

Abstract
Gestational choriocarcinomas are derived from placental trophoblast cells, with HLA-C being the only class I polymorphic molecule expressed. However, choriocarcinomas have not been profiled for endoplasmic reticulum aminopeptidase 2 (ERAP2) expression. ERAP2 trims peptides presented by human leukocyte antigens (HLA) that have shown to modulate immune response. Over 50% of choriocarcinomas we screened lack ERAP2 expression, which suggests that the absence of ERAP2 expression allows immune evasion of choriocarcinoma cells. We demonstrate that the ability of choriocarcinoma cells to activate lymphocytes was lowest with cells lacking ERAP2 (JEG-3) or HLA-C (JAr). This observation suggests that activation is dependent on expression of both ERAP2 and HLA-C molecules. In addition, an ERAP2 variant in which lysine is changed to asparagine (K392N) results in increased trimming activity (165-fold) for hydrophobic peptides and biologically never been detected. We hypothesize that homozygosity for the N392 ERAP2 variant is prohibited because it modulates the immune recognition of placental trophoblasts. We demonstrate that NK-cell activation and killing were significantly dependent on forced expression of the N392 ERAP2 isoform in JEG-3 cells. Cytotoxicity was confirmed by 7AAD killing assays showing that N392 ERAP2-isoform expressing JEG-3 cells had the highest percentage of apoptotic cells independent of the expression level of CD11a on lymphocytes. This is the first report showing that N392 ERAP2 promotes an immune clearance pathway for choriocarcinoma cells, and provides an explanation for why embryonic homozygosity for the N392 ERAP2 variant is not detected in any population.
Keywords: ERAP2, reproductive immunology, trophoblast, uterine cancer, apoptosis, choriocarcinoma
Summary Sentence
The N392 ERAP2 isoform expression promotes immune cell activation and apoptotic immune clearance of choriocarcinoma cells.
Introduction
Gestational choriocarcinomas arise from trophoblast cells in hydatidiform moles (50%), normal pregnancies (25%), and ectopic pregnancies or spontaneous abortions (25%). Although more common in Asia, Africa, and Central America, it is a rare disease (prevalence of 1 in 40,000 pregnancies and 1 in 40 hydatidiform cases) in Western countries. Individuals with hydatidiform moles have a relative risk of 2000–4000 for choriocarcinoma, making it one of the largest single risk factors for any known disease [1–3].
Another unique feature of choriocarcinoma is how it is recognized by the immune system. Human trophoblast cells express an unusual repertoire of human leukocyte antigen (HLA) molecules including the invariant HLA-G and HLA-E molecules and the polymorphic HLA-C molecule. They do not express the highly polymorphic HLA-A or HLA-B molecules [4]. The trophoblast cell lines in this study (BeWo, JAr, JEG-3, and T3M-3) are derived from choriocarcinomas with a mixture of villous and extravillous trophoblast, and they have a unique HLA phenotype of HLA-C, -G, and -E.
In the normal pregnancy, trophoblast cells may, depending upon genotype, express endoplasmic reticulum aminopeptidase 2 (ERAP2), which could play a role in immune tolerance. This enzyme trims antigenic peptide precursors in the endoplasmic reticulum (ER) to generate mature peptides that are bound and presented by HLA class I molecules. It is thus involved in determining the peptide repertoire presented for T-cell recognition [5–9]. ERAP2 has two haplotypes (haplotypes A and B). Andre et al. reported that ERAP2 transcripts associated with haplotype B undergo differential splicing to encode a truncated protein, which results in nonsense-mediated decay of the mRNA [10]. This study also demonstrated that haplotype B homozygotes had lower HLA class I expression on the cell surface, suggesting that naturally occurring ERAP2 deficiency affects HLA presentation and the immune response. Among the ERAP2 variants, a single nucleotide polymorphism (SNP) rs2549782 results in an amino acid substitution of lysine (K392) to asparagine (N392) near the catalytic center of the enzyme. This single amino acid change increases ERAP2-mediated peptide trimming of hydrophobic molecules by up to 165-fold, altering which peptides are available for binding to HLA molecules [11]. Thus, the N392 ERAP2 isoform could make cells more susceptible to immune surveillance, and increase immune-based destruction. Our hypothesis is that perturbation of immune surveillance by introduction of N392 ERAP2 prevents tumor and fetal growth by promoting apoptosis triggered by immune cells. However, there is no direct evidence linking evolutionary selection against N392 ERAP2 to tumor and fetal immunity.
ERAP2’s functional role in determining the peptide repertoire suggests that T cells would be responsible for the immune response. However, NK cells cannot be ignored because of their dominant presence in the uterus (70%), and their immediate response to any HLA changes. Thus, investigating if different ERAP2 variants affect the activation of NK and T cells could reveal whether one or both of the cell types promote an unfavorable tumor and fetal environment.
In contrast, ERAP2 deficiency contributes to immune evasion by tumor cells in vivo [10, 12]. Loss of the ERAP2 protein has been observed in renal carcinoma, colon adenocarcinoma, melanoma, and ovarian cancers, suggesting that the lack of ERAP2 benefits cancer growth and/or maintenance [12]. To promote cancer cell clearance by immune surveillance, affected cells must properly process the tumor-associated peptides by transporting, trimming, and presenting the peptide on the surface of the cells. Cancer cells evade the immune response by skipping one or more of these steps, as shown in previous studies. The role that ERAP2 plays in trimming of tumor-associated peptides provides potential therapeutic opportunities. However, this process is poorly understood in cancer.
ERAP1 and ERAP2 are zinc metallopeptidases of the oxytocinase M1 subfamily, which share consensus zinc-binding motifs essential for their enzymatic activity. ERAP enzymes trim amino acid residues from NH2 terminus of polypeptides [13]. The human ERAP1 and ERAP2 genes are located on chromosome 5q15 in the opposite orientation. Human ERAP2 has no orthologs in rodents, and evolutionary studies suggest that ERAP2 originates from a relatively recent duplication of ERAP1 [10]. Protein expression is seen in many tissues, and is strongly induced by type I and type II interferon (IFNs) [8] and tumor necrosis factor-alpha [14].
The concerted action of ERAP1 and ERAP2 determines the efficiency of peptide editing. However, as noted above studies with rodent models are limited because an ERAP2 orthologous gene is not present [15]. In our JEG-3 choriocarcinoma cell model, ERAP1 expression is constant, and ERAP2 variant expression is altered. This allows us to assess immune modulation determined by the combined actions of ERAP1 and ERAP2 variants.
The ERAP2 association in cancer supports the need to clarify the biological role of ERAP2 in modulating NK and T-cell-mediated immune responses in a choriocarcinoma model. The aim of this study was to elucidate the mechanism by which ERAP2 determines the fate of choriocarcinoma cells, NK cells, and T cells. We describe a novel in vitro model system that directly affects the immune response to HLA-C in the presence or absence of ERAP2 variants. Moreover, we demonstrate that introduction of the N392 ERAP2 variant into choriocarcinoma cells significantly increases their recognition and killing by NK cells.
Materials and methods
Human subjects
The studies were approved by the Virginia Commonwealth University IRB (HM20001364).
Trophoblast cell lines
The BeWo (ATCC CCL-98), JAr (ATCC HTB-144), and JEG-3 (ATCC HTB-36) choriocarcinoma cell lines were obtained from the ATCC. T3M-3 (RCB1018) is a gestational choriocarcinoma cell line of placental origin, obtained from Riken BioResource Center, Japan.
Cell culture and treatments
Cell lines were cultured in F-12K, RPMI-1640, MEM, or Ham's F-10 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cells were incubated at 37°C with 5% CO2. Cells were treated with IFN-γ (20 ng/ml) for 48 h at 37°C with 5% CO2.
DNA and RNA extraction
DNA was extracted from trophoblast cell lines BeWo, JAr, JEG-3, and T3M-3 using the Autopure system according to the manufacturer's instructions (Autogen). RNA was extracted from cell lines by using the Trizol method. Homogenized samples were removed from the flasks, centrifuged, mixed with chloroform, and then centrifuged. The aqueous layer was removed to an RNase-free tube where isopropyl alcohol was added. After centrifugation, the supernatant was removed from the tube containing the RNA gel-like pellet. The pellet was then washed with ethyl alcohol and allowed to dry before being resuspended in DEPC-treated water.
Genotyping
Single nucleotide polymorphism analysis for ERAP2 was performed using VIC- and FAM-labeled TaqMan Genotyping assays for SNP rs2248374 and SNP rs2549782 according to the manufacturer's protocol (Applied Biosystems). Real-time PCR was performed on extracted DNA samples by employing an ABI 7500 Fast Real-Time PCR Machine (Applied Biosystems) under the following conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles of amplification (92°C for 15 s and 60°C for 1 min).
HLA-C genotyping was performed at the VCU HLA core using PROTRANS AmpliPUR-Fast kit (Heidelberg, Germany) with genomic DNA from all four cell lines and blood donors.
RT-PCR
Complementary DNA for each cell line was prepared from 1 μg extracted RNA using Promega M-MLV Reverse Transcriptase (#M1701) with 1 μl of 10 U/μl Placental RNase Inhibitor, 2 μl Promega M-MLV 10x buffer, 2 μl oligo primer, 4 μl Invitrogen dNTP mix, and sufficient water added to bring the total reaction volume to 20 μl. An Eppendorf Mastercycler thermocycler was used with the following settings: 44°C for 1 h, 92°C for 10 min, and 4°C until ready. PCR reactions containing 1 μl cDNA template, 0.1 μl Invitrogen Platinum Taq DNA polymerase 2.5 μl 10x buffer, 0.75 μl 50 mM MgCl2, and between 7 and 10 μl 10 μM of the following primers with water up to 25 μl per reaction:
ERAP2 f2549782: 5΄- GGGGCCTCATTACATATAGGG -3΄
ERAP2 r2549782: 5΄- TTCCGCTGGTTTGGAGATAG -3΄
HLA-C forward: 5΄- GCCGCGAGTCCRAGAGG -3΄
HLA-C reverse P3: 5΄- GTTGTAGTAGCCGCGCAGG -3΄
An Eppendorf Mastercycler thermocycler was used with the following settings for each reaction:
ERAP2: 94°C for 5 min, 35 cycles of amplification (94°C for 30 s, 60°C for 30 s, 72° for 90 s), 72°C for 7min, and 4°C until ready.
HLA-C: 96°C for 1 min, 4 cycles of (96°C for 25 s, 70°C for 45 s, 72°C for 30 s), 26 cycles of (96°C for 25 s, 65°C for 45 s, 72°C for 30 s), 5 cycles of (96°C for 25 s, 58°C for 1 min, 72°C for 2 min), 72°C for 10 min, and 4°C until ready.
Western blotting
For protein extraction, cultured cells were pelleted and incubated in RIPA lysis buffer for 30 min at 4°C. Protein concentrations were determined by Lowry assay (BioRad). Total cellular protein (50 μg) was resolved on a 7.5% SDS-PAGE gel and transferred to PVDF membrane using the semidry transfer method. Membranes were blocked with 5% milk for 1 h at room temperature, and they were then incubated with primary antibody ERAP2 (R&D systems; #AF3830, dilution 0.1–0.2 μg/ml), HLA-C (AbCam; #ab126722, dilution 1:1000), and β-actin (Cell Signaling; #4967L, dilution 1:4000) overnight at 4°C. Blots were probed with HRP-conjugated secondary antibodies and visualized using Super Signal West PICO or Femto Luminal/Enhancer solution and West PICO or Femto stable peroxide solution (Thermo Scientific).
Immunofluorescence
Trophoblast cells were washed with phosphate-buffered saline (PBS) and then permeabilized with methanol (7 min, –80°C). The cells were then incubated overnight with ERAP2 (R&D Systems; #AF3830) antibody or anti-HLA-C (AbCam; #ab126722) and anti-E-Cadherin (Life Technologies; #131700) for detection. Cells were washed and incubated with Alexa Fluor 594 (Jackson ImmunoResearch; #715–585-150) and Alexa Fluor 488 (Jackson ImmunoResearch; #705–545-147) conjugated secondary antibodies. Nuclear stain DAPI was used as counterstain. The PFC region of sections was imaged using a Zeiss LSM 700 confocal laser-scanning microscope with the Ziess Axioimager. M2 microscope stand and a 20x/0.8 Plan-ApoChromat objective. Images were recorded using ZEN 2 (black edition) Carl Zeiss Microscopy GmbH, 2011 64-bit software.
Immunohistochemistry
Paraffin embedded tissue sections (4–7 μm) were heat fixed on to positively charged slides. Paraffin was removed from the slides using a Histo-Clear nontoxic histological clearing reagent (National Diagnostics; #HS-200) and hydrated by a series of alcohol to water washes. Sections were washed in 1× PBS for 10 min before being quenched for 30 min in a methanol/3% hydrogen peroxide solution. Sections being stained with ERAP2 antibody underwent antigen retrieval in a 10 mM citrate buffer for 5 min in a low-pressure cooker. The slides were then allowed to cool before resuming processing with a wash in 1 x PBS and blocking in 5% BSA for 20 min at room temperature. Sections stained with HLA-C antibody did not undergo antigen retrieval, but a blocking step was performed with Vectastain Goat Serum in 1× PBS.
Goat anti-human aminopeptidase LRAP/ERAP2 antibody (R&D Systems; #AF3830; diluted 1:20) was used for 1 h at room temperature or rabbit anti-HLA-C antibody [EPR6749] (Abcam; #ab126722; diluted 1:2000) was used overnight at 4°C. The slides were developed using the VECTASTAIN Elite ABC HRP Kit (Vector Laboratories; #PK-6100) and ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories; #SK-4105). Sections were counterstained using Vector Hematoxylin QS (Vector Laboratories; #H-3404) and destained with 0.05% acetic acid in acetone. Sections were finally rehydrated and mounted using VectaMount permanent mounting medium (Vector Laboratories; #H-5000).
Slides were examined using an Olympus BH-2 microscope. Pictures and scoring were taken with Olympus Q color5 camera and CellSens Dimension 1.15 ink software.
ImageJ ROI (region of interest) was used to perform quantitative analysis of histological staining as previously described [16].
Immune activation and cytotoxicity assays
JEG-3 and T3M-3 cells were cultured at 3.0 × 104 cells per well on 96-well plates with MEM+ media and Ham's F-10 media supplemented with penicillin/streptomycin and FBS. Transfection of the pTracer-CMV2-RFP plasmid expressing ERAP2N or ERAP2K was performed using FuGENE Transfection Reagent (Promega; #E2311). The expression plasmid was constructed such that ERAP2 protein carried a red florescent protein (RFP) tag for the immune activation assay or green florescent protein (GFP) tag for the cytotoxicity assay. Controls were treated with FuGENE Reagent alone. After 48 h, the transfection reagent was removed and fresh RPMI media supplemented with 10% FBS (heat inactivated) was added. Image acquisition was accomplished using a Zeiss Axiovert 40 CFL microscope with a 10×/0.25 Ph1 A-Plan objective and 20×/0.30 Ph 1 LD A-Plan objective. The images were taken using an Olympus Magnafire SP camera and MagnaFire SP 2.1B 1998–2001 software. After efficiency was established, peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of healthy pregnant and nonpregnant women using Histopaque (Sigma; #11191, #10771). Histopaque 1119 was added, and then Histopaque 1077 was layered on top. The preparation was centrifuged at 700 × g at room temperature for 30 min. Cells in the visible opaque layer on top of the Histopaque 1077 were harvested, washed twice with PBS, and then the cells were counted and 1.3 × 105 cells were added to each well. Plated cells were incubated overnight at 37°C for immune activation and 6 h for cytotoxicity. At the appropriate time points, lymphocytes were removed from wells and labeled with appropriate antibody listed below for flow cytometry analysis for immune activation. For the cytotoxicity assay, total PBMC and trophoblast cells were removed and stained with APC/Fire 750 mouse anti-human CD3 (BioLegend; #344840), PE anti-human CD56 (BioLegend; # 318306), and Brilliant Violet 421 anti-human CD154 (BioLegend; # 310824). They were then stained with 10 μM 7AAD (Cayman Chemical Company; #600120) for 20 min before the cells were photographed and analyzed with an Amnis ImageStreamX MarkII based on previously described protocols [17–20].
Caspase 3/7 cytotoxic assay
Immune activation assays as described above were assembled and incubated at 37°C for 4–5 h. After the lymphocytes were removed, the protocol for the Caspase-Glo 3/7 Assay (Promega; G8090) was followed according to the manufacturer's specifications. Chemiluminescence was detected using a POLARstar OPTIMA luminometer (BMG LabTECH).
Flow cytometry
FITC mouse anti-human CD3 (BioLegend; #300306), PerCP anti-human CD8 (BioLegend; #344708), PE anti-human CD56 (BioLegend; #318306), APC anti-human CD69 (BioLegend; #310910), PE/Cy7 anti-human CD11a (BioLegend; #301220), and corresponding IgG isotype controls were used for staining the lymphocytes. The lymphocytes were twice washed with FACS Buffer (1× PBS, 2% heat inactivated FBS) and centrifuged at 1600 RPM for 10 min at room temperature. The washed lymphocytes were blocked for 10 min in FACS Buffer and Human Trustain FcX Fc receptor blocking solution (Biolegend; #422302) at room temperature. The primary antibodies were used as per the manufacturer's instructions and incubated with the cells for 1 h at room temperature. The cells were then washed three times with FACS buffer, and they were then analyzed by flow cytometry at the VCU Flow Cytometry Core.
Statistical analysis
Lymphocyte activation and cytotoxic experiments were carried out in triplicate. Experiments were repeated three to six times. Two-tailed t-tests were performed with excel software or one-way ANOVA was performed with PRIZM software. A P value of less than 0.05 was considered to be a statistically significant difference versus control. The ERAP2 genotype and protein expression association was calculated using the Fisher exact test for count data.
Results
Most choriocarcinoma tumors lack ERAP2 expression
We evaluated 10 different choriocarcinoma specimens collected from women of South Korean origin and 4 choriocarcinoma cell lines (BeWo, JAr, JEG-3, and T3M-3) for the expression of ERAP2 and HLA-C to determine the association of these molecules with choriocarcinoma (Supplemental Table I). The expression level of ERAP2 was distinctive in each choriocarcinoma tissues analyzed. More than half of the tumors lacked ERAP2 as demonstrated by immunohistochemistry (Figure 1 and Supplemental Figure S1A). The ERAP2 expression level was significantly greater in positive tissues compared to negative tissues (Supplemental Figure S1B). However, HLA-C expression level or detection using immunohistochemistry analysis of choriocarcinoma tumor samples was not conclusive (data not shown).
Figure 1.
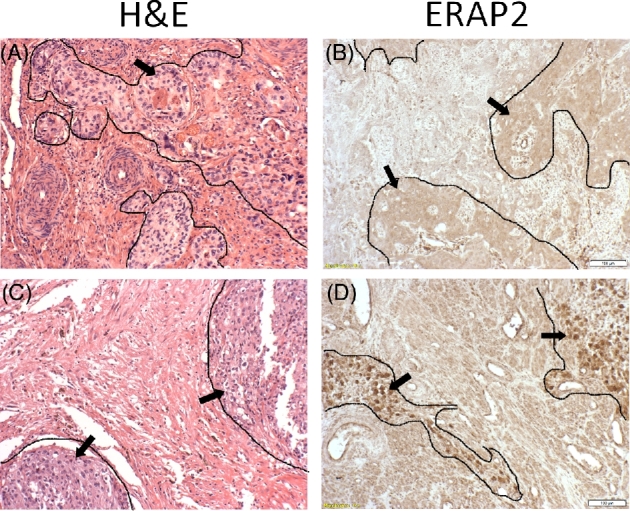
ERAP2 expression in choriocarcinoma. Tumor tissue sections obtained from Korean women were evaluated by immunohistochemistry which demonstrated absence or presence of ERAP2 expression. Outlined areas and arrows indicate areas of tumor tissue. Two representative samples are shown: H&E stains (A, C) of both samples show characteristic cells of choriocarcinoma tissue. ERAP2− (B), and ERAP2+ (D).
The cell lines were able to show distinctively that JAr and T3M-3 showed ERAP2 mRNA expression (Figure 2A), but surprisingly the immunofluorescence analysis and western blotting did not detect any ERAP2 protein expression in all the cell lines (Figure 2B and C). On the other hand, HLA-C mRNA and protein expression pattern nicely correlated that BeWo, JEG-3, and T3M-3 show detection in both molecules.
Figure 2.

ERAP2 and HLA-C profiles in choriocarcinoma cell lines. (A) Reverse transcriptase analysis of choriocarcinoma cell lines. RNA was extracted from BeWo, JAr, JEG-3, and T3M-3 cells and converted to cDNA. ERAP2 mRNA was detected in JAr and T3M-3 cells only, whereas HLA-C mRNA expression was detected in all cell lines tested, except JAr. (B) Protein localization analysis by immunofluorescence using primary antibodies against E-cadherin (red), ERAP2, and HLA-C (green). ERAP2 expression was undetectable in all cell lines. HLA-C expression detected in BeWo, JEG-3, and T3M-3. HLA-C was not detected in JAr cells. (C) Western blot analysis of cells stimulated by IFNγ. Protein was extracted from cell lines with or without IFNγ treatment. Western blot analysis showed that T3M-3 is the only one undetectable level of ERAP1. Noticeably, the unstimulated cells lacked ERAP2 detection and only the JAr and T3M-3 cells produce the detectable level of ERAP2 protein when stimulated with IFNγ. A representation of β-Actin loading control is shown. (D) Densitometry analysis of the ERAP2 expression level. Only JAr and T3M-3 showed a significant increase of ERAP2 expression level with IFNγ treatment (*P = 0.0253, **P = 0.0127). (E) Densitometry analysis of the HLA-C expression level. BeWo and JEG-3 showed a significant increase of HLA-C expression level with IFNγ treatment (*P = 0.0031, **P = 0.0077) but T3M-3 did not show a significant increase of HLA-C expression level with IFNγ treatment (ns P = 0.0619). Each experiment was repeated three times with three replicates in each experiment.
Both mRNA and protein expression analysis revealed that only JAr cells did not express HLA-C. HLA-C mRNA was observed to be more than 80-fold high in BeWo and JEG-3, in comparison to JAr and T3M-3 cells (Figure 2A and Supplemental Figure S2A). Interestingly, even though T3M-3 had relatively low HLA-C mRNA, the protein detection was the strongest in immunofluorescence analysis (Figure 2B). Regardless, all three (BeWo, JEG-3, and T3M-3) cell lines expressed HLA-C protein detected by both immunofluorescence and western blotting analyses (Figure 2B and C).
An ERAP2 genetic variant affects protein expression in trophoblast cell lines
To determine why some of the choriocarcinoma cell lines screened lacked expression of ERAP2 protein, we genotyped the ERAP2 SNPs that affect mRNA stability and the amino acid at position 392. The G allele of rs2248374 is a “loss-of-function” variant leading to RNA instability and loss of ERAP2 protein expression [10]. The T allele of rs2549782 is a “change-in-function” variant that translates into asparagine (N392), leading to substrate-specific changes in enzymatic activity [11]. This information allowed us to identify the optimal cell line to use to determine the function of ERAP2 variants and the consequent immune responses.
DNA genotyping revealed that T3M-3 and JAr cells are heterozygous for both rs2248374 (A/G) and rs2549782 (G/T), while BeWo and JEG-3 cells are homozygous rs2248374 (G) and rs2549782 (T) for both (Table 1). T3M-3 and Jar cells are heterozygous for the two SNPs, and because of the strict linkage disequilibrium (LD) between the two they are predicted to express K392 ERAP2, but not N392 due to RNA instability of the encoding transcript [10]. On the other hand, lack of protein expression in BeWo and JEG-3 in the face of some detectable message can be explained by nonsense-mediated RNA decay that leaves modest ERAP2 available for PCR amplification (Figure 2A) [10, 21]. There is a perfect correlation between G/G rs2248374 and T/T rs2549782 genotypes and the absence of ERAP2 protein expression in BeWo and JEG-3 cells (Figure 2B and C). The odds ratio is infinite due to a zero denominator. All expected cell count frequencies are <5. Fisher exact test yields a significant association (P = 0.000333).
Table 1.
ERAP2 genotype and protein expression in cell lines.
| Cell | SNP rs2248374 | SNP rs2549782 | ERAP2 without |
|---|---|---|---|
| line | (A/G) | (G/T) | IFNγ/with γ |
| BeWo | GG | TT | ND/ND |
| JAr | AG | GT | ND/++ (*P = 0.0233) |
| JEG-3 | GG | TT | ND/ND |
| T3M-3 | AG | GT | ND/+ (*P = 0.0127) |
ND = not detected.
ERAP2 protein is not detected until cells are stimulated with IFNγ. Cell lines that are heterozygous for SNP rs2248374 produce detectable levels of ERAP2 protein.
Differential expression of ERAP1, ERAP2, and HLA-C in BeWo, JAr, JEG-3, and T3M-3 cells with IFNγ stimulation
According to our genotype data, JAr and T3M-3 cells should express the ERAP2 protein, and BeWo and JEG-3 cells should not due to nonsense-mediated mRNA decay. In contrast to the mRNA expression patterns noted above, none of these cell lines expressed ERAP2 protein without IFNγ stimulation demonstrated by immunofluorescence analysis and western blotting (Figure 2B and C). The ERAP2 immunofluorescence labeled column only shows blue DAPI staining of the nucleus and red E-cadherin labeling outlines of the trophoblast cells. The discordance between the mRNA and protein expression levels of ERAP2 was also reported in melanoma cell lines suggesting that protein expression is controlled not only by transcription but also translational or post-translational mechanisms [5].
To determine if ERAP2 and HLA-C expression patterns change during immune activation when exposed to cytokines, we treated the choriocarcinoma cells with IFNγ, known to upregulate ERAP2 and HLA-C expression [22, 23]. We observed that 48 h of IFNγ treatment elevated protein expression of ERAP2 only in JAr and T3M-3 cells. However, IFNγ treatment could not stimulate BeWo and JEG-3 cells to express ERAP2 (Figure 2C), correlating the ERAP2 rs2248374 (“loss-of-function”) genotype analysis (Table 1). The absence of ERAP2 protein expression in BeWo and JEG-3 cell lines despite low levels of RNA can be explained by nonsense-mediated decay of the mRNA [24]. T3M-3, BeWo, and JEG-3 cells with endogenous HLA-C protein showed a distinct augmented protein expression post-IFNγ treatment, while JAr cells did not, suggesting that HLA-C protein expression in JAr cells is not inducible (Figure 2C, Table 2). The densitometry analysis of ERAP2 and HLA-C western blots revealed that IFNγ stimulation of JAr and T3M-3 cell lines significantly increased ERAP2 protein expression and HLA-C expression in BeWo and JEG-3 cells (Figure 2D and E).
Table 2.
HLA-C genotype and protein expression in cell lines.
| Cell | HLA-C | HLA-C protein expression without |
|---|---|---|
| line | genotype | IFNγ/with IFNγ |
| BeWo | Cw04/Cw07 | +/++ *P = 0.0031 |
| JAr | Cw06/Cw015 | ND/ND |
| JEG-3 | Cw04/Cw7 | +/+++ #P = 0.0077 |
| T3M-3 | Cw01 | +/++ @P = 0.0619 |
ND = not detected.
The JAr cell line does not produce a detectable level of HLA-C protein even when stimulated with IFNγ. All other cell lines express HLA-C in varying isoforms.
A previous study demonstrated that both ERAP1 and ERAP2 are required for proper trimming of the peptides [7]. Therefore, we screened for ERAP1 protein expression in these cell lines to assess how it might cooperatively play a role in immune modulation. Western blot analysis revealed that only T3M-3 did not express ERAP1, even after IFNγ stimulation. BeWo cells expressed a modest amount of ERAP1 in the presence of IFNγ. JAr and JEG-3 cells showed the highest level of ERAP1 expression (Figure 2C). We analyzed ERAP1 SNPs that are reported to be associated with functional differences. Supplemental Table II presents data on the three ERAP1 SNPs screened in these cell lines (rs27895, rs30187, and rs27044). JEG-3 cells are heterozygous (A/G) for rs30187, which is associated with less efficient trimming [25].
To determine if IFNγ had any effect on other HLA molecules that could affect immune modulation, HLA-E or -G molecules were detected by western blotting. We observed that HLA-E expression levels increased with IFNγ as expected in all cell lines. BeWo and JAr cells did not express HLA-G (Supplemental Figure S2).
In summary, when cell lines were analyzed post-IFNγ treatment, every cell line lacked either ERAP1, ERAP2, or HLA-C. The BeWo and JEG-3 cell lines lacked ERAP2 expression, the JAr cell line lacked HLA-C expression, and the T3M-3 cell line lacked ERAP1 expression. The presence of only HLA-C class I cell surface markers on JEG-3 and absence of endogenous ERAP2 protein make it an ideal in vitro model for elucidating the interplay between exogenous ERAP2, HLA-C-mediated antigen presentation and the subsequent immunological response.
HLA-C genotype of the cell lines and the immune cell donors
We genotyped HLA-C alleles of the cell lines (Table 2) and the donors (Table 3) to distinguish immune response due to HLA mismatch or due to the effect of ERAP2 variant expression. JAr and T3M-3 are heterozygous for Cw04/Cw07. BeWo is hemizygous for Cw01, and JEG-3 is heterozygous for Cw06/Cw15.
Table 3.
Donor lymphocyte HLA-C genotype.
| Donor | HLA-C | Source |
|---|---|---|
| H1 | Cw04/Cw16 | Cord blood |
| H2 | Cw16/Cw17 | Cord blood |
| H8 | Cw03/Cw07 | NP peripheral |
| H9 | Cw03/Cw06 | NP peripheral |
| H10 | Cw06/Cw07 | Peripheral |
| H14 | Cw06/Cw06 | Peripheral |
NP = nonpregnant donor.
A sample of lymphocyte donor sources and the very different HLA-C genotype of each used in lymphocyte activation assays and cytotoxicity assays.
We had three different sources of blood donors for the lymphocyte activation assays: cord blood, peripheral blood from nonpregnant donors, and peripheral blood from pregnant donors. Table 3 lists the HLA-C genotypes of the donors. Most of the lymphocyte activation assays were performed with HLA-C mismatches. However, the lymphocyte activation level due to mismatch was consistently low, and independent of different HLA-C allele combinations.
Differential NK and T-cell activation in trophoblast cell lines dependent on ERAP2 and HLA-C expression
A lymphocyte activation assay was performed using the choriocarcinoma cell lines as stimulators to detect lymphocyte activation level. Peripheral blood mononuclear cells were incubated with naïve JAr (ERAP2+, HLA-C–), JEG-3 (ERAP2–, HLA-C+), and T3M-3 (ERAP2+, HLA-C+) cells and then stained with fluorescently labeled antibodies specific for all T cells (CD3), cytotoxic T cells (CD8), NK cells (CD56), and an early activation (CD69) cell surface marker. The lymphocyte population exposed to T3M-3 cells positive for both ERAP2 and HLA-C molecules showed highest percentage of total number of activated T cells (CD8+CD69+) and NK cells (CD56+ CD69+) in comparison to those exposed to JEG-3 (ERAP2 negative) or JAr (HLA-C negative) cells (Figure 3A). When the subpopulation of lymphocytes was analyzed, small but not significant differences were observed in T cells (CD8+), with significantly higher numbers of NK cells (CD56+) (Figure 3B, P < 0.05). The presence of both ERAP2 and HLA-C protein in T3M-3 cells appears to trigger an exaggerated immune response toward choriocarcinoma cells by NK cells.
Figure 3.

Lymphocyte activation by choriocarcinoma cells. (A) PBMCs with HLA-C haplotype Cw03 and Cw07 were exposed to JAr (ERAP2+, HLA-C−), JEG-3 (ERAP2−, HLA-C+), or T3M-3 (ERAP2+, HLA-C+) cells, and they were then analyzed by flow cytometry for the detection of CD3 (all T cells), CD8 (T cells), CD56 (NK cells), and CD69 (activation). Percentages in the dot plots represent the active CD3+CD8+CD69+ T cells or CD3–CD56+CD69+ NK cells. (B) The histogram depicts the calculated percentage of subpopulations of T-cell activation (CD8+, CD69+) and NK-cell activation (CD56+, CD69+) in response to incubation with each cell preparation. Each experiment was repeated three times with three replicates in each experiment. There was significant activation of NK cells exposed to T3M-3 cells (one way ANOVA statistical analysis: * = P < 0.05), while T-cell activation was not significant compared to T cells exposed to other cell lines.
N392 ERAP2 isoform expressing JEG-3 cells manifest a differential immune response
The JEG-3 cell line appears to be an ideal cellular model to demonstrate immune modulation by the ERAP2 variants with four distinct characteristics: (1) it only lacks ERAP2 (other antigen process molecules such as the proteasome LMP7, peptide transporter TAP-1/TAP-2, and ERAP1 are present) [26]; (2) ERAP2 expression cannot be induced with IFNγ; (3) HLA-C is expressed in addition to HLA-G and HLA-E; and (4) it provokes the lowest immune response in the absence of ERAP2 protein expression. The lymphocyte activation assays were performed using JEG-3 cells devoid of endogenous ERAP2, cells transfected with empty vector, and with JEG-3 cells transfected with either K392 ERAP2 or N392 ERAP2 expressing pTracer-RFP plasmids to demonstrate the immune effects of these two ERAP2 variants. The transfection was successful for both JEG-3(K) and JEG-3(N) cells (Figure 4A). ERAP2 and HLA-C protein expression was detected by western blot analysis (Figure 4B). The relative protein expression levels according to densitometry analysis showed that the HLA-C expression level was constant among treatment groups (Figure 4C, P > 0.05). However, both ERAP2 variant transfected JEG-3 cells showed significantly higher expression of ERAP2 compared to untransfected JEG-3 cells (Figure 4C, P < 0.05). N392 ERAP2 protein expression levels in transfected cells were not significantly different compared to the K392 ERAP2 protein levels, but immune-induced apoptosis was consistently higher with N392-expressing cells compared to K392-expressing cells.
Figure 4.

Transfected JEG-3 cells express ERAP2 isoforms, and HLA-C expression level is unchanged. ERAP2-transfected JEG-3 cells were used as the stimulator cells in a lymphocyte activation assay. (A) A plasmid employing the RFP and either K392 or N392 ERAP2 protein transfected into JEG-3 cells was employed for lymphocyte activation assays. (B) Western blot analysis shows overexpression of K392 and N392 ERAP2 protein level, which did not produce any change in HLA-C expression. (C) Bar graph of densitometry of transfected JEG-3 cells shows relative protein expression of ERAP2 and HLA-C. One-way ANOVA of ERAP2 expression of JEG-3(K) and JEG-3(N) indicates a significant difference compared to untransfected JEG-3 cells (* and # P < 0.05, n = 3). There were no statistically significant differences in HLA-C expression among experimental groups JEG-3(K) or JEG-3(N) compared to the control group of JEG-3 cells without transfection (P > 0.05, n = 3).
A shift in lymphocyte activation was observed with stimulator JEG-3 cells expressing N392 ERAP2 protein compared to K392 ERAP2 or empty vector (Figure 5A). Specifically, only the activation of CD56+ NK cell was increased when stimulated by N392 ERAP2-expressing JEG-3 cells. However, there was not a significant percent activation of CD69+ cells either by NK or T cells compared to the empty vector (Figure 5B). This observation suggests that the empty vector induces some response. Therefore, it is critical to determine if activation specific to ERAP2 causes immune-induced apoptosis. First, we determined if the activated lymphocytes have upregulated CD11a, which indicates the ability to engage and kill the target cells. We detected relatively similar CD11a levels on NK and T cells (Figure 5A and B), indicating all experimental JEG-3 cell groups stimulated lymphocytes.
Figure 5.

N392 ERAP2 causes differential lymphocyte activation. (A) Representative dot plot of the flow cytometry analysis performed with PBMCs with HLA-C haplotype Cw03 and Cw06. The average percentage of T cells (CD3+, CD8+), NK cells (CD3–, CD56+), and both for CD69+ activation marker from the total number of lymphocytes is demonstrated. CD11a+ T and NK cells surface marker indicative of attachment and killing was also observed. There was no significant difference in CD11a expression level between the lymphocytes exposed to cells expressing ERAP2 isoforms and those that were not transfected. (B) The bar graphs show the percent activation of T and NK-cell populations exposed to transfected and nontransfected JEG-3 cells. There were no statistically significant differences across experimental groups (P > 0.05, n = 3). (C) Overlay histogram of PBMCs exposed to transfected and nontransfected JEG-3 cells shows that activated T cells are relatively the same between treatments. However, NK cells exposed to N392 ERAP2-expressing JEG-3 cells show a shift in intensity of the CD69+ activation maker. All experiments repeated at least three times with three replicates in each experimental group.
NK cells preferentially target and kill N392 ERAP2-expressing JEG-3 cells
We investigated the ability of NK and T cells to kill target cells using a 7AAD-apoptosis detection system to determine the specific population responsible for the killing of the ERAP2 variant expressing JEG-3 cells compared to JEG-3 cells transfected with an empty vector [11]. Even though NK cells proliferated more than T cells, they both expressed CD11a at similar levels, suggesting that both NK and T cells were able to attach and kill the N392 ERAP2 protein expressing JEG-3 target cells as demonstrated in Figure 6A and Supplemental Figure S3A [27, 28]. However, corresponding with the higher proliferation rate of NK cells, NK-cell-bound 7-ADD apoptotic N392 ERAP2 positive JEG-3 cells were almost 10 times (n = 3, 23.9% vs. 2.71%) more prevalent compared to T-cell-bound 7-AAD apoptotic JEG-3 cells (Figure 6D, P = 0.0000213).
Figure 6.

Lymphocytes attach and kill trophoblasts expressing ERAP2. (A) A transfected GFP-producing JEG-3 trophoblast cell can be seen with a T cell and NK cell attached. The T cell is labeled with APC/Fire 750 anti-CD3 (dark pink) and the NK cell is labeled with PE–CD56 (yellow) and BV421 anti-CD40L (purple). Cells transfected with the empty vector (GFP+ = green) are larger and healthier and do not have T or NK cells attached. (B) Dot plots from flow cytometry showed differences in the healthy populations between cells transfected with N392 or K392 ERAP2, or an empty vector and nontransfected cells when exposed to PBMCs. Live, rounded, and healthy cells appear on the top left plot along the left vertical axis. The percentage represents the amount of gated “healthy” cells. Red cells have a varying degree of 7AAD stain. (C) The bar graph shows the percentage of apoptotic cells stained positive with 7AAD in transfected in all experimental groups compared to JEG-3 nontransfected and JEG-3 transfected with a vector without ERAP2. Cytotoxicity assays were run three times with up to three replicates of each experimental groups. The significant cell death observed in JEG-3 cells expressing N392 ERAP2 isoform (*P = 0.040). (D) The table displays the percentage of NK or T cell bound 7AAD positive N392 ERAP2 JEG-3 cells and the significant difference between the two cell types was calculated using the Student t-test (P = 0.0000213). SD, standard deviation.
The higher cell death observed in N392 ERAP2 JEG-3 group was supported by the aspect ratio dot plot which demonstrated a lower amount of healthy cells in N392 ERAP2 protein expressing JEG-3 cells compared to JEG-3 cells transfected with empty vector and K392 ERAP2 (Figure 6B). More debris and irregular shaped cells resulting from cell death were observed with N392 ERAP2 transfected JEG-3 cells exposed to lymphocytes compared to untransfected JEG-3 cells or JEG-3 cells transfected with an empty vector. 7-AAD-positive dying cells were measured, and a significant level was observed only with N392 ERAP2 protein expressing JEG-3 cells compared to JEG-3 cells transfected with empty vector (Figure 6C: *P = 0.0013).
To confirm the increased apoptotic death of N392 ERAP2-expressing JEG-3 cells, we repeated the lymphocyte activation and killing experiment and measured the caspase3/7 levels as a marker of apoptosis. We used the total PBMC which included all mononuclear cells. The caspase 3/7 detection level was significantly higher luminescence in experimental groups expressing N392 ERAP2 compared to empty vector (Supplemental Figure S3B: *P = 0.0013).
Discussion
Cancer cells can avoid inducing an immune response by a variety of mechanisms including the following: (1) inhibiting tumor antigen transport to the ER, (2) improper trimming of peptides in the ER, and (3) inadequate peptide presentation on HLA class I molecules. Currently, there is evidence that disruptions in antigen transport and antigen presentation are used in immune evasion [29, 30]. However, the role of the antigen-trimming step by ER-resident amino peptidases, specifically ERAP2, in the immune escape mechanism has yet to be fully elucidated. The data presented here demonstrate that 8 out of 14 choriocarcinoma samples do not express ERAP2, suggesting that a lack of the antigen-processing enzyme could allow the cancer cell to escape immune surveillance. The genetic analysis of JEG-3 and BeWo choriocarcinoma cell lines indicated that the lack of expression is due to homozygosity for a genetic variation resulting in a transcript that undergoes nonsense-mediated mRNA decay [10]. This mode of immune escape may also be ideal for promoting pregnancy tolerance since it is documented that normal patients have lower ERAP2 transcripts compared to patients suffering from preeclampsia (PE). The placenta of PE patients had higher mRNA expression of ERAP2 and associated with clinical severity [31]. Unfortunately, the absence of ERAP2 expression in trophoblast cells, which could be beneficial to a successful pregnancy, may increase the chance of developing choriocarcinoma.
The potential mechanisms that determine the fate of choriocarcinoma cells related to ERAP2 activity are presented in Figure 7. In this study, we addressed one hypothesis that the fate is changed by forced expression of N392 ERAP2 variant by altering immune activation and killing ability. When the ERAP2 peptide-trimming activity increased in these cells by N392 isoform, the T and NK-cell activation and killing ability of these lymphocytes increased as well. Interestingly, the activation of immune cells was only apparent when endogenous HLA-C and ERAP2 both were present as seen in T3M-3 cells. ERAP2 expression in the absence of HLA-C in JAr cells results in only minimal lymphocyte activation simply due to an HLA-mismatch response. This observation suggests that both molecules are required for immune recognition in trophoblast cells. Further investigation is needed to determine the ratio of activating and inhibiting HLA-C molecules on these cell lines to confirm observation on HLA-C-associated differences in immune response.
Figure 7.

Illustration of ERAP2 immune evasion or anti-tumor cytotoxic mechanism hypothesis. Immune evasion is illustrated here with ERAP1 alone trimming limited antigenic peptides preventing immune clearance, whereas ERAP1 and N392 ERAP2 combined efficiently trimming antigenic peptides inducing cytotoxic immune response.
Based on our findings, the absence of ERAP2-mediated changes in antigen presentation via HLA-C may contribute to the complex molecular mechanisms that result in tolerance of choriocarcinoma cells and promoting a safe environment for pregnancy.
Our JEG-3 cell model also suggests that the susceptibility of cancer cells to cell-mediated immunity is influenced by the peptide trimming effects of different ERAP1/2 dimers that may have altered this repertoire. In JEG-3 cells, ERAP1 expression was constant, and we varied only the expression of the K392 ERAP2 and N392 ERAP2 proteins. Our immune activation assay clearly demonstrated a functional difference between ERAP1/K392 and ERAP1/N392. Even though the ERAP1 genotype data (Supplemental Table II) indicated that JEG-3 cells have ERAP1 protein with less trimming efficiency, the ERAP1/N392 combination strikingly demonstrated increased apoptosis of targeted cells by NK and T cells. This observation suggests that the peptide repertoire and presentation was dependent on ERAP2 isoform expression to determine the level of different immune responses. Similarly, with our low immune response with ERAP1/K392 combination, Bouvier et al. have shown that the ERAP1/2 (inactive/active) combination results in poor trimming activity of ERAP2 to both free and bound precursors [32]. Both of these observations imply that not only do ERAP2 variants determine its ability to trim peptides, but that peptide trimming also depends on ERAP1 isoforms forming either heterodimers or homodimers. Because we were able to keep ERAP1 expression constant in JEG-3 cells, we were able to clearly demonstrate that N392 ERAP2 played a major role in modulating immune response possibly by more efficient peptide trimming. These results warrant further investigation to define how different ERAP1/2 variant combinations influence the peptide repertoire and subsequent immune response.
The T-cell modulation by N392 ERAP2 was minimal compared to NK-cell activation level; however, the CD11a killing marker was expressed in equal amount on the surface of both lymphocytes (Figure 5A and B). This may explain why we observed significant apoptotic cell death of JEG-3 cells expressing N392 ERAP2 by both NK and T-cell killing (P < 0.05). Most of the cells visualized using ImageStream had NK and T cells attached, suggesting that both NK and T cells were inducing apoptosis. However, when the direct comparison of NK or T cell bound N392 JEG-3 + 7AAD was made independently, it was evident that more NK cells were bound and, therefore, responsible for inducing apoptosis.
The most striking observation was that apoptosis was significantly higher with JEG-3 cells expressing N392 ERAP2, even though CD11a was equally expressed on lymphocytes stimulated by JEG-3 cells transfected with an empty vector. This suggests that CD11a is not the only marker indicative of NK and T-cell ability to kill, and that additional molecules must be involved that distinctively target JEG-3 cells expressing N392 ERAP2 isoform.
It would not be surprising that limited T-cell killing may have been due to alteration of the peptide repertoire by “hyper” trimming of N392 ERAP2. It is likely that a shift in tumor epitope generation presented by HLA-C is responsible for an elevated immune response. Further investigation with peptide stripping is necessary to determine if a difference in the level of tumor-specific peptide presentation is responsible for the slight elevation of T-cell activation observed in this study. Equally interesting is the significant increase in NK-cell activation that could be explained by the alternative hypothesis that hyper trimming destroys the self-epitope lowering the HLA-C surface expression, marking the cell as “nonself” as illustrated in Figure 7. Both hypotheses warrant further investigation.
The apoptotic cell death in N392 ERAP2-expressing cells suggests a significant role in the immunological response during early pregnancy by potentially altering allogeneic antigen presentation by HLA-C. This is the first evidence to explain why evolutionary selection against N392 ERAP2 is observed in the Chilean population we have previously studied [21]. We never detected the homozygous TT genotype of rs2549782 in Chilean neonates or mothers with the homozygous AA genotype for rs2248374 that could result in a N392 ERAP2 homodimer. In addition, in all previous studied populations, the five SNP haplotype structures of ERAP2 pair with G allele of rs2248374 (loss of function) with the major T allele of rs2549782 (change in function), such that N392 ERAP2 isoform will never be expressed [10]. This strong LD preventing ERAP2 expression or elimination of N392ERAP-expressing embryos by immune surveillance may drive evolution. Our studies support this notion and for the first time demonstrate the N392 ERAP2’s ability to turn on the killing mode of T and NK cells providing an immunological explanation for why we never detect homozygosity for the N392 ERAP2 isoform in populations studied.
Our discovery of an immunological response leading to increased apoptotic death of choriocarcinoma cells expressing the N392 ERAP2 variant provides a possible explanation for why homozygosity for the N392 ERAP2 variant has not been detected in humans. The ability of N392 ERAP2 to elicit a strong NK-cell immune response could be employed as a novel therapy to clear choriocarcinoma cells and other difficult to treat cancers.
Supplementary data
Supplementary data are available at BIOLRE online.
Supplemental Table I. ERAP2 and HLA-C expression in choriocarcinoma samples and cell lines. Out of 10 tumor tissue samples and four cell line samples, only T3M-3 cell line expressed both ERAP2 680 or HLA-C proteins.
Supplemental Table II. Cell line ERAP1 SNP genotype. Three different SNPs of ERAP1 were genotyped to determine the cell lines with different trimming ability of ERAP1.
Supplemental Table III. List of all antibodies utilized in this study.
Online supplemental material. Figures S1–S3 include ERAP2 expression patterns and quantitative analysis of all Korean choriocarcinoma samples, quantitative PCR of ERAP2 and HLA-C transcripts of the choriocarcinoma cell lines, and HLA-E/-G expression level in choriocarcinoma cell lines. Lastly, additional apoptotic images of N392 ERAP2-expressing JEG-3 cell compared to healthy JEG-3 cell with a vector without N392 ERAP2 and validation of apoptosis detection by caspase 3/7 activation.
Acknowledgments
We thank VCUHS HLA Laboratory Director Dr Pam Kimball for assisting us in HLA-C genotyping. We especially thank the Labor and Delivery nurse and staff, Gloria Anderson and Karen Smith, for their efforts in recruiting patients for the study. RFP and GFP-labeled plasmids used in the research discussed above were engineered and generously supplied by the Amalfitano Lab at Michigan State University. Our lab would also like to acknowledge the help of Dr Maria Teves with the arrangement of figures and statistical analysis, Julie S. Farnsworth with experimental set up of the FLOW based assays, and Anthony Rodriguez with the gathering together of data files. The authors thank Dr Kevin T. Hogan and Dr Daniel Conrad for editorial assistance in the preparation of the manuscript.
Footnotes
Grant support: Research for this paper was supported by funds from NIH-NICHD grant R01 HD073555-S1 and supplemented by the Massey Cancer Center Cores by NCI P30 CA016059 and the NIH-NINDS Center Core Grant 5P30 NS047463.
References
- 1. Yoon A, Verma S, Birnbaum A. Exudative retinal detachment caused by metastatic choriocarcinoma to the choroid. J Emerg Med 2013; 44:617–619. [DOI] [PubMed] [Google Scholar]
- 2. Iverson L. Choriocarcinoma and related conditions in Southeast Asia. Schweiz Z Pathol Bakteriol 1958; 21:581–586. [DOI] [PubMed] [Google Scholar]
- 3. Bracken MB, Brinton LA, Hayashi K. Epidemiology of hydatidiform mole and choriocarcinoma. Epidemiol Rev 1984; 6:52–75. [DOI] [PubMed] [Google Scholar]
- 4. Apps R, Murphy SP, Fernando R, Gardner L, Ahad T, Moffett A. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology 2009; 127:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kamphausen E, Kellert C, Abbas T, Akkad N, Tenzer S, Pawelec G, Schild H, van Endert P, Seliger B. Distinct molecular mechanisms leading to deficient expression of ER-resident aminopeptidases in melanoma. Cancer Immunol Immunother 2010; 59:1273–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanioka T, Hattori A, Masuda S, Nomura Y, Nakayama H, Mizutani S, Tsujimoto M. Human leukocyte-derived arginine aminopeptidase. J Biol Chem 2003; 278:32275–32283. [DOI] [PubMed] [Google Scholar]
- 7. Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, Schomburg L, Fruci D, Niedermann G, van Endert PM. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol 2005; 6:689–697. [DOI] [PubMed] [Google Scholar]
- 8. Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M, Goldberg AL. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol 2002; 3:1169–1176. [DOI] [PubMed] [Google Scholar]
- 9. York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol 2002; 3:1177–1184. [DOI] [PubMed] [Google Scholar]
- 10. Andres AM, Dennis MY, Kretzschmar WW, Cannons JL, Lee-Lin SQ, Hurle B, Schwartzberg PL, Williamson SH, Bustamante CD, Nielsen R, Clark AG, Green ED. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet 2010; 6:e1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Evnouchidou I, Birtley J, Seregin S, Papakyriakou A, Zervoudi E, Samiotaki M, Panayotou G, Giastas P, Petrakis O, Georgiadis D, Amalfitano A, Saridakis E, Mavridis IM, Stratikos E. A common single nucleotide polymorphism in endoplasmic reticulum aminopeptidase 2 induces a specificity switch that leads to altered antigen processing. J Immunol 2012; 189:2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fruci D, Giacomini P, Nicotra MR, Forloni M, Fraioli R, Saveanu L, van Endert P, Natali PG. Altered expression of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in transformed non-lymphoid human tissues. J Cell Physiol 2008; 216:742–749. [DOI] [PubMed] [Google Scholar]
- 13. Hattori A, Matsumoto H, Mizutani S, Tsujimoto M. Molecular cloning of adipocyte-derived leucine aminopeptidase highly related to placental leucine aminopeptidase/oxytocinase. J Biochem 1999; 125:931–938. [DOI] [PubMed] [Google Scholar]
- 14. Forloni M, Albini S, Limongi MZ, Cifaldi L, Boldrini R, Nicotra MR, Giannini G, Natali PG, Giacomini P, Fruci D. NF-kappaB, and not MYCN, regulates MHC class I and endoplasmic reticulum aminopeptidases in human neuroblastoma cells. Cancer Res 2010; 70:916–924. [DOI] [PubMed] [Google Scholar]
- 15. Cifaldi L, Lo Monaco E, Forloni M, Giorda E, Lorenzi S, Petrini S, Tremante E, Pende D, Locatelli F, Giacomini P, Fruci D. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res 2011; 71:1597–1606. [DOI] [PubMed] [Google Scholar]
- 16. Jensen EC. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec 2013; 296:378–381. [DOI] [PubMed] [Google Scholar]
- 17. Zuba-Surma EK, Kucia M, Abdel-Latif A, Lillard JW Jr, Ratajczak MZ. The ImageStream System: a key step to a new era in imaging. Folia Histochem Cytobiol 2007; 45:279–290. [PubMed] [Google Scholar]
- 18. George TC, Basiji DA, Hall BE, Lynch DH, Ortyn WE, Perry DJ, Seo MJ, Zimmerman CA, Morrissey PJ. Distinguishing modes of cell death using the ImageStream® multispectral imaging flow cytometer. Cytometry 2004; 59:237–245. [DOI] [PubMed] [Google Scholar]
- 19. Jiang L, Tixeira R, Caruso S, Atkin-Smith GK, Baxter AA, Paone S, Hulett MD, Poon IK. Monitoring the progression of cell death and the disassembly of dying cells by flow cytometry. Nat Protoc 2016; 11:655–663. [DOI] [PubMed] [Google Scholar]
- 20. Zembruski NC, Stache V, Haefeli WE, Weiss J. 7-Aminoactinomycin D for apoptosis staining in flow cytometry. Anal Biochem 2012; 429:79–81. [DOI] [PubMed] [Google Scholar]
- 21. Vanhille DL, Hill LD, Hilliard DD, Lee ED, Teves ME, Srinivas S, Kusanovic JP, Gomez R, Stratikos E, Elovitz MA, Romero R, Strauss JF 3rd. A novel haplotype structure in a chilean population: implications for erap2 protein expression and preeclampsia risk. Mol Genet Genomic Med 2013; 1:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shin EC, Seifert U, Urban S, Truong KT, Feinstone SM, Rice CM, Kloetzel PM, Rehermann B. Proteasome activator and antigen-processing aminopeptidases are regulated by virus-induced type I interferon in the hepatitis C virus-infected liver. J Interferon Cytokine Res 2007; 27:985–990. [DOI] [PubMed] [Google Scholar]
- 23. Keskinen P, Ronni T, Matikainen S, Lehtonen A, Julkunen I. Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells. Immunology 1997; 91:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andres AM, Dennis MY, Kretzschmar WW, Cannons JL, Lee-Lin SQ, Hurle B, Schwartzberg PL, Williamson SH, Bustamante CD, Nielsen R, Clark AG, Green ED. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet 2010; 6:e1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reeves E, Colebatch-Bourn A, Elliott T, Edwards CJ, James E. Functionally distinct ERAP1 allotype combinations distinguish individuals with Ankylosing Spondylitis. Proc Natl Acad Sci USA 2014; 111:17594–17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roby KF, Fei K, Yang Y, Hunt JS. Expression of HLA class II-associated peptide transporter and proteasome genes in human placentas and trophoblast cell lines. Immunology 1994; 83:444–448. [PMC free article] [PubMed] [Google Scholar]
- 27. Lantos J, Grama L, Orosz T, Temes G, Roth E. Leukocyte CD11a expression and granulocyte activation during experimental myocardial ischemia and long lasting reperfusion. Exp Clin Cardiol 2001; 6:72–76. [PMC free article] [PubMed] [Google Scholar]
- 28. Bose TO, Pham QM, Jellison ER, Mouries J, Ballantyne CM, Lefrancois L. CD11a regulates effector CD8 T cell differentiation and central memory development in response to infection with Listeria monocytogenes. Infect Immun 2013; 81:1140–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tai W, Chen Z, Cheng K. Expression profile and functional activity of peptide transporters in prostate cancer cells. Mol Pharm 2013; 10:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinial relevance. Immunol Today 1995; 16:487–494. [DOI] [PubMed] [Google Scholar]
- 31. Yong HE, Murthi P, Borg A, Kalionis B, Moses EK, Brennecke SP, Keogh RJ. Increased decidual mRNA expression levels of candidate maternal pre-eclampsia susceptibility genes are associated with clinical severity. Placenta 2014; 35:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen H, Li L, Weimershaus M, Evnouchidou I, van Endert P, Bouvier M. ERAP1-ERAP2 dimers trim MHC I-bound precursor peptides; implications for understanding peptide editing. Sci Rep 2016; 6:28902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.