

Abstract
We report the clinical, neuroimaging, and neuropathologic characteristics of 2 patients who developed early onset dementia after a moderate-severe traumatic brain injury (TBI). Neuropathological evaluation revealed abundant β-amyloid neuritic and cored plaques, diffuse β-amyloid plaques, and frequent hyperphosphorylated-tau neurofibrillary tangles (NFT) involving much of the cortex, including insula and mammillary bodies in both cases. Case 1 additionally showed NFTs in both the superficial and deep cortical layers, occasional perivascular and depth-of-sulci NFTs, and parietal white matter rarefaction, which corresponded with decreased parietal fiber tracts observed on ex vivo MRI. Case 2 additionally showed NFT predominance in the superficial layers of the cortex, hypothalamus and brainstem, diffuse Lewy bodies in the cortex, amygdala and brainstem, and intraneuronal TDP-43 inclusions. The neuropathologic diagnoses were atypical Alzheimer disease (AD) with features of chronic traumatic encephalopathy and white matter loss (Case 1), and atypical AD, dementia with Lewy bodies and coexistent TDP-43 pathology (Case 2). These findings support an epidemiological association between TBI and dementia and further characterize the variety of misfolded proteins that may accumulate after TBI. Analyses with comprehensive clinical, imaging, genetic, and neuropathological data are required to characterize the full clinicopathological spectrum associated with dementias occurring after moderate-severe TBI.
Keywords: α-Synuclein, β-Amyloid, Hyperphosphorylated tau, Neurodegeneration, Neurofibrillary tangle, Proteinopathy, Traumatic brain injury
INTRODUCTION
It has long been recognized that severe traumatic brain injury (TBI) in early and mid-life is associated with an increased risk of late life dementia (relative risk 1.5–5.0) (1, 2). This association between TBI and dementia is based primarily on epidemiological investigations of patients with remote TBI and late-onset (over 65 years of age) probable Alzheimer disease (AD) and other dementias. In epidemiological studies, the diagnosis of AD is usually based on clinical grounds (probable AD) rather than definite (postmortem) examination (3–17). In a recent population-based study of individuals over age 65 who were dementia-free at initial assessment, a documented history of prior TBI was not associated with an elevated risk of subsequent AD or dementia; however, individuals with early-onset (i.e., before age 60) TBI-associated dementia were excluded from this study (18). Discordance across epidemiologic studies is probably due to methodological heterogeneity as well as to diagnostic uncertainty (11, 12), compounded by the current absence of validated clinical criteria for TBI-associated dementia. Further, the pathophysiologic mechanisms underlying this association have not been established. It is generally believed that TBI may initiate or accelerate molecular cascades involved in several different neurodegenerative processes associated with dementia, including AD, Parkinson disease, amyotrophic lateral sclerosis (ALS), and chronic traumatic encephalopathy (CTE) (19–21).
There have been few studies examining the neuropathological features of early-onset dementia among long-term survivors of single TBI (22–25). Furthermore, among the cases with long-term survival reported to date, comprehensive data pertaining to the timing, characteristics and progression of cognitive impairment (including behavioral and neuropsychological assessments) have not been available. Here, we report 2 cases of early-onset dementia after mid-life moderate-severe TBI and associated cognitive, behavioral, neuroimaging, and neuropathologic characteristics.
MATERIALS AND METHODS
For each case, we performed a medical chart review to characterize the mid-life TBI, its clinical presentation and recovery, followed by the clinical course and progression of dementia. Two brains received a comprehensive neuropathological evaluation, including microscopic examination of sections from 26 brain regions stained with hematoxylin and eosin (H&E), modified Bielschowsky, Luxol fast blue (LFB), and immunohistochemically for ß-amyloid (Aß 1–42, EMD Millipore [St. Louis, MO], 1:2000, pretreated with 88% formic acid for 2 minutes; or Aβ-4G8, BioLegend [San Diego, CA], 1:1000, pretreated with formic acid), hyperphosphorylated tau (p-tau) (AT8, Thermo Fisher Scientific/Pierce [Rockford, IL], 1:2000), TDP-43 (anti-TDP-43, rabbit monoclonal anti-TDP43, Abcam [Cambridge, UK], 109535, dilution 1:100), α-synuclein (rabbit monoclonal antiphosphor S129, dilution 1:100, Abcam 51253), and CD68 (mouse antihuman monoclonal antibody clone 514H12, Leica Biosystems [Wetzlar, Germany] PA0273).
Accumulations of abnormal protein were identified and their abundance was assessed semiquantitatively on a scale of 0–3 as follows: 0 = none, 1 = mild, 2 = moderate, and 3 = severe. Additionally, amyloid and tau deposits were assessed according to CERAD and Braak staging criteria (26, 27). In Case 1, the fixed right hemisphere underwent ex vivo 7 Tesla MRI using a multi-echo flash sequence at 200 µm resolution and 3 Tesla MRI using a diffusion-weighted sequence with 90 diffusion-weighted measurements at 750 µm resolution. Ex vivo 7 Tesla MRI was performed using previously described methods (28, 29). Ex vivo diffusion data acquisition, processing, and deterministic tractography was performed using previously described methods (30).
RESULTS
Case 1: Clinical History
The patient, a retired male senior military officer with multiple postgraduate degrees, had a history of concussion with brief loss of consciousness (LOC) during combative training in his second decade of life and no family history of neuropsychiatric diseases. At age 46, he experienced a second TBI from a motor vehicle accident with LOC of unknown duration. His initial Emergency Department (ED) Glasgow Coma Scale (GCS) score was 11. There he was agitated and combative, and experienced a single generalized tonic-clonic seizure. Computed tomography (CT) scan of the head did not show any intracranial abnormalities. He was hospitalized for 12 days (3 in the intensive care unit) and discharged home for outpatient rehabilitation. The duration of his posttraumatic amnesia (PTA) was 18 days. Based on Department of Defense (DoD)-Veterans Administration (VA) criteria (31), his TBI was classified as moderate/severe (moderate by GCS score and severe by PTA duration).
One month postinjury, he continued to experience significant cognitive impairment (e.g. he could name only the current president and 4 animals in 1 minute). Brain MRI showed a single focus of increased signal intensity in the left periventricular white matter (WM). Over the next 18 months, he made a near-complete cognitive recovery but reported persistent dysnomia, irritability, and diminished short-term memory. Twenty-one months after injury, he performed within the normal range on a comprehensive neuropsychological examination with only mild cognitive deficits and returned to full active duty, performing well in a major military command position.
At age 51 (4.5 years after his TBI), he retired from the military with normal cognitive exam (MMSE 30/30) but decline in performance skills from 1 year after his TBI. He was employed full time in the private sector until the age of 59 (12.5 years after his TBI), when he complained of increasing fatigue and persistent, worsening depression and memory difficulties. He underwent serial neuropsychological testing, which demonstrated a progressive decrement in performance IQ with initial preservation of verbal IQ (Supplementary Data Table S1a). From age 59, he experienced progressive neurocognitive decline characterized by behavioral/affective symptoms (depression, irritability, agitation, and fatigability), abnormal cognition in multiple domains (memory, visuospatial, calculations, and executive functions), and recurrent brief spells of hyperventilation and confusion, which were never associated with epileptiform activity on multiple scalp electroencephalogram recordings and which continued unabated until his death. Gross motor functions were preserved until very late in his dementia. At the age of 63 (17 years after his TBI), he recalled 0/3 items at 5 minutes and scored 20/30 on the MMSE. At age 67 (20 years after his TBI), his MMSE decreased to 11/30 with marked decrement in serial clock drawings across these time points (Supplementary Data Fig. S1). He had 3 1.5 Tesla brain MRIs performed 5, 7 and 10 years before his death. The last MRI obtained at age 67 (5 years before his death) reported global volume loss (based on clinical read by a neuroradiologist) and multifocal hyperintense signals in the subcortical and periventricular WM with no change from prior scans (Supplementary Fig. S2). His level of agitation increased and included public outbursts of violence requiring intervention. He was moved to a secure nursing facility for his safety and care. He died at the age of 72 with a diagnosis of severe dementia, 25 years after his TBI and 12 years after his cognitive decline became clinically evident. During his neurocognitive decline, he was variably diagnosed with “TBI-related frontotemporal dementia”, “probable AD”, or “cortical dementia”.
Neuropathological Findings
Gross examination of the brain showed marked diffuse atrophy (formalin-fixed brain weight: 930 gm) and a very thinned posterior corpus callosum (CC) with preservation of the anterior CC, preserved hippocampal volume, and a remote left inferior frontal gyrus contusion (Fig. 1A–C).
FIGURE 1.

Macroscopic appearance of the brain of a subject (Case 1) with dementia after a moderate-severe TBI. Thinning of the posterior corpus callosum and myelin loss. Gross appearance of brain specimen from Case 1. (A) The intact brain, left cerebral (contusion labeled with dashed arrows) and cerebellar hemispheres. (B) Coronal cross-section of the left hemisphere at the level of the caudate/putamen and anterior corpus callosum (aCC). (C) Coronal large cross-section of left hemisphere at the level of the hippocampus (Hip) and posterior corpus callosum (pCC). Of note, the marked intrasulcal space enlargement (as per diffuse cortical atrophy) (arrows, A) and marked thinning of the posterior corpus callosum (pCC) (arrow, C) contrasts the relatively preserved thickness of the anterior corpus callosum (aCC) (arrow, B), and the relatively normal appearance of the hippocampus (C). Panel C shows a LFB-hematoxylin stain of the left posterior corpus callosum (pCC). Panel D shows a LFB-hematoxylin stain of the left superior temporal cortex. These stains demonstrate extensive loss of myelin in the pCC and adjacent centrum semiovale (CS). Of note, the patchy myelin loss in temporal lobe spares the U-fibers (U). Cortical pathology associated with contusions are indicated by red arrows in panels C and D. The images were obtained using a 2.5× objective.
LFB-stained sections showed marked loss of myelin in the thinned posterior CC (Fig. 1D), patchy distributions of myelin loss in diffuse subcortical WM sections (Fig. 1D, E), and micro-contusions in the inferior orbitofrontal cortex (Fig. 1D). At higher magnification, there was minimal myelin staining with LFB in the posterior CC and scattered CD68-immunopositive amoeboid microglial cells. High levels of amyloid-ß neuritic plaques (Aß-NP), primarily diffuse rather than core Aß plaques, were observed in nearly all regions examined (olfactory bulb, frontal, orbito-frontal-cortex, parietal, superior, middle and inferior temporal, anterior cingulate, anterior insular, occipital, amygdala, basal ganglia, thalamus, mammillary bodies, and hypothalamus, hippocampus; Fig. 2A–E). In the occipital cortex, perivascular amyloid accumulation and cerebral amyloid angiopathy was found (Fig. 2D). Frequent NFTs and tau neurites were observed across almost all cortical and subcortical examined areas. In the parietal cortex there was superficial and deep cortical layer tau staining, as well as isolated regions of tau staining at the depths of sulci and perivascularly (Fig. 3A–D), the latter the pathognomonic lesion of CTE. NFTs were found predominantly in superficial layers of the insula and in the mammillary bodies. Both the rare perivascular and extensive superficial cortical and mammillary body tau deposits are very unusual neuropathological findings in late-onset AD. Subpial and perivascular astrocytosis (GFAP-positive lesions) was observed across all cortical regions, but was less significant than the total cortical tau and amyloid burden. No α-synuclein-positive lesions (Lewy bodies or Lewy neurites) were found in the cortex or brainstem. TDP-43 staining was negative. Apolipoprotein E (APOE) genotype was ɛ3/ɛ3. Supplementary Data Table S2 summarizes the neuropathological findings for this case (Case 1) in semiquantitative fashion.
FIGURE 2.

Extracellular β-amyloid accumulation in the cortex of a subject (Case 1) with dementia after moderate-severe TBI. High levels of extracellular β-amyloid accumulation detected by 4G8 antibody (1–42 β-amyloid) across various regions of the brain: olfactory bulb (A), middle frontal cortex (B), parietal cortex (C), occipital cortex (D), and hippocampus (E). Other notable findings include β-amyloid-positive cerebral congophilic angiopathy (CCA) in the vessels of the middle frontal cortex (B) and high frequency of diffuse β-amyloid plaques in the middle frontal cortex (B), parietal cortex (C), occipital cortex (D), and hippocampus (E). In the occipital cortex (D), immunohistochemistry (4G8) shows peculiar and unusually dense accumulation of β-amyloid around intracortical vessels. Objective 20 × (olfactory bulb; A), 5 × (middle frontal [B], parietal [C], occipital [D] cortices, and hippocampus [E]).
FIGURE 3.

AD- and CTE-like p-tau accumulation in the cortex of a subject (Case 1) with dementia after moderate-severe TBI. High levels of p-tau accumulation were seen across different regions of the brain: olfactory bulb, parietal cortex, occipital cortex, and hippocampus. Importantly, we observed the coexistence of 2 different types of p-tau pathology: AD-like and CTE-like p-tau pathology (red arrows, parietal cortex). This is a rare finding that is difficult to identify when diffuse and high levels of p-tau deposition are present in a brain. Tau immunohistochemistry with AT8 antibody. Objective 5 × (olfactory bulb, occipital cortex); 2.5 × (parietal cortex, hippocampus). (A) A portion of the olfactory bulb and tract positive for p-tau lesions; (B) p-tau lesions in the occipital cortex (note the perivascular distribution of p-tau pathology); (C) rarely observed copresence of 2 types of p-tau accumulation in the same region (parietal cortex): AD pathology (across all deeper cortical layers) and p-tau sulcal pathology (more superficial cortical layers), which is pathognomonic of CTE; (D) Diffuse p-tau pathology across all hippocampus.
Ex Vivo Neuroimaging
Axial images from ex vivo 7 Tesla MRI proton density, synthesized FLASH and T2* parameter maps demonstrated right parietal WM rarefaction, including in the region of the transcallosal fibers that cross the thinned splenium (Fig. 4A). Diffusion tractography of the CC revealed focal areas of WM injury, with disruption of parietal fibers crossing the splenium of the CC and frontal fibers crossing the genu of the CC, but relative preservation of frontal fibers crossing the body of the CC and occipito-temporal fibers crossing the splenium (Fig. 5A). To address the possibility of a technical artifact causing the CC tractography findings, tractography analyses of the ipsilateral corticospinal tract and cingulum bundle were performed as internal controls. These control tractography analyses revealed relative preservation of the corticospinal tract and cingulum bundle (Fig. 5B, C), as compared to the extensive transcallosal tract disruptions, with near-complete absence of parietal fibers crossing the splenium. MRI performed 5 years before his death show a thinned posterior CC and atrophy but not the extensive subcortical WM gliosis that was evident on ex vivo imaging (Fig. 5D).
FIGURE 4.
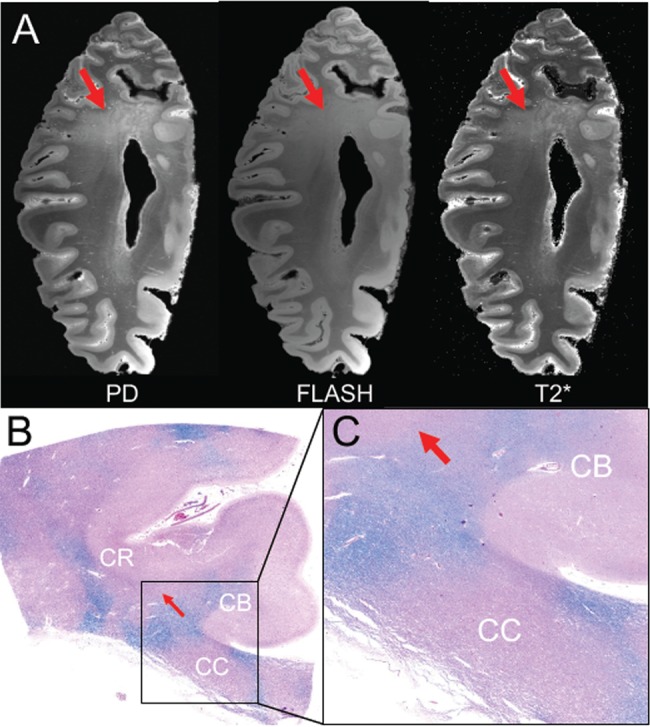
Imaging-histopathological correlations in a subject (Case 1) with dementia after a moderate-severe TBI. (A) Axial images from the proton density (PD) map, FLASH acquisition and T2* parameter map (T2*) derived from the ex vivo 7T MRI scan demonstrate rarification of myelin in the parietal white matter (red arrows). (B) Corresponding section from the parietal region that underwent ex vivo MRI. The histological section was stained with hematoxylin and eosin (H&E) and counter-stained with Luxol fast blue (LFB) for detection of myelin. This histological section (B), which is shown in zoomed view in panel (C), reveals patches of myelin loss throughout the coronal radiata (CR) and corpus callosum (CC). The LFB stain is more prominent in the cingulum bundle (CB), indicating that this white matter pathway is relatively spared.
FIGURE 5.

Diffusion tractography findings of white matter in a subject (Case 1) with dementia after a moderate-severe TBI. Diffusion tractography results indicate focal regions of white matter injury. (A) Right lateral view of interhemispheric, transcallosal fiber tracts generated with a region of interest traced in the corpus callosum (CC). Fiber tracts from nearby white matter bundles (e.g., fornix, cingulum bundle) have been eliminated to optimize visualization. Transcallosal tracts are intact in parts of the frontal and temporal lobes, but they are disrupted in other regions such as the parietal lobe (arrow). These parietal tractography findings are consistent with the ex vivo 7T images seen in Figure 7A that show rarification of the parietal hemispheric white matter, as well as the histopathologic data in Figures 7B and C showing white matter injury in this region. To exclude the possibility that global loss of white matter and/or technical artifact is responsible for the transcallosal tractography results, we show relative preservation of corticospinal tract (CST) fibers from a right lateral view (B) and cingulum bundle (CB) fibers from a right lateral view (C). Corresponding imaging from an MRI (D) obtained 4 years before his death does not show the extensive gliosis visible in the ex vivo imaging (arrow).
Case 2: Clinical History
The patient was a female college graduate with a family history of early onset AD (mother). Prior to her TBI, she worked in a daycare center, as a waitress and as a trademark researcher. At the age of 39, she was hospitalized following a motor vehicle accident resulting in a severe TBI with complex polytrauma. Head CT revealed multiple intracranial abnormalities including right brainstem injury, multifocal cerebral contusions, possible hypoxic injury, and multiple intracerebral hemorrhages (bilateral throughout the cerebral parenchyma with major focal areas within the left temporal lobe and the anterior right cerebellum) and subarachnoid hemorrhage in the left Sylvian fissure. The patient had LOC for approximately 24 hours, followed by a posttraumatic confusional state with fluctuating levels of alertness, sleep disruption and periods of severe agitation. PTA resolved at approximately 6 weeks postinjury. She fulfilled DoD-VA criteria for a severe TBI based on the duration of LOC and PTA.
Within 2 years of her TBI, she recovered full independence at home and in the community, working as a volunteer in a library but unable to return to her previous job. In the years following the TBI, she reported increased levels of anxiety, depression, headaches, and memory problems. She began noticing a progressive decline in memory and concentration approximately 4–5 years after the TBI. Previously she functioned independently, managing her own finances, actively volunteering at a library and driving. Twelve years post-TBI, at age 51, she underwent neuropsychological evaluation. No deficits of attention or verbal ability were observed, but she had significant deficits in executive function and semantic memory. Subsequent serial neuropsychological assessments reflected a progressive decline in these domains until her death at age 63 years. MMSE scores declined from 26 at 13 years post-TBI to 18 at 18 years post-TBI with a rapid decline to 5, 3 years later (Supplementary Data Table S1b).
At the age of 54 (15 years post-TBI) she was diagnosed with possible AD. She continued to experience anxiety and depression. By age 58 (19 years post-TBI) she was unable to live alone, requiring help with activities of daily living, and misusing previously familiar objects. She began exhibiting mild parkinsonism, which progressed to moderate rigidity, stiffness, flexed posture, and shuffling gait by age 61. She developed periods of confusion, paranoia, irritability and agitation beginning at age 57, and by age of 60 she became more withdrawn, passive and apathetic. She died at age 63 (24 years after her TBI) with a clinical diagnosis of probable AD.
Neuropathological Findings
The formalin-fixed brain weighed 920 grams. There was severe atrophy of the frontal lobes and anterior medial temporal lobes bilaterally. The parietal, temporal, and occipital lobes showed mild–moderate atrophy. The cerebral WM was diffusely atrophied and the CC was severely thinned at the level of the posterior thalamus. There were small (1.5-cm diameter) contusions in the anterior frontal poles and anterior left medial temporal pole. The ventricles were severely enlarged, especially the frontal horns of the lateral ventricles. Bilaterally, the entire hippocampal formations were atrophic. The amygdala and entorhinal cortex were moderately atrophic. The thalamus and hypothalamus were atrophied; the mammillary bodies were very atrophic and displayed a brown discoloration (Fig. 6A). There was severe cellular depigmentation of the substantia nigra and locus coeruleus (Fig. 6B).
FIGURE 6.
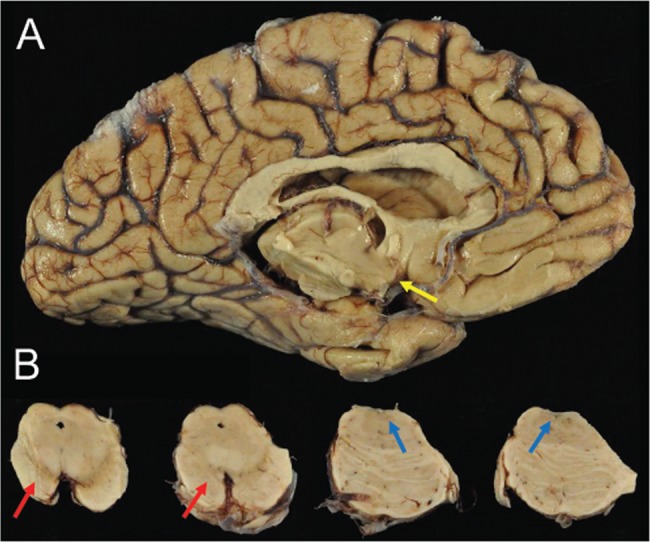
Macroscopic appearance of the brain of a subject (Case 2) with dementia after a single severe TBI. (A) There was marked atrophy of cerebral cortex, corpus callosum, and medial temporal lobe with severe neuronal loss in amygdala, mammillary bodies (yellow arrow), entorhinal cortex, and hypothalamus. (B) Images show (from left to right) mesencephalon including substantia nigra (SN) and locus coeruleus (LC) with pigmented neuronal loss in SN (red arrows) and LC (blue arrows).
Microscopic neuropathologic examination demonstrated severe neuronal loss in the amygdala, entorhinal cortex, hypothalamus, substantia nigra, and locus coeruleus. In addition, marked neuronal loss with spongiform change and gliosis was present in the temporal lobe (Fig. 7A, B) and there was neuronal loss and gliosis in CA1 hippocampus (Fig. 8) with evidence of focal remote microhemorrhages (Fig. 7C, D). There was also widespread perivascular hemosiderin-laden macrophage deposition and microgliosis in WM and focal remote microhemorrhage in subinsular WM and CA1 hippocampus (Fig. 7E–G). Large axonal spheroids were present in the hypothalamus. Abundant NFT and tau dot-like lesions were predominantly found in the superficial layers of cerebral cortex but were found throughout the deep gray and brainstem nuclei (Fig. 8A–L) and were especially prominent in the mammillary bodies (Fig. 8E), both atypical locations for tau deposition in late-onset AD. AT8 immunoreactivity was observed in thorned-shaped astrocytes in the subpial regions and occasionally around small blood vessels in a CTE like pattern (Fig. 8B). There were dense NFTs and tau-neurites in the lateral dorsal and periventricular thalamus, hypothalamus, nucleus basalis of Meynert, mammillary bodies, brainstem tegmentum, substantia nigra, median raphe, and locus coeruleus (Fig. 8A–L). Abundant diffuse Aß plaques and Aß-NP were also found (Fig. 9A–D). There was also vascular amyloid deposition in the leptomeninges (Fig. 9C). Unusually large α-synuclein intraneuronal inclusions were found in mammillary bodies, substantia nigra, and hypothalamus, with widespread intraneuronal Lewy bodies and Lewy neurites in the medial temporal lobe, cingulate cortex, medulla, and olfactory bulb (Fig. 10A–H). Scattered extracellular α-synuclein deposits were also seen in the locus coeruleus, diencephalon, and mammillary bodies. The size and shape of the α-synuclein deposits, particularly in the temporal cortex, locus coeruleus, and Edinger–Westphal nucleus are highly atypical features in LBD. Additionally, TDP-43-positive intraneuronal inclusions and dot-like structures were found in the frontal and temporal cortices, amygdala, entorhinal cortex, caudate nucleus, and hippocampus, including the dentate gyrus (Fig. 11A–D). APOE genotype was ɛ4/ɛ4. Supplementary Data Table S3 summarizes the pathological findings for this case (Case 2) in a semiquantitative fashion.
FIGURE 7.

Neuronal loss, spongiform change, vascular lesions, and hemosiderin-laden macrophages in a subject (Case 2) with dementia after a single severe TBI. (A, B) Images showing temporal cortex neuronal loss and spongiosus cortex; 20× magnification with hematoxylin and eosin (H&E) staining (A) and GFAP immunohistochemistry (B). (C, D) Images showing neuronal loss and gliosis in hippocampus; 10× magnification of CA1 region stained with H&E and Luxol-fast blue (LFB) myelin staining (C), and 40× magnification of GFAP immunohistochemistry (D). (E–G) Images showing microhemorrhage in the hippocampus and white matter with perivascular hemosiderin-laden macrophages; 20× magnification with H&E of CA1 region of hippocampus (E); 10× magnification with H&E of white matter (F); and 40× magnification with LFB-H&E of white matter (G).
FIGURE 8.

p-Tau accumulation in the brain of a subject (Case 2) with dementia after a single severe TBI. Atypical pattern and distribution of p-tau neurofibrillary pathology, predominantly involving superficial layers of cerebral cortices (B), with dotlike neurites (A, B, D, G, J), but also throughout midbrain, deep gray and brainstem nuclei (C, E, F, H, I, K, L). Immunostaining for p-tau with AT8 histochemistry at 10×–40× magnification.
FIGURE 9.

β-Amyloid accumulation in the cortex and leptomeninges of a subject (Case 2) with dementia after a severe TBI. Sparse Aβ-plaques, predominantly diffuse plaques with vascular amyloid deposits in the leptomeninges (C, arrow). β-Amyloid immunostain (A, C) and Bielschowsky silver stain (B, D), 10× magnification.
FIGURE 10.

α-Synuclein pathology in the brain of a subject (Case 2) with dementia after a severe TBI. α-Synuclein-positive Lewy bodies and Lewy neurites in entorhinal, parietal and frontal cortices, mammillary body, diencephalon and substantia nigra (SN), medial temporal cortex (A–H) with unusually large, voluminous α-synuclein-positive axonal spheroids in mammillary bodies, SN, medulla and diencephalon (A, B, C, F, H). Immunostaining with anti-phosphorylated α-synuclein at 20×–40× magnification.
FIGURE 11.

TDP43 pathology in the cerebral cortex of a subject (Case 2) with dementia after a severe TBI. (A–D) TDP-43-positive inclusions in temporal lobe, amygdala and hippocampus, including dentate gyrus. Immunostaining with anti-TDP-43 antibody at 20×–40× magnification.
DISCUSSION
These 2 cases of early-onset dementia (59 and 51 years of age for Case 1 and 2, respectively, based on formal neuropsychological testing) after moderate-severe TBI highlight the diverse pathophysiological mechanisms and multiple proteinopathies that can be associated with posttraumatic neurodegeneration. Neuropathological evaluation revealed several findings shared by both cases, including abundant Aβ-NP, Aβ diffuse plaques, and frequent NFTs involving much of the cortex (especially the superficial layers), insula, and unusually, mammillary bodies. However, several distinct neuropathological findings were observed in each case. Case 1 had prominent WM rarefaction as well as focal perivascular and sulcal tau suggestive of CTE in 1 region (parietal cortex), whereas Case 2 had abundant as well as giant and bizarrely shaped Lewy bodies throughout the cortex, amygdala and brainstem. Case 2 also revealed frequent NFTs in the superficial layers of the cortex, hypothalamus and brainstem, as well as intraneuronal TDP-43 inclusions. These distinct and unusual pathological findings resulted in different neuropathological diagnoses: atypical AD (because of the hippocampal sparing of amyloid deposition and superficial cortical and mammillary body tau deposition) with features of CTE and WM loss in Case 1, and atypical DLB-limbic predominant (because of the highly atypical size and shape of both cortical and brainstem α-synuclein deposits), atypical AD (because of the superficial cortical and mammillary tau deposition) and coexistent TDP-43 pathology in Case 2. Collectively, these observations suggest that moderate-severe TBI may be associated with multiple proteinopathies that individually are classically characteristic of AD, DLB, frontotemporal dementia, and CTE, but are differentiated because of distinct morphologies, unusual distributions, and anatomic locations (e.g., mammillary bodies) and unique combinations of proteinopathy depositions in single patients (as in Case 2) that are more often singularly observed.
Both cases conformed to National Institute of Aging (NIA) criteria for definite AD, but both were diagnosed as atypical AD because the “classic” pathologic pattern of AD was only a portion of a more complex picture. Consistent with the atypical pathological findings, the clinical course and progression of behavioral and cognitive deficits in both cases were also not absolutely characteristic of AD. Both AD and CTE share p-tau immunoreactivity, but the distribution of NFT in the frontal, insular and temporal cortices, WM, diencephalon, brainstem, and spinal cord in CTE differs from that in AD (32). Further, in CTE, p-tau is found in NFT and astrocytes, and the pattern is distinctly perivascular with a tendency to be distributed irregularly at the depths of the cortical sulci and predominantly in superficial cortical layers (32). In Case 1, the preservation of hippocampal volume and paucity of NFT in the hippocampus relative to the significant NFT burden in supragranular layers of prefrontal cortices, the anterior insular cortex and mammillary bodies are highly atypical for AD and suggest an overlap with CTE. The occasional perivascular and sulcal tau further suggests CTE (Fig. 3C) (32–34). Likewise, Case 2 was also diagnosed with atypical AD because of the burden and distribution of NFT. In this case, however, there were no CTE features.
Case 2 had 2 additional misfolded protein accumulations (α-synuclein and TDP-43) and was diagnosed with DLB, but several features of these pathologic protein accumulations were atypical for DLB (e.g., amounts, size, and anatomical distribution of α-synuclein Lewy bodies and TDP-43 inclusions). The distribution of misfolded protein accumulations in both cases is inconsistent with the anatomical and histological distributions in classical AD, DLB, or FTD. Rather, the patterns of NFT, α-synuclein, and TDP-43 deposition do not conform to any typical neurodegenerative disease patterns described to date, and seem to suggest that a single severe TBI may promote deposition of multiple pathologic aggregated proteins that collectively contribute to dementia. Ubiquinated TDP-43, identified as the common pathologic proteinopathy linking frontotemporal dementia and ALS (35), has not been uniformly evaluated in neurodegenerative brain banks, but has been shown to accumulate in CTE (36) and to be upregulated and redistributed in experimental models of diffuse axonal injury (DAI) (37), suggesting it may play a role in TBI pathogenesis both acutely and chronically (38). In case series, TDP-43 is reported in up to 36% of cognitively normal aged individuals (39) between the ages of 50 and 102, and 59% of 188 pathologically confirmed AD cases (40). Its role in neurodegenerative disorders remains to be clarified.
Importantly, the absence of the APOE ɛ4 allele in the genotype of Case 1 excluded the possibility of a β-amyloid deposition enhancer due to a genetic risk factor, such as APOE ɛ4. By contrast, Case 2 was homozygous for the APOE ɛ4 allele and had a family history of early-onset dementia in a first degree relative (mother). A genetic contribution (APOE ɛ4/ɛ4) to the multiple pathologic protein accumulations in Case 2 cannot be totally excluded. However, a genetic predisposition for neurodegeneration does not exclude either that a TBI could be an accelerator or an enhancer of disease also, or even more, in genetically predisposed subjects. It is possible that Case 2 represents a familial dementia with isolated midlife severe TBI that could have accelerated or induced neurodegenerative processes at an earlier age. The precise role of APOE genotype in dementia pathogenesis and TBI-related dementia is not fully understood, but likely impacts type, rate, and distribution of misfolded protein accumulation, especially β-amyloid (6, 41). The APOE ɛ4 allele, though, has also been associated with pure synucleinopathies, dual AD-DLB pathology and FTD with motor neuron disease (FTLD-MND) (42, 43).
Both cases had marked global atrophy associated with widespread WM loss. The extensive WM loss by routine neuropathological staining, ex vivo 7 Tesla FLASH MRI, and ex vivo 3 Tesla diffusion tractography MRI, was a striking feature of Case 1. The pathogenesis of chronic posttraumatic WM loss is not completely understood, but has been previously described and likely contributed to both patients’ cognitive dysfunction (44). The severe and diffuse WM loss that we observed in these 2 cases of posttraumatic neurodegeneration, including with ex vivo MRI, may help distinguish posttraumatic neurodegeneration from AD, DLB, or frontotemporal dementia (FTD). Diffuse axonal injury is associated with cortical neuronal loss, but the exact mechanisms are not known. Experimental data implicate dysregulation of intracellular calcium metabolism, enhanced calpain proteolysis, reduced cellular energetics from mitochondrial dysfunction and persistent and even prolonged inflammatory responses (45). Microglial activation has been shown to persist for years, even decades, and may, once triggered, underlie delayed, and seemingly de novo neurodegeneration after TBI. Further, it is not possible to exclude that WM loss and associated DAI could trigger, contribute or accelerate the formation of β-amyloid and tau accumulation and deposition in the same anatomical regions to which injured WM tracts project. Both anterograde and retrograde intraneuronal transportation systems could be involved in the pathogenetic mechanisms since bidirectional intraneuronal transportation cellular systems are affected in DAI.
Autopsy studies have reported conflicting neuropathological findings in cases of late-life dementia and remote TBI. A recent large clinico-neuropathological correlation study of late-life dementias after TBI with LOC showed an association with Lewy body accumulation (α-synucleinopathy) and microinfarcts, but not Aβ-NP or NFTs (46). In contrast, in a retrospective study of specimens in a neurodegenerative brain bank, individuals with a history of TBI had higher than expected prevalence of AD pathology in a study of 115 autopsy cases (47). In other reports, repetitive TBI and potentially even single mild TBI was associated with CTE (32, 34). A recent reexamination of 1,721 specimens in a neurodegenerative brain bank found CTE changes in 21 of 66 former athletes with repetitive mild TBIs during sports participation ranging from high school to professional levels and no CTE changes among 198 controls or 33 cases with a history of remote single TBI. The majority (20/21) of the CTE cases were also diagnosed with at least 1 other neurodegenerative disorder (6 with AD, 4 with AD and DLB, 3 with ALS, 3 with DLB, 3 with FTLD-tau, and 1 with FTLD-FUS) (48).
Currently, there are a small number of pathological studies examining the association between single TBI and dementia (1, 44, 46, 49), and reports examining the pathological findings in early-onset dementia after single midlife TBI remain rare (22–25). Johnson et al examined a cohort of brain specimens from individuals with a history of remote (range 1–47 years) single TBI and performed imunohistochemical staining for NFT, Aβ deposition, and TDP-43 in 39 TBI survivors (49). They found widespread NFT and Aß plaques in up to a third of the cohort, but no association between a history of single TBI and abnormally phosphorylated TDP-43 inclusions (49).
The mechanisms by which neurodegeneration and pathologic protein accumulation occur after TBI is largely unknown, but they are likely influenced by the metabolic cascade of events that occur early after injury. Ikonomovic et al characterized AD-related changes in surgically resected cortex from 18 severe TBI subjects and found diffuse cortical Aβ immunostaining in one-third of subjects, diffuse tau immunostaining in a larger subset and tau-positive NFT-like changes in resected tissue from 2 older subjects (50). Aβ extracellular accumulation might be explained by the fact that Aß precursor protein (β-APP) temporarily accumulates in response to acute TBI as shown in genetically susceptible mice, and the cleavage of β-APP results in the characteristic Aß plaques of AD (51, 52). In support of this, Ikonomovic et al also found diffuse cortical Aβ-deposits in surgically resected cortex as early as 2 hours after injury, along with ß-APP and apolipoprotein E immunostaining (50). In a recent study of chronic severe TBI survivors without dementia, Scott et al found increased amyloid accumulation by 11 C-Pittsburgh compound B (11 C-PiB) PET imaging in 9 TBI subjects imaged between 1 and 17 years after severe TBI compared to age-matched controls and, importantly, in a pattern distinct from the Aß accumulation seen in AD (53).
Despite the widespread acceptance of the epidemiologic association between single TBI and dementia, the pathogenesis and type of brain pathologies underlying this association remain unclear. Because acute TBI is a heterogeneous and complex pathologic process resulting from varying combinations of neuronal and axonal injuries, vascular compromise, ionic shifts, loss of metabolic homeostasis, and neuroinflammation (44, 54), there are multiple potential proteins that can accumulate and independently trigger different neurodegenerative processes. While a limited sample, the pathologic findings in these 2 cases support the hypothesis that post-TBI dementia can be characterized as a polypathology “ensemble” with features that overlap with, but may be distinct from the currently established dementia subtypes. Advanced imaging technologies, including ex vivo MRI will likely play an important diagnostic role, given their ability to provide high spatial resolution of focal lesions and WM connectivity without the sampling limitations inherent in histopathological analysis. These 2 cases add to the growing literature of case series and case reports of dementia after mid-life TBI followed decades later by dementia and expand the number, types, and distribution of pathologic protein accumulations that may be associated with TBI. Finally, these 2 cases highlight the critical need for complete clinicopathological correlation and large-scale prospective studies of posttraumatic dementias to determine the full spectrum of clinical features, neuropathology, and pathogenetic mechanisms underlying TBI-associated dementia.
Supplementary Material
REFERENCES
- 1. Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000; 55:1158–66 [DOI] [PubMed] [Google Scholar]
- 2. Himanen L, Portin R, Isoniemi H, et al. Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology 2006; 66:187–92 10.1212/01.wnl.0000194264.60150.d3 [DOI] [PubMed] [Google Scholar]
- 3. Fleminger S, Oliver DL, Lovestone S, et al. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 2003; 74:857–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Meara ES, Kukull WA, Sheppard L, et al. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am J Epidemiol 1997; 146:373–84 [DOI] [PubMed] [Google Scholar]
- 5. Mehta KM, Ott A, Kalmijn S, et al. Head trauma and risk of dementia and Alzheimer’s disease: the Rotterdam study. Neurology 1999; 53:1959–62 [DOI] [PubMed] [Google Scholar]
- 6. Katzman R, Galasko DR, Saitoh T, et al. Apolipoprotein-epsilon4 and head trauma: Synergistic or additive risks?. Neurology 1996; 46:889–91 [PubMed] [Google Scholar]
- 7. Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer’s disease. Neurology 1995; 45:555–7 [DOI] [PubMed] [Google Scholar]
- 8. Washington PM, Villapol S, Burns MP.. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy?. Exp Neurol 2016; 275:381–8 10.1016/j.expneurol.2015.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gardner RC, Burke JF, Nettiksimmons J, et al. Dementia risk after traumatic brain injury vs. nonbrain trauma: the role of age and severity. JAMA Neurol 2014; 71:1490–7 10.1001/jamaneurol.2014.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mendez MF, Paholpak P, Lin A, et al. Prevalence of traumatic brain injury in early versus late-onset Alzheimer’s disease. J Alzheimers Dis 2015; 47:985–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sayed N, Culver C, Dams-O’Connor K, et al. Clinical phenotype of dementia after traumatic brain injury. J Neurotrauma 2013; 31:1117–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dams-O’Connor K, Spielman L, Hammond F, et al. An exploration of clinical dementia phenotypes among individuals with and without traumatic brain injury. Neurorehabilitation 2013; 32:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Katzman R, Aronson M, Fuld P, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol 1989; 25:317–24 10.1002/ana.410250402 [DOI] [PubMed] [Google Scholar]
- 14. Fratiglioni L, Ahlbom A, Viitanen M, et al. Risk factors for late-onset Alzheimer’s diease: a population-based, case-control study. Ann Neurol 1993; 33:258–66 [DOI] [PubMed] [Google Scholar]
- 15. Tsolaki M, Fountoulakis K, Chantzi E, et al. Risk factors for clinically diagnosed Alzheimer’s disease: a case-control study of a Greek population. Int Psychogeriatr 1997; 9:327–41 [DOI] [PubMed] [Google Scholar]
- 16. LoBue C, Wilmoth K, Cullum CM, et al. Traumatic brain injury history is associated with earlier age of onset of frontotemporal dementia. J Neurol Neurosurg Psychiatry 2016; 87:817–20 10.1136/jnnp-2015-311438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tolppanen A-M, Taipale H, Hartikainen S.. Head or brain injuries and Alzheimer’s disease: a nested case-control register study. Alzheimers Dement 2017; [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18. Dams-O’Connor K, Gibbons LE, Landau A, et al. Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: a population-based study. J Neurol Neurosurg Psychiatry 2013; 84:177–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mortimer JA, van Duijn CM, Chandra V, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991; 20 Suppl 2:S28–35 [DOI] [PubMed] [Google Scholar]
- 20. Goldman SM, Tanner CM, Oakes D, et al. Head injury and Parkinson's disease risk in twins. Ann Neurol 2006; 60:65–72 10.1002/ana.20882 [DOI] [PubMed] [Google Scholar]
- 21. Chen H, Richard M, Sandler DP, et al. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol 2007; 166:810–6 10.1093/aje/kwm153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kenney K, Massetti S, Shively S, et al. Clinicopathological Correlation of a Case of Dementia after TBI. Poster presentation at the 2014 Alzheimer's Association International Conference (AAIC), Copenhagen, Denmark, July 12–17, 2014.
- 23. Daneshvar DH, Katz DI, Stein TD, et al. Atypical AD, Lewy body disease and TDP 43 proteinopathy following single TBI. Poster presentation at the 2014 Alzheimer's Association International Conference (AAIC), Boston, MA, July 13–18, 2013.
- 24. Tateno A, Sakayori T, Takizawa Y, et al. A case of Alzheimer’s disease following mild traumatic brain injury. Gen Hosp Psychiatry 2015; 37:97.e7–9 [DOI] [PubMed] [Google Scholar]
- 25. Johnson VE, Stewart W, Trojanowski JQ, et al. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol 2011; 122:715–26 10.1007/s00401-011-0909-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41:479–86 [DOI] [PubMed] [Google Scholar]
- 27. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112:389–404 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Augustinack JC, van der Kouwe AJW, Blackwell ML, et al. Detection of entorhinal layer II using 7 Tesla magnetic resonance imaging. Ann Neurol 2005; 57:489–94 10.1002/ana.20426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fischl B, Salat DH, van der Kouwe AJ, et al. Sequence-independent segmentation of magnetic resonance images. Neuroimage 2004; 23:S69–84 [DOI] [PubMed] [Google Scholar]
- 30. Edlow BL, Haynes RL, Takahashi E, et al. Disconnection of the ascending arousal system in traumatic coma. J Neuropathol Exp Neurol 2013; 72:505–23 10.1097/NEN.0b013e3182945bf6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.VA/DoD Clinical Practice Guideline for the Management of Concussion-Mild Traumatic Brain Injury. Prepared by the Management of Concussion-mild TBI Working Group, Dept of VA & DoD, Version 2.0–2016 (February 2016). Available at: http://www.healthquality.va.gov/guidelines/Rehab/mtbi/mTBICPGFullCPG50821816.pdf, page 7.
- 32. McKee AC, Cairns NJ, Dickson DW, the TBI/CTE group, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75–86, and 10.1007/s00401-015-1515-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hof PR, Bouras C, Buée L, et al. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol 1992; 85:23–30 [DOI] [PubMed] [Google Scholar]
- 34. McKee A, Stein TD, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013; 136:43–64 10.1093/brain/aws307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130–3 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 36. McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropath Exp Neurol 2010; 92:608–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moisse K, Mepham J, Volerning K, et al. Cytsolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL-I-mice: support for a role for TDP-re in the physiological response to neuronal injury. Brain Res 2009; 1296:176–86 10.1016/j.brainres.2009.07.023 [DOI] [PubMed] [Google Scholar]
- 38. Blennow K, Hardy J, Zetterberg H.. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012; 76:886–9 10.1016/j.neuron.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 39. Elobeid A, Libard S, Leino M, et al. Altered proteins in the aging brain. J Neuropathol Exp Neurol 2016; 75:316–25 10.1093/jnen/nlw002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Josephs KA, Whitwell JL, Tosakulwong N, et al. TDP-re and pathological subtype of Alzheimer’s disease impact clinical features. Ann Neurol 2015; 78:697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hartman RE, Laurer H, Longhi L, et al. Apolipoprotein e4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer’s disease. J Neurosci 2002; 22:10083–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsuang D, Leverenz JB, Lopez OF, et al. APOE ɛ4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 2013; 70:223–8 10.1001/jamaneurol.2013.600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vossel KA, Bien-Ly N, Bernardo A, et al. ApoE and TD-43 neuropathology in two siblings with familial FTLD-motor neuron disease. Neurocase 2013; 19:295–301 10.1080/13554794.2012.667124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson VE, Stewart JD, Begbie FD, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013; 136:28–42 10.1093/brain/aws322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hill CS, Coleman MP, Menon DK.. Traumatic axonal injury: mechanisms and translational opportunities. Trends Neurosci 2016; 39:311–24 10.1016/j.tins.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crane PK, Gibbons LE, Dams-O’Connor K, et al. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol 2016; 73:1062–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jellinger KA, Paulus W, Wrocklage C, et al. Traumatic brain injury as a risk factor for Alzheimer disease. Comparison of two retrospective autopsy cohorts with evaluation of ApoE genotype. BMC Neurol 2001; 1:3. 10.1186/1471-2377-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015; 130:877–99 10.1007/s00401-015-1502-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson VE, Stewart W, Smith DH.. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012; 22:142–9 10.1111/j.1750-3639.2011.00513.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ikonomovic MD, Uryu K, Abrahamson EE, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 2004; 190:192–203 [DOI] [PubMed] [Google Scholar]
- 51. Corrigan F, Pham CL, Vink R, et al. The neuroprotective domains of the amyloid precursor protein, in traumatic brain injury, are located in the two growth factor domains. Brain Res 2011; 1378:137–43 10.1016/j.brainres.2010.12.077 [DOI] [PubMed] [Google Scholar]
- 52. Smith DH, Uryu K, Saatman KE, et al. Protein accumulation in traumatic brain injury. Neuromolecular Med 2003; 4:59–72 10.1385/NMM:4:1-2:59 [DOI] [PubMed] [Google Scholar]
- 53. Scott G, Ramlackhansingh AF, Edison P, et al. Amyloid pathology and axonal injury after brain trauma. Neurology 2016; 86:821–8 10.1212/WNL.0000000000002413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hay JR, Johnson VE, Young AMH, et al. Blood-brain disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 2015; 74:1147–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.