

Summary
Objective
To evaluate the prevalence of various etiologies of epilepsies and epilepsy syndromes and to estimate cognitive function in cases of childhood‐onset epilepsy.
Methods
A population‐based retrospective registry study. We identified all medically treated children with epilepsy born in 1989–2007 in Finland's Kuopio University Hospital catchment area, combining data from the birth registry and the national registry of special‐reimbursement medicines. We reevaluated the epilepsy diagnoses and syndromes and gathered data on etiologies and cognitive impairment.
Results
We identified 289 children with epilepsy. The annual incidence rate of epilepsies and epilepsy syndromes was 38 in 100,000, and the misdiagnosis rate was 3%. A specific etiology was identified in 65% of the cases, with a structural etiology accounting for 29% and a genetic or presumed genetic etiology for 32%. Most patients with unknown‐etiology epilepsy had focal epilepsy and were of normal intelligence. Intellectual disability was detected in 35% of cases, and only 17% in this group had an unknown etiology for the epilepsy. Electroclinical syndromes (mainly West syndrome) were recognized in 35% of the patients.
Significance
Epilepsy is a complex disease that encompasses many etiologies and rare syndromes. The etiology and specific epilepsy syndrome are important determinants of the outcome and key factors in treatment selection. Etiological diagnosis can be achieved for the majority of children and syndromic diagnosis for only a third.
Keywords: Epilepsy, Epilepsy syndrome, Etiology, Prevalence, Childhood, Adolescence, Pediatric, Classification, Cognition, Intelligence
Key Points.
Epilepsy is a complex disease with many etiologies and rare syndromes
Mental impairment and subtle intellectual difficulties exist frequently in epilepsy
Patient‐specific etiology and epilepsy syndrome are important determinants of the outcome and treatment selection
Epilepsy is one of the most prevalent neurological diseases worldwide. About 65 million people have epilepsy, roughly 25% of whom are children younger than 15 years of age.1, 2, 3, 4 The annual incidence of epilepsy in children ranges from 33 to 82 in 100,000 persons per year in developed countries. The incidence is invariably reported to be highest in the first year of life and falls to adult levels by the end of the first decade.1, 5, 6, 7 Recent studies in Finland have found a steady decline in the incidence of epilepsy in all age groups except the elderly over the past two decades. This improvement might be due to progress in obstetric and neonatal care/treatment and an extensive national immunization program.8, 9, 10
The incidence and prevalence of specific seizure types and epilepsy syndromes in children are not as well documented as the incidence of epilepsy.7 Population‐based studies have found a slight predominance of focal seizures as compared with generalized seizures. Only about a third of children with epilepsy can be classified as having a specific epilepsy syndrome.7, 11 Though causes of epilepsy are increasingly being identified, 36–80% of cases still have an unknown etiology.5, 6, 12, 13 Recent advances in the International League Against Epilepsy (ILAE) classification system emphasize the role of evaluating the underlying neurological cause and etiology of epilepsy in the diagnostic work‐up. Etiology is important, as a major determinant of treatment, prognosis, and clinical course. Neuroimaging and genetic advances have improved accuracy levels in diagnosis of epilepsies.14, 15, 16, 17
Cognitive and behavioral comorbidities, including some degree of intellectual disability, are commonplace in children with epilepsy and can have an even greater impact on quality of life than do seizures.18 Comorbidities influence language skills, cognitive functioning, and psychosocial outcomes. With their effects on academic achievements and professional skills, cognitive abnormalities and difficulties have a long‐lasting impact on children's lives even after seizures cease. Cognitive disability is the major comorbidity in these cases.14, 19, 20, 21, 22 In population‐based studies, intellectual disability has been verified in 20–50% of cases,23, 24 and a Finnish population‐based study25 found cognitive function to be within normal or borderline range for 50%, to show mild retardation for 22%, and to evidence moderate to severe retardation in 28%.
The aim for our study was to evaluate the prevalence of various etiologies of epilepsies and epilepsy syndromes and to estimate cognitive function in a population‐based birth registry. In previous work, we have published an evaluation of perinatal and reproductive risk factors for childhood epilepsy in this cohort.26
Materials and Methods
The study population
We analyzed data gathered from the prospective Kuopio University Hospital (KUH) Birth Registry between January 1, 1989, and December 31, 2007, which corresponded to total‐population births (n = 42,865) (for infants either born after the 22nd week of pregnancy or weighing 500 g or more). Stillborn fetuses (n = 193) and neonatal deaths (death in liveborn within the 28 completed days of life) (n = 177) were excluded from the analysis. Total population of live births was n = 42,495 (Fig. 1).
Figure 1.
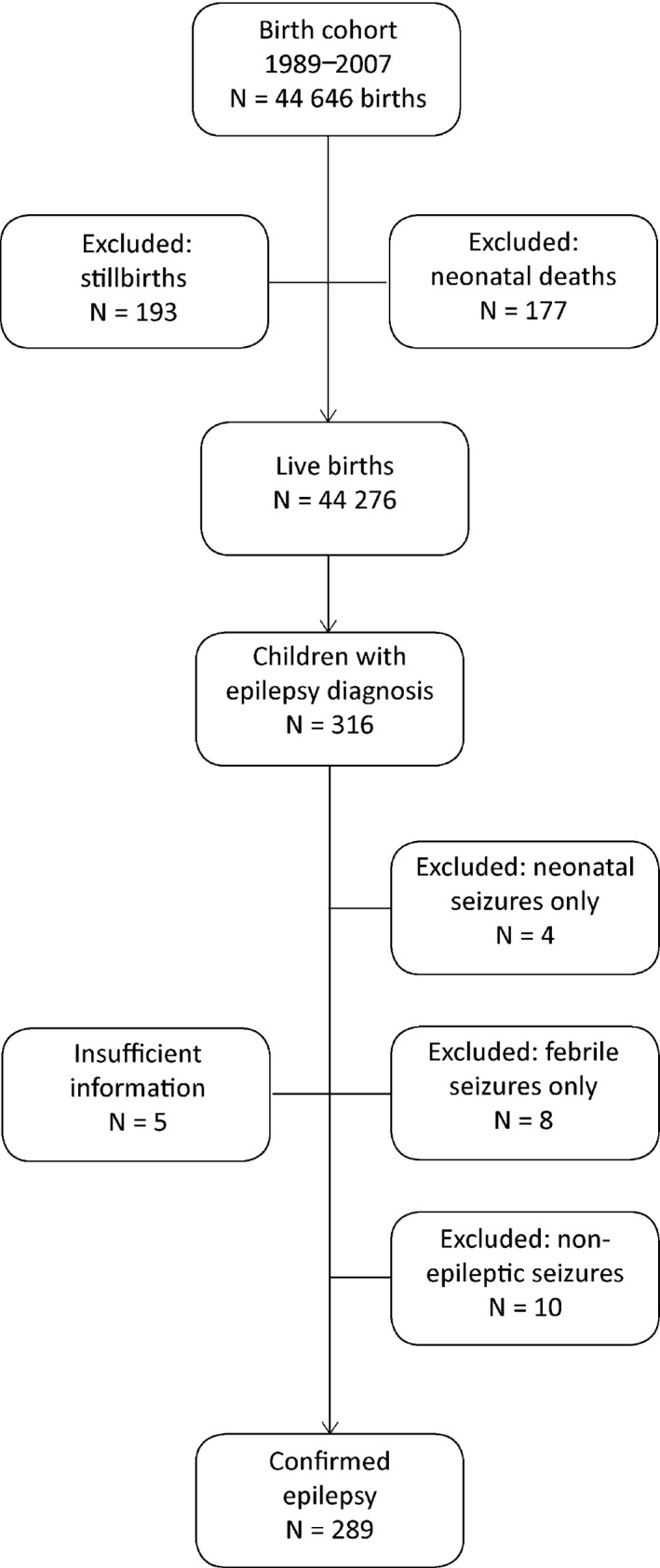
Flowchart of the data collection.
Kuopio University Hospital is a tertiary‐level perinatal center in eastern Finland and the only delivery hospital in the North Savo district; hence, the data covered all women giving birth in the area. Children with a diagnosis of epilepsy were identified from the register of the Social Insurance Institution of Finland, which was linked to the Kuopio University Birth Registry through children's unique identification numbers. The Social Insurance Institution (SII) maintains a national registry of medicines that are subject to special reimbursement. A full refund is given for the costs of antiepileptic drugs (AEDs), and qualification for this is based on clinical diagnosis (ICD‐10) and a statement provided in semistructured form by a pediatric neurologist. The right to receive a full refund for AEDs is granted by the SII for noninstitutionalized patients. The SII data on full‐reimbursement AEDs between January 1, 1989, and December 31, 2007, were synthesized with KUH Birth Registry data in January 2009. We found that 316 of the children received AEDs for epilepsy linked to refunds. The medical records of each identified patient were thoroughly reviewed retrospectively for purpose of the current study (by researcher AS). The clinical factors recorded were onset of seizures, seizure types and syndromes, etiology of epilepsy, electroencephalograms (EEGs), imaging, and neuropsychology results. Final classification was performed by two neurologists (AS and RK).
Definitions
For our research, epilepsy was defined as a disease of the brain if any of the following conditions were fulfilled: (1) at least two unprovoked (or reflex) seizures occurred more than 24 h apart; (2) one unprovoked (or reflex) seizure occurred and the likelihood of further seizures was similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years; and (3) there was a diagnosis of epilepsy syndrome.27
We excluded patients with any of the following: (1) febrile seizures only, (2) neonatal seizures only, (3) single unprovoked seizures or acute symptomatic seizures without a need for further AED treatment, and (4) nonepileptic seizures only. Seizures were classified as either focal or generalized. The etiology of the epilepsy was classified as genetic/presumed genetic, structural, metabolic, immune, infectious, or unknown, as suggested by the ILAE.28 Specific electroclinical syndromes were identified whenever possible.
Cognitive assessment
The level of cognitive function was categorized as normal (IQ ≥ 70), mild cognitive impairment (IQ 50–69), moderate cognitive impairment (IQ 35–49), or severe/profound cognitive impairment (IQ < 34).29 Evaluation was done by neuropsychological assessment, in case there had been any doubt of learning difficulties. Neuropsychological assessment was not available for part of the children performing at normal level, and their evaluation was done on the basis of review of medical records, including school performance and everyday adaptive functioning. Also, part of the children having profound cognitive impairment were only evaluated on the basis of medical records, because they were too disabled to perform in formal neuropsychological assessment.
Statistical methods
The data management and analysis was performed with the Statistical Package for the Social Sciences (SPSS), version 22 (SPSS, Inc., Chicago, IL, U.S.A.). Descriptive statistics are reported upon here as numbers and/or percentages. Cumulative incidence was calculated by means of the Kaplan–Meier method via the log‐rank test. A chi‐squared test was applied for comparison of cognition between genders. For all results, p values < 0.05 were considered significant.
Ethics approval
The study was approved by the Ethics Committee of Kuopio University Hospital. All women giving birth to children gave informed consent for the collection of study data and for subsequent use of said data in scientific research, at the time of data collection. The SII granted permission to link its data on epilepsy to the birth registry and for use of the data in this study. Authorization for using the hospital medical records was granted by the regulatory authority responsible for the administration of said data in Finland, that is, the respective hospital districts.
Results
The study population
The original study population consisted of 316 children. After reanalysis of all available data from medical records of the pertinent investigations and from follow‐up on the children, we were able to verify the epilepsy diagnosis of 289 children; reevaluation revealed that 10 (3%) had nonepileptic seizures, 8 (3%) turned out to have had febrile seizures only, and 4 (1%) had neonatal seizures. For 5 children, there was not enough information for a new analysis (Fig. 1).
The age at the time of epilepsy diagnosis was median 4.7 years (mean 5.5 years, standard deviation [SD] ± 0.39, range 0–19) with no difference evident between sexes (Fig. 2). Duration of follow‐up of the cohort members was median 13.9 years (mean 14.8 years, SD ± 5.09 median 13.9 years, range 7.4–26.4). The annual incidence rate was 38 per 100,000, and the cumulative incidence of epilepsy, 0.7%. The figure was highest for those diagnosed before the age of 12 months, at 141 per 100,000 persons per year.
Figure 2.
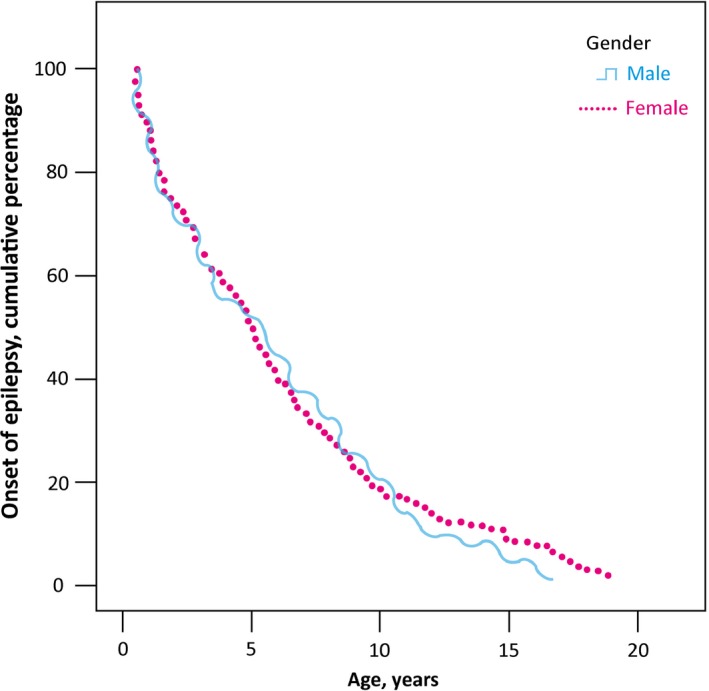
Distribution of epilepsy diagnosis by age separately for males and females.
Seizure types
In 202 patients (70%), seizure onset was focal only, whereas 53 (18%) had generalized‐onset seizures only. Six (2%) had both focal and generalized‐onset seizures. For 28 subjects (10%), including 25 patients with epileptic spasms, the seizure type could not be classified as focal or generalized.
Etiology
The etiology of the epilepsy could be identified in 188 patients, that is, in 65% of cases (Table 1). A genetic or presumed genetic etiology was identified in 91 cases (32%), with most being generalized genetic epilepsies. In 12 cases (4%), the mother too had epilepsy. Brain MRI was performed for 92% of the patients and brain CT for 5%; thus, brain imaging was employed for 97% of cases. A structural etiology was found in 85 patients (29%), for whom perinatal insult and malformation in cortical and brain development were the main causes of epilepsy.
Table 1.
Etiology of epilepsies (N = 289)
| Genetic, N = 91 (32%) | |
| Genetic | |
| Chromosomal or gene abnormalities | 20 |
| Presumed genetic | |
| Epileptic encephalopathy | 5 |
| Generalized genetic epilepsies | 39 |
| Focal genetic epilepsy | 27 |
| Structural, N = 84 (29%) | |
| Malformation of cortical and brain development | 29 |
| Hypothalamic hamartoma | 1 |
| Tuberous sclerosis | 3 |
| Vascular malformation | 2 |
| Trauma | 7 |
| Perinatal insult | 35 |
| Stroke | 1 |
| Tumor | 5 |
| Angioma | 1 |
| Metabolic, N = 4 (1%) | |
| Congenital hyperinsulinism | 3 |
| 3‐methylglutaricaciduria | 1 |
| Infection, N = 9 (3%) | |
| Congenital infection | 2 |
| Encephalitis | 4 |
| Meningitis | 2 |
| Unspecified | 1 |
| Immunological, N = 0 (0%) | 0 |
| Unknown, N = 101 (35%) | 101 |
Electroclinical syndromes
Electroclinical syndromes were recognized in 35% of the patients (Table 2). The five main syndromes identified were West syndrome (25 patients; incidence 60/100,000/year), benign epilepsy with centrotemporal spikes, childhood and juvenile absence epilepsy, and juvenile myoclonic epilepsy. Eight patients developed Lennox‐Gastaut syndrome during the follow‐up period.
Table 2.
Electroclinical epilepsy syndromes as a first diagnosis (N = 98)
| M | F | T | |
|---|---|---|---|
| Neonatal period | |||
| Benign familiar neonatal epilepsy (BNFN) | 1 | 0 | 1 |
| Early myoclonic encephalopathy (EME) | 0 | 0 | 0 |
| Ohtahara syndrome | 1 | 0 | 1 |
| Infancy | |||
| Epilepsy of infancy with migrating focal seizures | 0 | 0 | 0 |
| West syndrome | 15 | 10 | 25a |
| Myoclonic epilepsy of infancy (MEI) | 3 | 0 | 3 |
| Benign infantile epilepsy | 2 | 3 | 5 |
| Benign familial infantile epilepsy | 0 | 0 | 0 |
| Dravet syndrome | 0 | 0 | 0 |
| Myoclonic epilepsy in nonprogressive disorders | 0 | 0 | 0 |
| Childhood | |||
| Genetic febrile seizures plus (GEFS+) | 3 | 1 | 4 |
| Panayiotopoulos syndrome | 0 | 1 | 1 |
| Epilepsy with myoclonic atonic seizures (MAE) | 0 | 1 | 1 |
| Benign epilepsy with centrotemporal spikes | 10 | 9 | 19 |
| Autosomal‐dominant nocturnal frontal‐lobe epilepsy (ADNFLE) | 0 | 0 | 0 |
| Late‐onset childhood occipital epilepsy (Gastaut type) | 3 | 0 | 3 |
| Eyelid myoclonia | 0 | 1 | 1 |
| Epilepsy with myoclonic absences | 0 | 0 | 0 |
| Lennox‐Gastaut syndrome | 0 (5) | 0 (3) | 0 (8)a |
| Epileptic encephalopathy with continuous spikes and waves during sleep (CSWS) | 3 | 1 | 4 |
| Landau‐Kleffner syndrome (LKS) | 2 | 0 | 2 |
| Childhood absence epilepsy (CAE) | 5 | 8 | 13 |
| Adolescence–adulthood | |||
| Juvenile absence epilepsy (JAE) | 2 | 2 | 4 |
| Juvenile myoclonic epilepsy (JME) | 2 | 4 | 6 |
| Epilepsy with generalized tonic‐clonic seizures alone | 3 | 1 | 4 |
| Progressive myoclonic epilepsies (PMEs) | 1 | 0 | 1 |
| Autosomal‐dominant epilepsy with auditory features (ADEAF) | 0 | 0 | 0 |
| Other familial temporal‐lobe epilepsy | 0 | 0 | 0 |
| Less specific age relationship | |||
| Familial focal epilepsy with variable foci (childhood to adulthood) | 0 | 0 | 0 |
| Reflex epilepsies | 0 | 0 | 0 |
F, female; M, male; T, total number.
Ninety‐eight out of 289 patients (34%) had electroclinical epilepsy syndrome as a first diagnosis.
Six West syndrome patients, 1 patient with focal epilepsy, and 1 with generalized epilepsy and chromosomal anomaly were evaluated as having Lennox‐Gastaut syndrome (in all 8 patients).
Intellectual disability
Of the full cohort of 289 children, a psychological assessment was available for 192 children (66%). Ninety‐seven (34%) children were not formally assessed because they were either too impaired or showed normal school performance. Cognitive functioning (Fig. 3) was within the normal range for 188 of patients (65%). Mild mental impairment was found in 34 (11%), moderate mental impairment in 22 (8%), and severe/profound mental impairment in 45 (16%). In addition, 58 (20%) children with normal cognitive performance had a speech disorder or learning difficulties that necessitated assistance for academic achievements. Statistical analysis revealed no differences between sexes in cognitive functioning.
Figure 3.
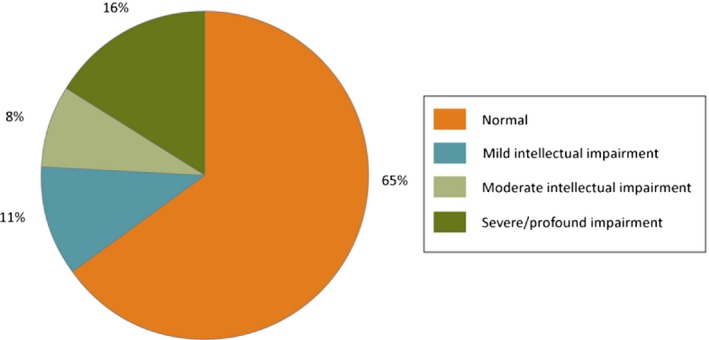
Cognition in childhood‐onset epilepsy.
Table 3 shows the epilepsy syndromes and the etiology of the epilepsy in the patients with cognitive impairment (mild, moderate, or severe/profound) (N = 101), accounting for 35% of all cases. The main epileptic syndrome associated with cognitive impairment was West syndrome, with 25 patients; there were only 1 to 2 patients in each of the other syndrome groups. For 21 cognitively impaired patients (21%), the etiology was cortical and brain‐development malformation, when we consider West syndrome patients among those with structural anomalies. Hypoxic ischemic encephalopathy existed in 8 children and perinatal insults in 3 (together, 11%). There were 9 cases of cerebral bleeding or stroke (9%), just 1 of them after the perinatal period, and there was 1 case of head trauma and 1 near‐drowning later in childhood. There were 17 cases (17%) with unknown etiology, and most of the remaining cases had a genetic or presumed genetic reason.
Table 3.
Epilepsy syndromes or the etiology of epilepsy in the patients with cognitive impairment (mild, moderate, severe/profound, N = 101)
| Epilepsy and epileptic syndromes | Number |
|---|---|
| Ohtahara syndrome | 1 |
| Hypoxic ischemic encephalopathy | |
| West syndrome | 21a |
| 2 hypoxic ischemic encephalopathies 5 chromosomal anomaly or syndrome 3 tuberous sclerosis complex | |
| 5 structural anomalies | |
| 1 perinatal insult and 1 congenital hyperinsulinism 5 unknown etiologies | |
| Myoclonic epilepsy of infancy | 1 |
| Genetic febrile seizures plus | 1 |
| Epilepsy with myoclonic atonic seizures | 1 |
| Eyelid myoclonia | 1 |
| Epileptic encephalopathy with continuous spikes and waves during sleep | 2 |
| 1 unknown etiology and 1 chromosomal anomaly | |
| Landau‐Kleffner syndrome | 1 |
| Gelastic seizures with hypothalamic hamartoma | 1 |
| Epilepsy with malformations of cortical and brain development | 16 |
| 3 focal cortical dysplasias 1 schizencephalia | |
| 3 porencephalia/holoprosencephalies | |
| 9 corpus callosum hypoplasia/aplasias or other structural anomalies | |
| Epilepsy with tumor | 1 |
| 1 Glioma | 1 |
| Epilepsy with infection | 4 |
| 2 congenital infections | |
| 1 meningitis and 1 encephalitis | |
| Epilepsy with trauma | 2 |
| 1 traumatic brain injury and 1 near‐drowning | |
| Epilepsy with perinatal insult | 15 |
| 2 birth asphyxias | |
| 5 hypoxic ischemic encephalopathies 1 cerebral infarction | |
| 7 cases of intracerebral bleeding | |
| Epilepsy with stroke | 1 |
| 1 case of cerebral bleeding | |
| Epilepsy with metabolic reason | 4b |
| 2 cases of congenital hyperinsulinism | |
| 1 Glut1‐deficiency syndrome and 1 3‐methylglutaric aciduria | |
| Epilepsy with chromosomal anomalies and syndromes | 17c |
| 5 chromosomal anomalies | |
| 3 Catch syndromes | |
| 2 fragile‐X syndromes | |
| 1 Peho syndrome | |
| 1 Angelmann syndrome and 1 Aicardi syndrome 1 Rett syndrome and 1 Masa syndrome | |
| 1 Coats syndrome and 1 Sotos syndrome | |
| Focal epilepsy and developmental delay | 8 |
| Generalized epilepsy and developmental delay | 2 |
| Unclassified epilepsy and epilepsy of unknown etiology | 1 |
| Total | 101 |
Of all the children with epilepsy, cognitive impairment was seen in 101 out of 289, or 35%.
The West syndrome group includes patients from other groups such as those with epilepsy with malformation in cortical and brain development, chromosomal anomalies and syndromes, perinatal insult, and metabolic reasons.
The West syndrome group includes one congenital‐hyperinsulinism patient.
The West syndrome group includes one Peho‐syndrome patient.
Discussion
Our population‐based birth cohort study documented the prevalence of various epilepsy syndromes and etiologies of epilepsy in 289 children. It showed that childhood‐onset epilepsy does not consist merely of recurrent seizures—there is a great burden of symptomatic etiology and cognitive impairment. This disease burden affects day‐to‐day life and casts independent coping into question for those worst affected.
Our study found the incidence of epilepsy in early childhood to be high, with the highest incidence among those younger than 12 months old, at 141 per 100,000 per year. Recent data from Finland10 show a high annual incidence in infants during the corresponding era in the whole of Finland. The overall annual incidence of epilepsy in our study was 36 per 100,000 people per year when all other reasons for antiepileptic treatment were excluded from analysis, a figure comparable to those found in other population‐based studies: annual incidence of between 33 and 88 cases in 100,000 persons per year. In a similarity to other studies, the incidence of epilepsy tended to decline in the course of childhood.7, 8, 9 In many studies, the incidence uncovered is slightly higher in boys,6 but incidence was balanced between boys and girls in our work.
The misdiagnosis rate was low, at 3%. It was accounted for by patients who later were diagnosed with nonepileptic psychogenic events or syncope. In recent review, the prevalence of epilepsy misdiagnosis has varied largely between 2% and 70%, depending on the study.30 The low misdiagnosis rate found in our work is probably related mostly to the fact that the case facility, although serving as the area's primary specialist diagnostic unit for pediatric neurology, also has a tertiary epilepsy service with experienced clinical neurophysiologists and full video‐EEG service, which can also be used for diagnostic purposes. Hence, two important common contributors to the misdiagnosis of epilepsy—namely, overinterpretation of EEGs and lack of ictal EEGs—have been largely avoided.
In our study, nearly three‐quarters of patients had focal epilepsy, while less than a fifth had generalized epilepsy and the rest had mostly epileptic spasms. In general terms, incidence studies have found focal epilepsy to be more common in children.5, 6, 7
Under the most recent ILAE classification, electroclinical syndromes were found to constitute a third of childhood epilepsy cases after review of medical records. This is consistent with earlier work.7, 11 Most electroclinical syndromes were rare, but West syndrome had an incidence rate of 59 in 100,000 live births and is slightly higher than the reported.31We found only one epilepsy constellation, hypothalamic hamartoma and gelastic seizures, without any findings of mesial temporal‐lobe epilepsy and hippocampal sclerosis (<1%).32
Earlier studies have identified a structural etiology for epilepsy in about 25% of cases.15 In our study, structural anomalies accounted for roughly 30%, with perinatal insults, malformation in cortical and brain development, and head trauma covering the majority. A genetic or presumed genetic source accounts for another third of the etiologies, and the etiology is unknown in the remaining cases. It should be noted here that although genetic etiologies can be clearly defined as chromosomal and gene anomalies, presumed genetic ones are harder to delineate; the genetic basis of “idiopathic” epilepsies in general is quite well established, but that of focal “idiopathic” epilepsies specifically is more debatable.7, 33
Intellectual disability appears frequently in children with epilepsy, but alongside these cases are the clear majority: epileptic children whose cognition is categorized as normal yet who still face subtle cognitive difficulties that affect their future achievements.34 In our study, the mental‐impairment group accounted for about one‐third of all cases, with the majority in this category having a severe or profound impairment, as seen in earlier studies. Subtle cognitive difficulties were exhibited by a fifth of the children in this group, a finding that has emerged also in earlier studies.25 In the mentally impaired group, West syndrome with multiple etiologies was remarkably frequent. Cortical and brain malformation was a cause for about one‐fifth of the cases in this group. Other frequent causes of epilepsy and intellectual disability are antenatal and perinatal problems such as hypoxic ischemic encephalopathy, other perinatal insults, perinatal intracerebral bleeding or stroke, and congenital infections.
Treatable conditions such as Glut‐1 deficiency (one case) and congenital hyperinsulinism (three cases) were found in our study. The etiology was unknown for 2 patients, with whom there was a strong suspicion of metabolic disorder. Additional genetic screening might be helpful with respect to inherited metabolic disorders. In the mental‐impairment group, the etiology was unknown in under a fifth of cases (5 West syndrome patients and 8 focal‐epilepsy and developmental‐delay patients), while most of the intellectually normal children—especially in the focal‐epilepsy group—had an unknown etiology. On account of the retrospective nature of the study, DNA microarray technology was not available for some of the patients. It is an inescapable conclusion that further genetic studies are needed, from targeted mutation analysis to extended genetic screening such as next‐generation sequencing and whole‐exome sequencing.35
Strengths and limitations
Being a geographical population, our study population is representative of the general pediatric epilepsy population. An additional strength is that the retrospective design allowed us to reevaluate the diagnosis on the basis of all evidence available after follow‐up and hence evaluate the misdiagnosis rate also.
It is clear, however, that among the children born in the latter part of the recruitment period, there are still subjects who are going to receive an epilepsy diagnosis only later and who therefore are not covered by the study. Accordingly, our incidence figures are accurate for early childhood and somewhat underestimated for later childhood and adolescence, as compared, for example, with some other earlier studies from the same area.9, 36
At the same time, new etiologies, especially genetic ones, are being identified constantly, and it is clear from our retrospective data that the diagnostic work‐up for some of the patients was done many years ago, and that modern imaging and molecular diagnostics tools would have revealed more precise etiologies.
Conclusion
Epilepsy is a complex disease encompassing many etiologies and rare syndromes. The patient‐specific etiology and epilepsy syndrome are important determinants of the outcome and treatment selection. Accurate etiological diagnosis can be achieved for most children, whereas syndromic diagnosis is possible for only one‐third of them.
Disclosure of Conflict of Interest
Reetta Kälviäinen has received speaker's honoraria from Eisai, UCB, and Orion; honoraria for membership of advisory boards from Eisai, Fennomedical, GW Pharmaceuticals, Pfizer, Sage Therapeutics, and UCB; and research support for her institute from the Academy of Finland, Vaajasalo Foundation, Saastamoinen Foundation, UCB, and Eisai. None of the remaining authors have any potential conflicts of interest to disclose. We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
The study was supported by the Arvo and Lea Ylppö Foundation and the Finnish Epilepsy Research Foundation. We thank neuropsychologist Marja Äikiä, neurophysiologist Sara Määttä, and biostatistician Tuomas Selander for their advice on the analysis of the study material.
Biography
Arja Sokka, MD, is a pediatric neurologist and PhD student in Epilepsy Group, University of Eastern Finland.

References
- 1. Forsgren L, Beghi E, Oun A, et al. The epidemiology of epilepsy in Europe—a systematic review. Eur J Neurol 2005;12:245–253. [DOI] [PubMed] [Google Scholar]
- 2. Brodie MJ, Barry SJ, Bamagous GA, et al. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012;78:1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moshe SL, Perucca E, Ryvlin P, et al. Epilepsy: new advances. Lancet 2015;385:884–898. [DOI] [PubMed] [Google Scholar]
- 4. Thurman DJ, Beghi E, Begley CE, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011;52(Suppl. 7):2–26. [DOI] [PubMed] [Google Scholar]
- 5. Cowan LD. The epidemiology of the epilepsies in children. Ment Retard Dev Disabil Res Rev 2002;8:171–181. [DOI] [PubMed] [Google Scholar]
- 6. Wirrell EC, Grossardt BR, Wong‐Kisiel LC, et al. Incidence and classification of new‐onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population‐based study. Epilepsy Res 2011;95:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camfield P, Camfield C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord 2015;17:117–123. [DOI] [PubMed] [Google Scholar]
- 8. Sillanpaa M, Kalviainen R, Klaukka T, et al. Temporal changes in the incidence of epilepsy in Finland: nationwide study. Epilepsy Res 2006;71:206–215. [DOI] [PubMed] [Google Scholar]
- 9. Sillanpaa M, Lastunen S, Helenius H, et al. Regional differences and secular trends in the incidence of epilepsy in Finland: a nationwide 23‐year registry study. Epilepsia 2011;52:1857–1867. [DOI] [PubMed] [Google Scholar]
- 10. Saarinen MM, Sillanpaa M, Schmidt D, et al. Long‐term changes in the incidence of childhood epilepsy. A population study from Finland. Epilepsy Behav 2016;58:81–85. [DOI] [PubMed] [Google Scholar]
- 11. Eriksson KJ, Koivikko MJ. Prevalence, classification, and severity of epilepsy and epileptic syndromes in children. Epilepsia 1997;38:1275–1282. [DOI] [PubMed] [Google Scholar]
- 12. Mann JR, McDermott S. Maternal pre‐eclampsia is associated with childhood epilepsy in South Carolina children insured by Medicaid. Epilepsy Behav 2011;20:506–511. [DOI] [PubMed] [Google Scholar]
- 13. Syvertsen M, Nakken KO, Edland A, et al. Prevalence and etiology of epilepsy in a Norwegian county—a population based study. Epilepsia 2015;56:699–706. [DOI] [PubMed] [Google Scholar]
- 14. Guerrini R. Epilepsy in children. Lancet 2006;367:499–524. [DOI] [PubMed] [Google Scholar]
- 15. Berg AT. Epilepsy, cognition, and behavior: the clinical picture. Epilepsia 2011;52(Suppl. 1):7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shorvon SD. The etiologic classification of epilepsy. Epilepsia 2011;52:1052–1057. [DOI] [PubMed] [Google Scholar]
- 17. Scheffer IE, French J, Hirsch E, et al. Classification of the epilepsies: new concepts for discussion and debate. Special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia Open 2016;1:37–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nickels KC, Zaccariello MJ, Hamiwka LD, et al. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol 2016;12:465–476. [DOI] [PubMed] [Google Scholar]
- 19. Hamiwka LD, Wirrell EC. Comorbidities in pediatric epilepsy: beyond “just” treating the seizures. J Child Neurol 2009;24:734–742. [DOI] [PubMed] [Google Scholar]
- 20. Chin RF, Cumberland PM, Pujar SS, et al. Outcomes of childhood epilepsy at age 33 years: a population‐based birth‐cohort study. Epilepsia 2011;52:1513–1521. [DOI] [PubMed] [Google Scholar]
- 21. Park J, Yum MS, Choi HW, et al. Determinants of intelligence in childhood‐onset epilepsy: a single‐center study. Epilepsy Behav 2013;29:166–171. [DOI] [PubMed] [Google Scholar]
- 22. Camfield P, Camfield C, Arts WF, et al. The outcome of childhood epilepsy: what improvements are needed? Epileptic Disord 2013;15:101–104. [DOI] [PubMed] [Google Scholar]
- 23. Camfield C, Camfield P. Preventable and unpreventable causes of childhood‐onset epilepsy plus mental retardation. Pediatrics 2007;120:e52–e55. [DOI] [PubMed] [Google Scholar]
- 24. Camfield PR, Camfield CS. What happens to children with epilepsy when they become adults? Some facts and opinions. Pediatr Neurol 2014;51:17–23. [DOI] [PubMed] [Google Scholar]
- 25. Rantanen K, Eriksson K, Nieminen P. Cognitive impairment in preschool children with epilepsy. Epilepsia 2011;52:1499–1505. [DOI] [PubMed] [Google Scholar]
- 26. Raisanen S, Sokka A, Georgiadis L, et al. Infertility treatment and umbilical cord length‐novel markers of childhood epilepsy? PLoS ONE 2013;8:e55394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–482. [DOI] [PubMed] [Google Scholar]
- 28. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 29. Greenspan S, Woods GW. Intellectual disability as a disorder of reasoning and judgement: the gradual move away from intelligence quotient‐ceilings. Curr Opin Psychiatry 2014;27:110–116. [DOI] [PubMed] [Google Scholar]
- 30. Xu Y, Nguyen D, Mohamed A, et al. Frequency of a false positive diagnosis of epilepsy: a systematic review of observational studies. Seizure 2016;41:167–174. [DOI] [PubMed] [Google Scholar]
- 31. Gaily E, Lommi M, Lapatto R, et al. Incidence and outcome of epilepsy syndromes with onset in the first year of life: a retrospective population‐based study. Epilepsia 2016;57:1594–1601. [DOI] [PubMed] [Google Scholar]
- 32. Korff CM, Scheffer IE. Epilepsy classification: a cycle of evolution and revolution. Curr Opin Neurol 2013;26:163–167. [DOI] [PubMed] [Google Scholar]
- 33. Pal DK, Ferrie C, Addis L, et al. Idiopathic focal epilepsies: the “lost tribe.” Epileptic Disord 2016;18:252–288. [DOI] [PubMed] [Google Scholar]
- 34. Berg AT, Langfitt JT, Testa FM, et al. Global cognitive function in children with epilepsy: a community‐based study. Epilepsia 2008;49:608–614. [DOI] [PubMed] [Google Scholar]
- 35. Ream MA, Patel AD. Obtaining genetic testing in pediatric epilepsy. Epilepsia 2015;56:1505–1514. [DOI] [PubMed] [Google Scholar]
- 36. Sillanpaa M, Gissler M, Schmidt D. Efforts in epilepsy prevention in the last 40 years: lessons from a large nationwide study. JAMA Neurol 2016;73:390–395. [DOI] [PubMed] [Google Scholar]