 2. Janz D. Die Epilepsien. Stuttgart, Germany: Thieme Medical Publishers; 1969. [Google Scholar]
- 3. Janz D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta Neurol Scand 1985;72:449–459. [DOI] [PubMed] [Google Scholar]
- 4. Delgado‐Escueta A, Enrile‐Bascal F. Juvenile myoclonic epilepsy of Janz. Neurology 1984;34:285–294. [DOI] [PubMed] [Google Scholar]
- 5. Escayg A, De Waard M, Lee DD, et al. Coding and noncoding variation of the 130 human calcium‐channel beta4‐subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet 2000;66:1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki T, Delgado‐Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet 2004;36:842–849. [DOI] [PubMed] [Google Scholar]
- 7. Cossette P, Lui L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 2002;31:184–189. [DOI] [PubMed] [Google Scholar]
- 8. Emberger W, Windpassinger C, Patek E, et al. Assignment of the human GABAA receptor delta‐subunit gene (GABRD) to chromosome band 1p36.3 distal to marker NIB1364 by radiation hybrid mapping. Cytogenet Cell Genet 2000;89:281–282. [DOI] [PubMed] [Google Scholar]
- 9. Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2 encoding a voltage‐gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet 2003;33:527–532. Note: Retraction: Nature Genet. 41: 1043 only, 2009. [DOI] [PubMed] [Google Scholar]
- 10. Bai D, Alonso ME, Medina MT, et al. Juvenile myoclonic epilepsy: linkage to chromosome 6p12 in Mexico families. Am J Med Genet 2002;113:268–274. [DOI] [PubMed] [Google Scholar]
- 11. Stogmann E, Lichtner P, Baumgarter C, et al. Mutations in the CLCN2 gene are a rare cause of idiopathic generalized epilepsy syndromes. Neurogenetics 2006;7:265–268. [DOI] [PubMed] [Google Scholar]
- 12. Stogmann E, Linchter P, Baumgarter C, et al. Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology 2006;67:2029–2031. [DOI] [PubMed] [Google Scholar]
- 13. Annesi F, Gambardella A, Michelucci R, et al. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia 2007;48:1686–1690. [DOI] [PubMed] [Google Scholar]
- 14. Medina MT, Suzuki T, Alonso ME, et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology 2008;70(22 Pt 2):2137–2144. [DOI] [PubMed] [Google Scholar]
- 15. Pinto D, Louwaars S, Westland B, et al. Heterogeneity at the JME 6p11‐12 locus: absence of mutations in the EFHC1 gene in linked Dutch families. Epilepsia 2006;47:1743–1746. [DOI] [PubMed] [Google Scholar]
- 16. Wallace R. Identification of a new JME gene implicates reduced apoptotic neuronal death as a mechanism of epileptogenesis. Epilepsy Curr 2005;5:11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Subaran Ryan L, Conte Juliette M, Stewart William CL, et al. Pathogenic EFHC1 mutations are tolerated in healthy individuals dependent on reported ancestry. Epilepsia 2015;56:188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Von Podewils F, Kowoll V, Schroeder W, et al. Predictive value of EFHC1 variants for the long‐term seizure outcome in juvenile myoclonic epilepsy. Epilepsy Behav 2015;44:61–66. [DOI] [PubMed] [Google Scholar]
- 19. Bailey JN, Patterson C, de Nijs L, et al. EFHC1 variants in juvenile myoclonic epilepsy: reanalysis according to NHGRI and ACMG guidelines for assigning disease causality. Genet Med Epub July 28, 2016. [DOI] [PubMed] [Google Scholar]
- 20. Jara‐Prado A, Martinez‐Juarez IE, Ochoa A, et al. Novel Myoclonin1/EFHC1 mutations in Mexican patients with juvenile myoclonic epilepsy. Seizure 2012;21:550–554. [DOI] [PubMed] [Google Scholar]