

Abstract
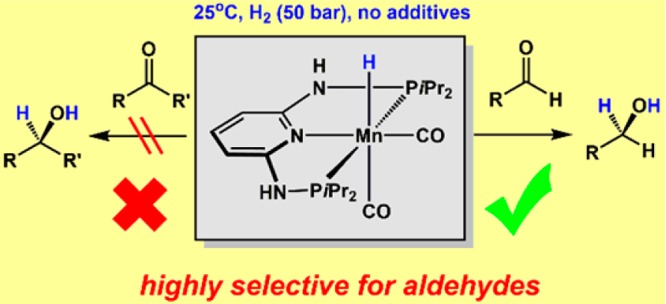
Several hydride Mn(I) and Re(I) PNP pincer complexes were applied as catalysts for the homogeneous chemoselective hydrogenation of aldehydes. Among these, [Mn(PNP-iPr)(CO)2(H)] was found to be one of the most efficient base metal catalysts for this process and represents a rare example which permits the selective hydrogenation of aldehydes in the presence of ketones and other reducible functionalities, such as C=C double bonds, esters, or nitriles. The reaction proceeds at room temperature under base-free conditions with catalyst loadings between 0.1 and 0.05 mol% and a hydrogen pressure of 50 bar (reaching TONs of up to 2000). A mechanism which involves an outer-sphere hydride transfer and reversible PNP ligand deprotonation/protonation is proposed. Analogous isoelectronic and isostructural Re(I) complexes were only poorly active.
Keywords: hydrogenation, aldehydes, manganese, pincer complexes, DFT calculations
Introduction
One environmentally friendly and sustainable method to prepare alcohols, which are valuable commodities for a large number of fine and bulk chemicals, is the catalytic hydrogenation of carbonyl compounds with dihydrogen.1 Over the years, many highly efficient and active homogeneous catalysts based on precious but also non-precious metals have been described for this purpose (Scheme 1).2 Especially catalysts which reveal full selectivity for aldehydes over ketones and/or alkenes3,4 are of practical importance for the synthesis of flavors,5 fragrances,3 and pharmaceuticals.6
Scheme 1. Well-Defined Catalysts for the Chemoselective Hydrogenation of Aldehydes.
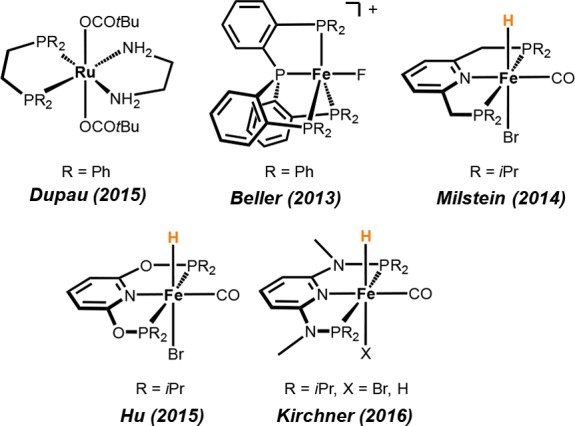
In the past couple of years, the development and advancement of hydrogenation catalysts based on earth-abundant, inexpensive non-precious metals experienced tremendous progress.7 In particular, iron- and manganese-based catalysts turned out to be highly active for the hydrogenation of carbonyl compounds, imines, and nitriles (Scheme 2).8−11 In the case of manganese, however, most hydrogenations proceed at relatively high catalyst loadings and elevated temperatures and, in addition, require large amounts of strong bases as additives. As yet, only iron-based systems proved to be reasonably chemoselective for the reduction of aldehydes, as shown in Scheme 1.12−14 We recently described the application of [Fe(PNPMe-iPr)(CO)(H)(Br)] and [Fe(PNPMe-iPr)(H)2(CO)] as highly active catalysts for the homogeneous hydrogenation of aldehydes (Scheme 1).15,16
Scheme 2. Manganese Catalysts for the Hydrogenation of Ketones and Aldehydes.

In this paper, we describe an experimental and theoretical investigation of the chemoselective hydrogenation of aldehydes with dihydrogen using several hydride Mn(I) and Re(I) PNP pincer complexes as catalysts (Scheme 3). To the best of our knowledge, this is the first example of an efficient manganese-based selective hydrogenation of aldehydes which proceeds under mild and base-free conditions with low catalyst loadings. It has to be noted that Re pincer complexes have rarely been used in (de)hydrogenation catalysis.17,18
Scheme 3. PNP Pincer Complexes Tested as Catalysts for the Hydrogenation of Aldehydes (R = iPr) and Structural View of Re1 Showing 30% Thermal Ellipsoids.
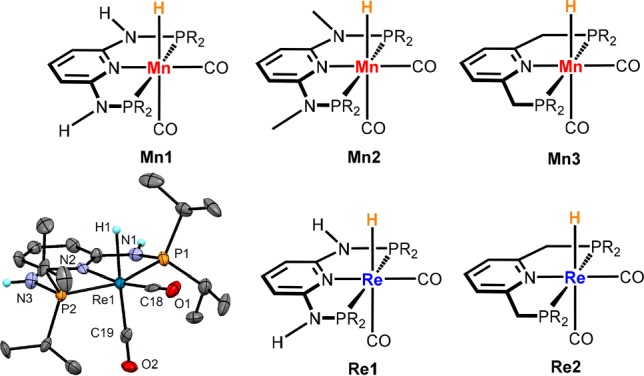
Selected bond lengths (Å) and angles (°): Re1–P1 2.347(3), Re1–P2 2.342(3), Re1–N2 2.162(8), Re1–C18 1.87(1), Re1–C19 1.94(1), Re1–H1 1.91(5), P1–Re1–P2 158.2(1).
Results and Discussion
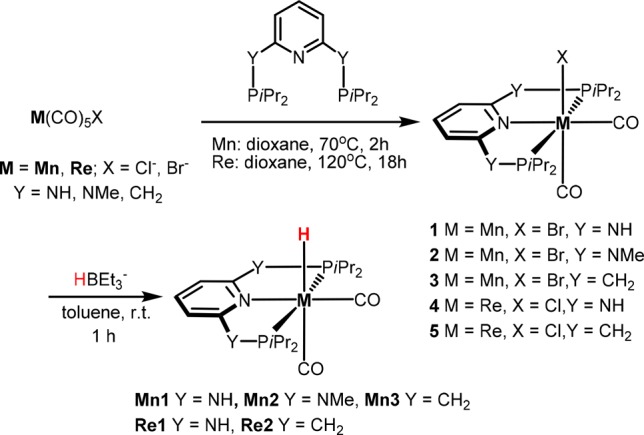
The reaction of [M(CO)5X] (M = Mn, X = Br; M = Re, X = Cl) with the respective PNP pincer ligands in dioxane at elevated temperatures afforded the neutral biscarbonyl complexes [M(PNP)(CO)2X] (1–5) (Scheme 4). Treatment of these intermediates with Na[HBEt3] (1.1 equiv) in toluene afforded complexes Mn1, Mn2, Mn3, Re1, and Re2. The synthesis of Mn1 and Mn2 was already reported previously.19 All new complexes could be isolated in 77–95% isolated yields and were fully characterized by a combination of elemental analysis, 1H, 13C{1H}, and 31P{1H} NMR, and IR spectroscopy (see Supporting Information (SI)). In addition, the molecular structure of Re1 was determined by X-ray crystallography (Scheme 3, bottom left).
Scheme 4. Synthesis of Hydride Mn(I) and Re(I) PNP Pincer Complexes.
The catalytic performance of Mn1, Mn2, Mn3, Re1, and Re2 was then investigated for the hydrogenation of aldehydes. The experiments were performed in EtOH as solvent using 4-fluorobenzaldehyde as model substrate to find the most active catalyst and optimal hydrogenation reaction conditions (Table 1). No reaction took place in aprotic solvents such as THF or toluene at 50 bar H2, a catalyst loading of 1.0 mol%, and a reaction time of 18 h. In the absence of dihydrogen, the hydrogenation of 4-fluorobenzaldehyde to yield 4-fluorobenzyl alcohol was not observed—no reaction took place. Thus, a possible transfer-hydrogenation mechanism in EtOH could be excluded. It has to be further emphasized that ketones, e.g., acetophenone and 4-fluoroacetophenone, did not react with any of the catalysts tested under the same reaction conditions described below.
Table 1. Hydrogenation of 4-Fluorobenzaldehyde with Several Manganese and Rhenium Catalystsa.

| entry | cat. | solvent | S/C | P (bar) | t (h) | conversion (%)b | TON |
|---|---|---|---|---|---|---|---|
| 1 | Mn1 | THF | 1000 | 50 | 18 | ||
| 2 | Mn1 | toluene | 1000 | 50 | 18 | ||
| 3 | Mn1 | EtOH | 1000 | 30 | 1 | 54 | 540 |
| 4 | Mn1 | EtOH | 1000 | 30 | 4 | >99 | 1000 |
| 5 | Mn1 | EtOH | 2000 | 50 | 18 | >99 | 2000 |
| 6c | Mn1 | EtOH | 20000 | 50 | 48 | 52 | 10400 |
| 7 | Mn2 | EtOH | 100 | 50 | 18 | ||
| 8 | Mn3 | EtOH | 100 | 50 | 18 | 21 | 21 |
| 9 | Re1 | EtOH | 100 | 50 | 18 | 86 | 86 |
| 10d | Re1 | EtOH | 100 | 50 | 18 | 95 | 95 |
| 11 | Re2 | EtOH | 100 | 50 | 18 | 76 | 76 |
Reaction conditions: catalysts (0.4–20.0 μmol), 4-fluorobenzaldehyde (2.0 mmol), EtOH (4 mL), 50 bar H2, 25 °C.
Determined by 19F NMR spectroscopy.
In the presence of DBU (1.2 μmol, 3 equiv).
Performed at 50 °C.
When Mn1 (0.1 mol%) was used as catalyst, complete conversion was observed after 4 h under a hydrogen pressure of 30 bar (Table 1, entry 4). By lowering the catalyst loading to 0.05 mol%, quantitative conversion was achieved after 18 h at a hydrogen pressure of 50 bar (Table 1, entry 5). If the reaction was performed in the presence of 3 equiv of DBU (1,8-diaza-bicyclo[5.4.0]undec-7-ene) as external base, 4-fluorobenzyl alcohol was obtained in 52% yield after 48 h under a hydrogen pressure of 50 bar and a catalyst loading of 0.005 mol% (Table 1, entry 6). This corresponds to a turnover number (TON) of 10400. Complexes Mn2 and Mn3 showed no or poor reactivity, even with a catalyst loading of 1 mol% (Table 1, entries 7 and 8). Surprisingly, the Re(I) complexes Re1 and Re2 with 1 mol% catalyst loadings were poorly active, affording only 45 and 76%, respectively, of 4-fluorobenzyl alcohol (Table 1, entries 9 and 11). At 50 °C, 4-fluorobenzyl alcohol was obtained in 95% yield (Table 1, entry 10).
Once Mn1 was determined to be the most active catalyst and its general applicability proved, various substrates were been tested to establish scope and limitations (Table 2). The catalytic experiments were conducted in the presence of 0.1–0.05 mol% of catalyst at 25 °C and 50 bar hydrogen pressure, for a reaction time of 18 h, without addition of any additives. The best results could be obtained for aromatic aldehydes bearing electron-withdrawing halogen substituents as well as electron-donating groups such as 4-anisaldehyde and 4-tolylaldehyde on the phenyl ring (Table 2, A1–A5) where catalyst loadings of 0.05 mol% were employed. Heteroaromatic substrates as well as aliphatic aldehydes could be reduced quantitatively under the same reaction conditions but with a catalyst loading of 0.1 mol% (Table 2, A6–A17). Substrates with conjugated and non-conjugated C=C double bonds were also selectively hydrogenated. For instance, citronellal or lyral, which are used in the flavor and fragrance industry (Table 2, A14–17), as well as the more challenging α,β-unsaturated substrate cinnamaldehyde (Table 2, A12) were not hydrogenated. In order to investigate the catalyst’s selectivity toward substrates with other unsaturated functionalities which can be easily hydrogenated, additional studies were carried out. Competitive experiments were carried out using equimolar mixtures of 4-fluorobenzaldehyde and the respective co-substrates at a catalyst-to-substrate ratio of 1:1000 with respect to the aldehyde. These studies showed that ketones, esters, alkynes, and nitrile groups were not hydrogenated. Moreover, these functionalities also did not interfere with the hydrogenation of the aldehyde moieties.
Table 2. Hydrogenation of Aldehydes A1–A17 with Catalyst Mn1a,b.

Reaction conditions: A1–A5 (1.0 μmol, 0.05 mol% Mn1), A6–A17 (2.0 μmol, 0.1 mol% Mn1), aldehyde (2 mmol), EtOH (4 mL), 50 bar H2, 25 °C, 18 h.
Yields (in parentheses) based on integration of 1H spectra using mesitylene as internal standard.
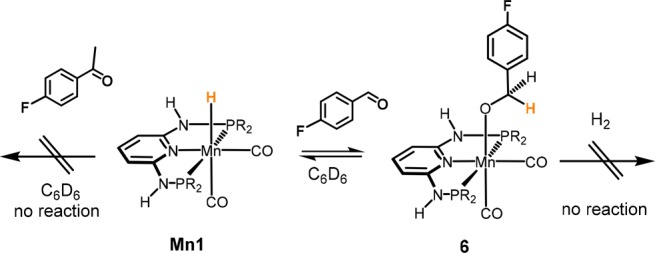
Stoichiometric experiments show that Mn1 reacts readily with aldehydes, even in aprotic solvents such as benzene or THF. The addition of 1 equiv of 4-fluorobenzaldehyde to a solution of the Mn(I) hydride Mn1 in C6D6 revealed the formation of a new but minor manganese species (Scheme 5). The concentration of this compound did not change over time but grew with increasing amount of added substrate. Thus, addition of up to 20 equiv of aldehyde was required to observe complete conversion of the manganese hydride complex. The new compound was tentatively assigned as the alkoxide complex 6, generated by insertion of the aldehyde into the metal hydride bond of Mn1. Compound 6 could not be isolated and exhibited singlet resonances at 115.8 and 140.9 ppm in the 19F{1H} and 31P{1H} NMR spectra, respectively (free 4-fluorobenzyl alcohol exhibits a singlet at 116.1 ppm in the 19F{1H} NMR spectrum). In the IR spectrum, 6 displays the expected two signals of the symmetric and asymmetric CO stretching frequency at 1925 and 1848 cm–1 (cf. 1873 and 1790 cm–1 in Mn1). However, no further reaction took place when a benzene (or THF) solution of the in situ-generated alkoxide complex 6 was exposed to dihydrogen. There was also no catalytic reaction if a 3:1 mixture of THF/EtOH was used. Accordingly, EtOH as solvent is not required for the insertion step but obviously plays a crucial role in the subsequent dihydrogen activation step. Moreover, Mn1 did not react with 4-fluoroacetophenone in both aprotic and protic solvents.
Scheme 5. Reaction of Mn1 with 4-Fluorobenzaldehyde and 4-Fluoroacetophenone in C6D6.
The reaction mechanism was explored in detail by means of DFT calculations.20 Benzaldehyde was taken as substrate and Mn1 (A in the calculations) as active catalyst. An explicit ethanol molecule (solvent) was considered, providing a proton shuttle and H-bond stabilization of the intermediates. Two different paths were considered, as shown in a simplified manner in Scheme 6. The more likely one proceeds via participation of the acidic N–H bond of the PNP ligand in a bifunctional mechanism (path I). This is supported by the fact that catalyst Mn2, bearing NMe linkers, is catalytically inactive and Mn3, featuring CH2 linkers which are less acidic than the NH linkers in Mn1, is only poorly active (Table 1, entry 8).
Scheme 6. Simplified Catalytic Cycles for Benzaldehyde Hydrogenation with Mn1.
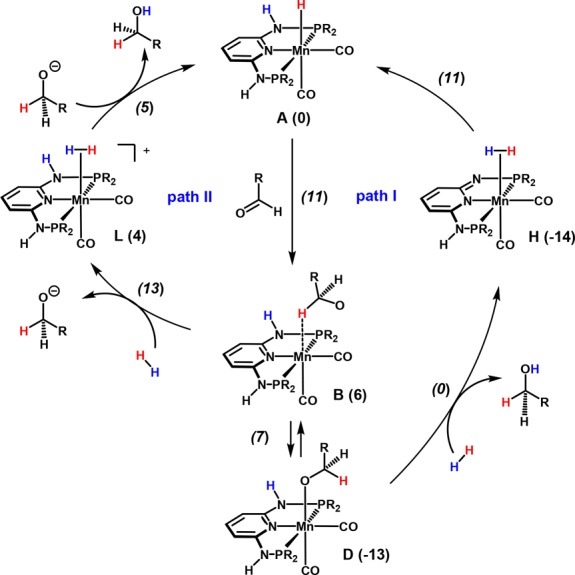
Free energies in kcal/mol are referred to A (Mn1 + EtOH + benzaldehyde); transition state energies are given in italics; R = iPr).
A reasonable mechanism has been established by means of DFT calculations. The free energy profile for path I is depicted in Figure 1. The first step is the attack of the hydride ligand in complex A to the carbonyl C-atom of a free benzaldehyde molecule. The result is intermediate B, a species with the resulting alkoxide weakly bonded to the metal by one C–H bond. This is a fairly easy step with a barrier of 11 kcal/mol and a free energy balance of ΔG = 6 kcal/mol, indicating that B is less stable than the initial reactants. The alkoxide in B can easily leave the metal following a dissociative path, through intermediate C. From here, the alkoxide may coordinate the metal by the O-atom, forming D through an easy process involving proton exchange with the solvent (SI, Figure S1). Importantly, the alkoxide complex D is 13 kcal/mol more stable than the initial reagents and represents the catalyst resting state. Naturally, there can be proton exchange between the solvent, EtOH, and benzyl alkoxide. Thus, the subsequent species may be either one. Following the profile in Figure 1, the coordinated alkoxide in D is protonated by the N–H proton of the PNP arm, with assistance of the ethanol molecule, from D to E. This process has a barrier of 13 kcal/mol and is endergonic, with ΔG = 7 kcal/mol. Intermediate F is 3 kcal/mol more stable than the reactants and features a dearomatized PNP ligand. The HOMO and LUMO of complex F are depicted in Figure 2. The HOMO corresponds to the ligand π-system, with a significant contribution of the lone pair of the deprotonated N-atom. The LUMO is essentially metal z2 pointing toward the empty coordination position.
Figure 1.

Free energy profile calculated for the hydrogenation of benzaldehyde catalyzed by the hydride complex Awith ligand N–H bond participation. Free energies (kcal/mol) are referred to the initial reactants (A), and relevant distances (Å) are presented.
Figure 2.
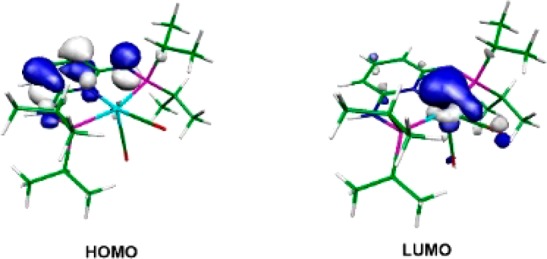
HOMO and LUMO of deprotonated Mn1 (F in calculations).
The reaction continues along the profile represented in Figure 3, F′ being equivalent to F with a different relative orientation of the three molecules. Exchange of benzyl alcohol by one H2 molecule produces intermediate G. Dihydrogen coordination is facile, with a barrier of only 1 kcal/mol (TSGH) in a clearly exergonic step, ΔG = −9 kcal/mol. The resulting intermediate H is an η2-H2 complex, which is 14 kcal/mol more stable than the initial reagents. Rearrangement of the H-bond network between the H2 complex and the nearby ethanol molecule changes H into I. In the final step, there is splitting of the H–H bond with re-protonation of the PNP N-atom and regeneration of the hydride ligand in J, corresponding to the initial reactant A and an ethanol molecule. The last step is exergonic, with J being 25 kcal/mol more stable than A. Despite the presence of an ethanol molecule acting as a proton shuttle, the associated barrier is significant (ΔG⧧ = 21 kcal/mol). The highest barrier along path I is 25 kcal/mol, corresponding to the difference between intermediate H, the most stable one, and transition state TSIJ.
Figure 3.

Free energy profile calculated for the hydrogenation of benzaldehyde catalyzed by the hydride complex A in a bifunctional mechanism with ligand N–H bond participation. The free energy values (kcal/mol) are referred to the initial reactants (A), and relevant distances (Å) are presented.
For comparison, the first step of the mechanism was also calculated for acetophenone as substrate. The barrier for the attack of the hydride ligand in complex A to the carbonyl C-atom of a free acetophenone molecule is significantly higher than the one calculated for benzaldehyde (18 vs 11 kcal/mol, respectively; SI, Figure S2). This trend is in accordance with the fact that ketones are not hydrogenated under the same reaction conditions. The remarkable substrate selectivity was recently also explained by the relative stability of alkoxide intermediates formed upon aldehyde insertion into the metal–H bond in the case of related iron PNP pincer complexes based on DFT calculations.21 It has to be noted that the related Mn(I) PNP pincer complex [Mn(PNP-iPr)(CO)3]Br (Scheme 2) was shown to act as a catalyst for the hydrogenation of ketones but at a catalyst loading of 5 mol%, a temperature of 130 °C in the presence of 10 mol% base, and a hydrogen pressure of 50 bar in toluene as solvent.10d
The alternative mechanism (path II) shares the first part in Figure 1 until formation of the cationic intermediate C. Following the profile represented in Figure 4, addition of H2 to C yields intermediate K. From here, coordination of dihydrogen is easy, with a barrier of merely 1 kcal/mol (TSKL) in an exergonic step (ΔG = −8 kcal/mol). The difference between the two mechanisms is that while here H2 coordinates to complex [Mn(PNP)(CO)2]+, producing the cationic dihydrogen complex [Mn(PNP)(η2-H2)(CO)2]+, in path I that process occurs with the neutral metallic species [Mn(PNP′)(CO)2], featuring a deprotonated PNP ligand (PNP′), and yields the corresponding neutral H2 complex: [Mn(PNP′)(η2-H2)(CO)2]. The mechanism proceeds from L with protonation of the free alkoxide by means of the coordinated H2. The associated barrier (TSLM) is negligible (1 kcal/mol), and the resulting species (M) is 13 kcal/mol more stable than A. The highest barrier in path II is 26 kcal/mol, measured between the O-coordinated alkoxide complex D and the highest following transition state TSKL. This is the transition state associated with H2 coordination and formation of the dihydrogen complex in L. It has to be noted that the same reaction pathway was recently established for the chemoselective hydrogenation of aldehydes catalyzed by [Fe(PNPMe-iPr)(CO)(H)(Br)], where metal–ligand cooperation was not possible due NMe linkers.15
Figure 4.

Free energy profile calculated for the hydrogenation of benzaldehyde catalyzed by the hydride complex Awithout ligand N–H bond participation. The free energy values (kcal/mol) are referred to the initial reactants (A), and relevant distances (Å) are presented.
In path I, alkoxide protonation is accomplished by the N–H proton in the PNP ligand, yielding a metallic fragment with a dearomatized PNP ligand. This corresponds to a bifunctional mechanism with participation of the PNP ligand that is further regenerated by the coordinated H2 molecule. In path II, there is no participation of the PNP ligand, and alkoxide protonation is made directly by the coordinated H2 molecule. The difference between the highest barriers calculated for the two mechanisms is only 1 kcal/mol (25 kcal/mol for path I, 26 kcal/mol for path II); thus, in principle, both could occur under the experimental conditions. If entropy corrections for non-standard conditions are considered, the total barrier for path I rises to 26.5 kcal/mol due to the lower molecularity of TSIJ when compared to TSKL and to the reaction conditions. This makes path I slightly less favorable than path II.
Conclusion
Several hydride Mn(I) and Re(I) PNP pincer complexes were prepared and tested as catalysts for the homogeneous chemoselective hydrogenation of aldehydes. [Mn(PNP-iPr)(CO)2(H)] (Mn1), based on the 2,6-diaminopyridine scaffold, where the PiPr2 moieties of the PNP ligand connect to the pyridine ring via NH linkers, was found to be the most efficient catalyst for this process. The reaction is highly chemoselective also in the presence of other functional groups which can be hydrogenated, such as ketones, esters, alkynes, olefins, nitriles, and α,β-unsaturated double bonds. The low catalyst loadings (0.1–0.05 mol%), mild and base-free reaction conditions (25 °C, 50 bar H2), and broad applicability make this catalyst interesting for the syntheses of fine and bulk chemicals. The catalysis works also with lower catalyst loadings (0.005 mol%) but requires then the addition of an external base. Based on experimental and computational studies, a bifunctional mechanism with participation of the PNP ligand (deprotonation/protonation) is proposed. An alternative mechanism without participation of the PNP ligand cannot be fully dismissed but seems to be less likely. Surprisingly, analogous isoelectronic and isostructural Re(I) complexes turned out to be only poorly active.
Acknowledgments
Financial support by the Austrian Science Fund (FWF) is gratefully acknowledged (Project No. P29584-N28). L.F.V. acknowledges Fundação para a Ciência e Tecnologia, Projecto Estratégico - PEst-OE/QUI/UI0100/2013.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b00153.
The authors declare no competing financial interest.
Supplementary Material
References
- a de Vries J. G., Elsevier C. J., Eds. Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, 2007. [Google Scholar]; b Dupau P. In Organometallics as Catalysts in the Fine Chemical Industry; Beller M., Blaser H. U., Eds.; Springer-Verlag: Berlin, 2012. [Google Scholar]; c Johnson N. B.; Lennon I. C.; Moran P. H.; Ramsden J. A. Industrial-Scale Synthesis and Applications of Asymmetric Hydrogenation Catalysts. Acc. Chem. Res. 2007, 40, 1291–1299. 10.1021/ar700114k. [DOI] [PubMed] [Google Scholar]; d Dub P. A.; Ikariya T. Catalytic Reductive Transformations of Carboxylic and Carbonic Acid Derivatives Using Molecular Hydrogen. ACS Catal. 2012, 2, 1718–1741. 10.1021/cs300341g. [DOI] [Google Scholar]
- a Noyori R.; Ohkuma T. Rapid, productive and stereoselective hydrogenation of ketones. Pure Appl. Chem. 1999, 71, 1493–1501. 10.1351/pac199971081493. [DOI] [Google Scholar]; b Noyori R.; Ohkuma T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem., Int. Ed. 2001, 40, 40–73. . [DOI] [PubMed] [Google Scholar]; c Noyori R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem., Int. Ed. 2002, 41, 2008–2022. . [DOI] [PubMed] [Google Scholar]; d Ohkuma H.; Ooka T.; Ikariya R.; Noyori R. Preferential hydrogenation of aldehydes and ketones. J. Am. Chem. Soc. 1995, 117, 10417–10418. 10.1021/ja00146a041. [DOI] [Google Scholar]; e Baldino S.; Facchetti S.; Zanotti-Gerosa A.; Nedden H. G.; Baratta W. Transfer Hydrogenation and Hydrogenation of Commercial-Grade Aldehydes to Primary Alcohols Catalyzed by 2-(Aminomethyl)pyridine and Pincer Benzo[h]quinoline Ruthenium Complexes. ChemCatChem 2016, 8, 2279–2288. 10.1002/cctc.201600420. [DOI] [Google Scholar]
- Bonomo L.; Kermorvan L.; Dupau P. Ruthenium-Catalyzed Highly Chemoselective Hydrogenation of Aldehydes. ChemCatChem 2015, 7, 907–910. 10.1002/cctc.201500006. [DOI] [Google Scholar]
- a Casey C. P.; Strotman N. A.; Beetner S. E.; Johnson J. B.; Priebe D. C.; Guzei I. A. PPh3-Substituted [2,5-Ph2-3,4-Tol2(η5-C4COH)]Ru(CO)(PPh3)H Exhibits Slower Stoichiometric Reduction, Faster Catalytic Hydrogenation, and Higher Chemoselectivity for Hydrogenation of Aldehydes over Ketones Than the Dicarbonyl Shvo Catalyst. Organometallics 2006, 25, 1236–1244. 10.1021/om050939z. [DOI] [Google Scholar]; b Diab L.; Smejkal T.; Geier J.; Breit B. Supramolecular Catalyst for Aldehyde Hydrogenation and Tandem Hydroformylation–Hydrogenation. Angew. Chem., Int. Ed. 2009, 48, 8022–8026. 10.1002/anie.200903620. [DOI] [PubMed] [Google Scholar]
- a Surburg H., Panten J., Eds. Common Fragrance and Flavor Materials; Wiley-VCH: Weinheim, 2006. [Google Scholar]; b Saudan L. A. Hydrogenation Processes in the Synthesis of Perfumery Ingredients. Acc. Chem. Res. 2007, 40, 1309–1319. 10.1021/ar700140m. [DOI] [PubMed] [Google Scholar]
- a Smith A. B.; Barbosa J.; Wong W.; Wood J. L. Total Syntheses of (+)-Trienomycins A and F via a Unified Strategy. J. Am. Chem. Soc. 1996, 118, 8316–8328. 10.1021/ja961401a. [DOI] [Google Scholar]; b Kobayakawa Y.; Nakada M. J. Enantioselective total synthesis of (A)-cyathin B2. J. Antibiot. 2014, 67, 483–485. 10.1038/ja.2014.23. [DOI] [PubMed] [Google Scholar]
- Bullock R. M., Ed. Catalysis Without Precious Metals; Wiley-VCH: Weinheim, 2010. [Google Scholar]
- a Maji B.; Barman M. K. Recent Developments of Manganese Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. Synthesis 2017, 49, 3377–3393. 10.1055/s-0036-1590818. [DOI] [Google Scholar]; b Garbe M.; Junge K.; Beller M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 2017, 4344–4362. 10.1002/ejoc.201700376. [DOI] [Google Scholar]; c Kallmeier F.; Kempe R. Manganese Complexes for (De)Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem., Int. Ed. 2018, 57, 46–60. 10.1002/anie.201709010. [DOI] [PubMed] [Google Scholar]; d Zell T.; Langer R. From ruthenium to iron and manganese - a mechanistic view on challenges and design principles of base metal hydrogenation catalysts. ChemCatChem 2018, 10.1002/cctc.201701722. [DOI] [Google Scholar]; e Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Catalytic (de)hydrogenation promoted by non-precious metals – Co, Fe and Mn: recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. 10.1039/C7CS00334J. [DOI] [PubMed] [Google Scholar]
- Fe-catalyzed hydrogenation reactions:; a Federsel C.; Boddien A.; Jackstell R.; Jennerjahn R.; Dyson P. J.; Scopelliti R.; Laurenczy G.; Beller M. A Well-Defined Iron Catalyst for the Reduction of Bicarbonates and Carbon Dioxide to Formates, Alkyl Formates, and Formamides. Angew. Chem., Int. Ed. 2010, 49, 9777–9780. 10.1002/anie.201004263. [DOI] [PubMed] [Google Scholar]; b Langer R.; Leitus G.; Ben-David Y.; Milstein D. Efficient hydrogenation of ketones catalyzed by an iron pincer complex. Angew. Chem., Int. Ed. 2011, 50, 2120–2124. 10.1002/anie.201007406. [DOI] [PubMed] [Google Scholar]; c Langer R.; Iron M. A.; Konstantinovski L.; Diskin-Posner Y.; Leitus G.; Ben-David Y.; Milstein D. Iron Borohydride Pincer Complexes for the Efficient Hydrogenation of Ketones under Mild, Base-Free Conditions: Synthesis and Mechanistic Insight. Chem. - Eur. J. 2012, 18, 7196–7209. 10.1002/chem.201200159. [DOI] [PubMed] [Google Scholar]; d Ziebart C.; Federsel C.; Anbarasan P.; Jackstell R.; Baumann W.; Spannenberg A.; Beller M. Well-Defined Iron Catalyst for Improved Hydrogenation of Carbon Dioxide and Bicarbonate. J. Am. Chem. Soc. 2012, 134, 20701–20704. 10.1021/ja307924a. [DOI] [PubMed] [Google Scholar]; e Fleischer S.; Zhou S.; Junge K.; Beller M. General and Highly Efficient Iron-Catalyzed Hydrogenation of Aldehydes, Ketones, and α,β-Unsaturated Aldehydes. Angew. Chem., Int. Ed. 2013, 52, 5120–5124. 10.1002/anie.201301239. [DOI] [PubMed] [Google Scholar]; f Srimani D.; Diskin-Posner Y.; Ben-David Y.; Milstein D. Iron Pincer Complex Catalyzed, Environmentally Benign, E-Selective Semi-Hydrogenation of Alkynes. Angew. Chem., Int. Ed. 2013, 52, 14131–14134. 10.1002/anie.201306629. [DOI] [PubMed] [Google Scholar]; g Wienhofer G.; Baseda-Kruger M.; Ziebart C.; Westerhaus F. A.; Baumann W.; Jackstell R.; Junge K.; Beller M. Hydrogenation of nitroarenes using defined iron–phosphine catalysts. Chem. Commun. 2013, 49, 9089–9091. 10.1039/c3cc42983k. [DOI] [PubMed] [Google Scholar]; h Bornschein C.; Werkmeister S.; Wendt B.; Jiao H.; Alberico E.; Baumann W.; Junge H.; Junge K.; Beller M. Mild and selective hydrogenation of aromatic and aliphatic (di)nitriles with a well-defined iron pincer complex. Nat. Commun. 2014, 5, 4111. 10.1038/ncomms5111. [DOI] [PubMed] [Google Scholar]; i Chakraborty S.; Dai H.; Bhattacharya P.; Fairweather N. T.; Gibson M. S.; Krause J. A.; Guan H. Iron-Based Catalysts for the Hydrogenation of Esters to Alcohols. J. Am. Chem. Soc. 2014, 136, 7869–7872. 10.1021/ja504034q. [DOI] [PubMed] [Google Scholar]; j Chakraborty S.; Lagaditis P. O.; Förster M.; Bielinski E. A.; Hazari N.; Holthausen M. C.; Jones W. D.; Schneider S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. 10.1021/cs5009656. [DOI] [Google Scholar]; k Lagaditis P. O.; Sues P. E.; Sonnenberg J. E.; Wan K. Y.; Lough A. J.; Morris R. H. Iron(II) Complexes Containing Unsymmetrical P-N-P′ Pincer Ligands for the Catalytic Asymmetric Hydrogenation of Ketones and Imines. J. Am. Chem. Soc. 2014, 136, 1367–1380. 10.1021/ja4082233. [DOI] [PubMed] [Google Scholar]; l Werkmeister S.; Junge K.; Wendt B.; Alberico E.; Jiao H.; Baumann W.; Junge H.; Gallou F.; Beller M. Hydrogenation of Esters to Alcohols with a Well-Defined Iron Complex. Angew. Chem., Int. Ed. 2014, 53, 8722–8726. 10.1002/anie.201402542. [DOI] [PubMed] [Google Scholar]; m Bertini F.; Mellone I.; Ienco A.; Peruzzini M.; Gonsalvi L. Iron(II) Complexes of the Linear rac-Tetraphos-1 Ligand as Efficient Homogeneous Catalysts for Sodium Bicarbonate Hydrogenation and Formic Acid Dehydrogenation. ACS Catal. 2015, 5, 1254–1265. 10.1021/cs501998t. [DOI] [Google Scholar]; n Rivada-Wheelaghan O.; Dauth A.; Leitus G.; Milstein D.; Diskin-Posner Y. Synthesis and Reactivity of Iron Complexes with a New Pyrazine-Based Pincer Ligand, and Application in Catalytic Low-Pressure Hydrogenation of Carbon Dioxide. Inorg. Chem. 2015, 54, 4526–4538. 10.1021/acs.inorgchem.5b00366. [DOI] [PubMed] [Google Scholar]; o Zhang Y.; MacIntosh A. D.; Wong J. L.; Bielinski E. A.; Williard P. G.; Mercado B. Q.; Hazari N.; Bernskoetter W. H. Iron catalyzed CO2 hydrogenation to formate enhanced by Lewis acid co-catalysts. Chem. Sci. 2015, 6, 4291–4299. 10.1039/C5SC01467K. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Gorgas N.; Stöger B.; Veiros L. F.; Pittenauer E.; Allmaier G.; Kirchner K. Efficient Hydrogenation of Ketones and Aldehydes Catalyzed by Well-Defined Iron(II) PNP Pincer Complexes: Evidence for an Insertion Mechanism. Organometallics 2014, 33, 6905–6914. 10.1021/om5009814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mn-catalyzed hydrogenations of ketones and aldehydes:; a Elangovan S.; Topf C.; Fischer S.; Jiao H.; Spannenberg A.; Baumann W.; Ludwig R.; Junge K.; Beller M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. 10.1021/jacs.6b03709. [DOI] [PubMed] [Google Scholar]; b Kallmeier F.; Irrgang T.; Dietel T.; Kempe R. Highly Active and Selective Manganese C=O Bond Hydrogenation Catalysts: The Importance of the Multidentate Ligand, the Ancillary Ligands, and the Oxidation State. Angew. Chem., Int. Ed. 2016, 55, 11806–11809. 10.1002/anie.201606218. [DOI] [PubMed] [Google Scholar]; c Widegren M. B.; Harkness G. J.; Slawin A. M. Z.; Cordes D. B.; Clarke M. L. A Highly Active Manganese Catalyst for Enantioselective Ketone and Ester Hydrogenation. Angew. Chem., Int. Ed. 2017, 56, 5825–5828. 10.1002/anie.201702406. [DOI] [PubMed] [Google Scholar]; d Bruneau-Voisine A.; Wang D.; Roisnel T.; Darcel C.; Sortais J.-P. Hydrogenation of ketones with a manganese PN3P pincer pre-catalyst. Catal. Commun. 2017, 92, 1–4. 10.1016/j.catcom.2016.12.017. [DOI] [Google Scholar]; e Garbe M.; Junge K.; Walker S.; Wei Z.; Jiao H.; Spannenberg A.; Bachmann S.; Scalone M.; Beller M. Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand. Angew. Chem., Int. Ed. 2017, 56, 11237–11241. 10.1002/anie.201705471. [DOI] [PubMed] [Google Scholar]
- a Perez M.; Elangovan S.; Spannenberg A.; Junge K.; Beller M. Molecularly Defined Manganese Pincer Complexes for Selective Transfer Hydrogenation of Ketones. ChemSusChem 2017, 10, 83–86. 10.1002/cssc.201601057. [DOI] [PubMed] [Google Scholar]; b Zirakzadeh A.; de Aguiar S. R. M. M.; Stöger B.; Widhalm M.; Kirchner K. Enantioselective Transfer Hydrogenation of Ketones Catalyzed by a Manganese Complex Containing an Unsymmetrical Chiral PNP′ Tridentate Ligand. ChemCatChem 2017, 9, 1744–1748. 10.1002/cctc.201700042. [DOI] [Google Scholar]; c Espinosa-Jalapa N. A.; Nerush A.; Shimon L. J. W.; Leitus G.; Avram L.; Ben-David Y.; Milstein D. Manganese-Catalyzed Hydrogenation of Esters to Alcohols. Chem. - Eur. J. 2017, 23, 5934–5938. 10.1002/chem.201604991. [DOI] [PubMed] [Google Scholar]; d Elangovan S.; Garbe M.; Jiao H.; Spannenberg A.; Junge K.; Beller M. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem. 2016, 128, 15590–15594. 10.1002/ange.201607233. [DOI] [Google Scholar]; e Bertini F.; Glatz M.; Gorgas N.; Stöger B.; Peruzzini M.; Veiros L. F.; Kirchner K.; Gonsalvi L. Carbon dioxide hydrogenation catalysed by well-defined Mn(I) PNP pincer hydride complexes. Chem. Sci. 2017, 8, 5024–5029. 10.1039/C7SC00209B. [DOI] [PMC free article] [PubMed] [Google Scholar]; f van Putten R.; Uslamin E. A.; Garbe M.; Liu C.; Gonzalez-de-Castro A.; Lutz M.; Junge K.; Hensen E. J. M.; Beller M.; Lefort L.; Pidko E. A. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem., Int. Ed. 2017, 56, 7531–7534. 10.1002/anie.201701365. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kar S.; Goeppert A.; Kothandaraman J.; Prakash G. K. S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. 10.1021/acscatal.7b02066. [DOI] [Google Scholar]
- Wienhofer G.; Westerhaus F. A.; Junge K.; Ludwig R.; Beller M. A Molecularly Defined Iron-Catalyst for the Selective Hydrogenation of α,β-Unsaturated Aldehydes. Chem. - Eur. J. 2013, 19, 7701–7707. 10.1002/chem.201300660. [DOI] [PubMed] [Google Scholar]
- Zell T.; Ben-David Y.; Milstein D. Highly efficient, general hydrogenation of aldehydes catalyzed by PNP iron pincer complexes. Catal. Sci. Technol. 2015, 5, 822–826. 10.1039/C4CY01501K. [DOI] [Google Scholar]
- Mazza S.; Scopelliti R.; Hu X. Chemoselective Hydrogenation and Transfer Hydrogenation of Aldehydes Catalyzed by Iron(II) PONOP Pincer Complexes. Organometallics 2015, 34, 1538–1545. 10.1021/acs.organomet.5b00105. [DOI] [Google Scholar]
- a Gorgas N.; Stöger B.; Veiros L. F.; Kirchner K. Highly Efficient and Selective Hydrogenation of Aldehydes: A Well-Defined Iron(II) Catalyst exhibits Noble Metal Activity. ACS Catal. 2016, 6, 2664–2672. 10.1021/acscatal.6b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brünig J.; Csendes Z.; Weber S.; Gorgas N.; Bittner R. W.; Limbeck A.; Bica K.; Hoffmann H.; Kirchner K. Chemoselective Supported Ionic Liquid Phase (SILP) Aldehyde Hydrogenation Catalyzed by an Fe(II) PNP Pincer Complex. ACS Catal. 2018, 8, 1048–1051. 10.1021/acscatal.7b04149. [DOI] [Google Scholar]
- Wei D.; Roisnel T.; Darcel C.; Clot E.; Sortais J.-B. Hydrogenation of Carbonyl Derivatives with a Well-Defined Rhenium Precatalyst. ChemCatChem 2017, 9, 80–83. 10.1002/cctc.201601141. [DOI] [Google Scholar]
- For rhenium-catalyzed (de)hydrogenation reactions, see:; a Piehl P.; Pena-Lopez M.; Frey A.; Neumann H.; Beller M. Hydrogen autotransfer and related dehydrogenative coupling reactions using a rhenium(I) pincer catalyst. Chem. Commun. 2017, 53, 3265–3268. 10.1039/C6CC09977G. [DOI] [PubMed] [Google Scholar]; b Schleker P. P. M.; Honeker R.; Klankermayer J.; Leitner W. Catalytic Dehydrogenative Amide and Ester Formation with Rhenium–Triphos Complexes. ChemCatChem 2013, 5, 1762–1764. 10.1002/cctc.201200942. [DOI] [Google Scholar]; c Jin H.; Xie J.; Pan C.; Zhu Z.; Cheng Y.; Zhu C. Rhenium-Catalyzed Acceptorless Dehydrogenative Coupling via Dual Activation of Alcohols and Carbonyl Compounds. ACS Catal. 2013, 3, 2195–2198. 10.1021/cs400572q. [DOI] [Google Scholar]; d Vogt M.; Nerush A.; Iron M. A.; Leitus G.; Diskin-Posner Y.; Shimon L. J. W.; Ben-David Y.; Milstein D. Activation of Nitriles by Metal Ligand Cooperation. Reversible Formation of Ketimido- and Enamido-Rhenium PNP Pincer Complexes and Relevance to Catalytic Design. J. Am. Chem. Soc. 2013, 135, 17004–17018. 10.1021/ja4071859. [DOI] [PubMed] [Google Scholar]; e Abdukader A.; Jin H.; Cheng Y.; Zhu C. Rhenium-catalyzed amination of alcohols by hydrogen transfer process. Tetrahedron Lett. 2014, 55, 4172–4174. 10.1016/j.tetlet.2014.05.068. [DOI] [Google Scholar]; f Vogt M.; Nerush A.; Diskin-Posner Y.; Ben-David Y.; Milstein D. Reversible CO2 binding triggered by metal–ligand cooperation in a rhenium(I) PNP pincer-type complex and the reaction with dihydrogen. Chemi. Sci. 2014, 5, 2043–2051. 10.1039/C4SC00130C. [DOI] [Google Scholar]; g Mazzotta M. G.; Xiong M.; Abu-Omar M. M. Carbon Dioxide Reduction to Silyl-Protected Methanol Catalyzed by an Oxorhenium Pincer PNN Complex. Organometallics 2017, 36, 1688–1691. 10.1021/acs.organomet.7b00223. [DOI] [Google Scholar]
- a Rao G. K.; Korobkov I.; Gabidullin B.; Richeson D. Employing a neutral ‘‘PN3P” pincer to access mer-Re(I) tricarbonyl complexes: Autoionization of a halo ligand and the role of an N-R (R = H, Me) substituent. Polyhedron 2018, 143, 62–69. 10.1016/j.poly.2017.08.020. [DOI] [Google Scholar]; b Choualeb A.; Maccaroni E.; Blacque O.; Schmalle H. W.; Berke H. Rhenium Nitrosyl Complexes for Hydrogenations and Hydrosilylations. Organometallics 2008, 27, 3474–3481. 10.1021/om7011024. [DOI] [Google Scholar]
- a Mastalir M.; Glatz M.; Gorgas N.; Stöger B.; Pittenauer E.; Allmaier G.; Veiros L. F.; Kirchner K. Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride MnI and FeII PNP Pincer Complexes. Chem. - Eur. J. 2016, 22, 12316–12320. 10.1002/chem.201603148. [DOI] [PubMed] [Google Scholar]; b Mastalir M.; Glatz M.; Pittenauer E.; Allmaier G.; Kirchner K. Sustainable Synthesis of Quinolines and Pyrimidines Catalyzed by Manganese PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 15543–15546. 10.1021/jacs.6b10433. [DOI] [PubMed] [Google Scholar]; c Mastalir M.; Pittenauer E.; Allmaier G.; Kirchner K. Manganese-Catalyzed Aminomethylation of Aromatic Compounds with Methanol as Sustainable C1 Building Block. J. Am. Chem. Soc. 2017, 139, 8812–8815. 10.1021/jacs.7b05253. [DOI] [PubMed] [Google Scholar]
- a Parr R. G.; Yang W.. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]; b Free energy values were obtained at the PBE0/VDZP level using the Gaussian 09 package. All calculations included solvent effects (ethanol) using the PCM/SMD model. A full account of the computational details and a complete list of references are provided as SI.
- Morello G. R.; Hopmann K. H. A Dihydride Mechanism Can Explain the Intriguing Substrate Selectivity of Iron-PNP-Mediated Hydrogenation. ACS Catal. 2017, 7, 5847–5855. 10.1021/acscatal.7b00764. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.