

Abstract
Background
There is an urgent need for blood-based molecular tests to assist in the detection and diagnosis of cancers at an early stage, when curative interventions are still possible, and to predict and monitor response to treatment and disease recurrence. The rich content of proteins in blood that are impacted by tumor development and host factors provides an ideal opportunity to develop noninvasive diagnostics for cancer.
Content
Mass spectrometry instrumentation has advanced sufficiently to allow the discovery of protein alterations directly in plasma across no less than 7 orders of magnitude of protein abundance. Moreover, the use of proteomics to harness the immune response in the form of seropositivity to tumor antigens has the potential to complement circulating protein biomarker panels for cancer detection. The depth of analysis currently possible in a discovery setting allows the detection of potential markers at concentrations of less than 1 μg/L. Such low concentrations may exceed the limits of detection of ELISAs and thus require the development of clinical assays with exquisite analytical sensitivity. Clearly the availability for discovery and validation of biospecimens that are highly relevant to the intended clinical application and have been collected, processed, and stored with the use of standard operating procedures is of crucial importance to the successful application of proteomics to the development of blood-based tests for cancer.
Summary
The realization of the potential of proteomics to yield blood biomarkers will benefit from a collaborative approach and a substantial investment in resources.
For disease investigation the profiling of blood constituents, notably serum and plasma, using protein characterization technologies holds long-standing interest because of the easy accessibility of this circulating fluid and its rich content of proteins that inform scientists about the health status of an individual. The available methodologies to analyze proteins have evolved dramatically over the past few decades. The initial method consisted of 1-dimensional protein separations, which was followed by the use of 2-dimensional polyacryl-amide gel electrophoresis coupled with Edman sequencing (1, 2). The advent of mass spectrometry, coupled with the sequencing of human and other genomes, has had a dramatic impact on the field of proteomics (1, 2). The capabilities of current proteomics technologies in terms of coverage of the proteome and depth of analysis that can be achieved in a quantitative manner are truly astounding compared with just a decade ago (3). Recent advances include substantial increases in speed, analytical sensitivity, and dynamic range, and the availability of multiple fragmentation techniques (3). Equally important is orthogonal sample fractionation before mass spectrometry. Yet there remains a perception that proteomics technologies are inadequate to address the protein complexities inherent in cells, tissues, and biological fluids. Here we outline strategies for the application of proteomics to the development of blood-based cancer markers.
Limitations of Current Modalities for Cancer Detection
An important issue to consider in developing markers for cancer detection using proteomics is the status of currently available modalities. At present the detection of particular common cancers relies heavily on procedures, notably imaging, that are specific to each cancer type, such as computerized axial tomography (CT)2 scans for lung cancer, mammograms for breast cancer, and pelvic ultrasounds for ovarian cancer. Advances in imaging technology have allowed improved detection of small lesions. These advances have also led to increases in false-positive findings, necessitating invasive procedures to make a definitive diagnosis (4, 5). The recently concluded National Lung Screening Trial led to a finding of a 20% reduction in lung cancer mortality with CT screening (6). However, there are substantial concerns regarding the economic burden of large-scale implementation of CT screening and the high rate of false positives (7). The frequency of detecting noncalcified nodules on a single CT varies from 5% to 60% in a lung cancer–screening population, with higher rates noted with the use of thinner slice widths (8–20). Given the high probability of false-positive findings associated with CT screening, there is a substantial need for additional modalities to discriminate between benign vs malignant nodules. There are similar challenges in imaging-based screening for other malignancies and a need for complementary diagnostic tests. In contrast, once protein-based biomarkers have been developed for each of the common cancer types, the resulting combined marker panels could all be assayed with 1 aliquot of a blood or plasma sample, thus allowing screening for common cancers on a single platform such as a microchip.
The Potential of Proteomics for Uncovering Alterations in Circulating Proteins Associated with Cancer
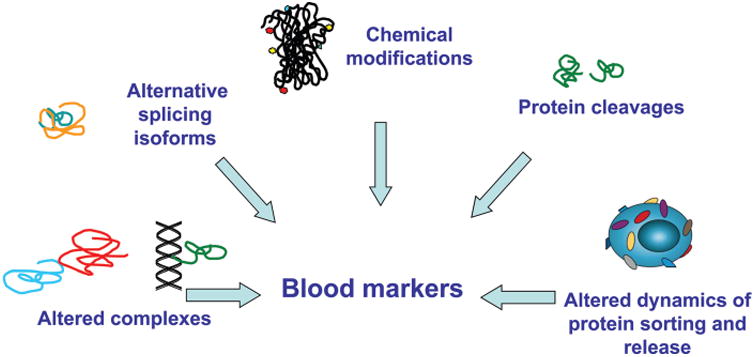
Protein markers currently in clinical use, which include CA125 for ovarian cancer, CA19-9 for pancreatic cancer, CEA (carcinoembryonic antigen) for colon cancer, and PSA (prostate-specific antigen) for prostate cancer, have important limitations with respect to their utility for screening. Other common cancers, notably breast and lung cancer, lack established biomarkers with demonstrated clinical utility in a screening setting. Thus, there is a need for biomarkers with prerequisite diagnostic sensitivity and specificity for detecting common cancer types. The rich content of proteins in blood and the multitude of modified forms of proteins that can inform scientists about the health status of an individual and of virtually most organs in the body provides an ideal opportunity to develop noninvasive diagnostics for cancer (Fig. 1). However, despite a voluminous literature going back decades pertaining to protein and other types of biomarkers for common cancers, blood-based diagnostic tests applicable to cancer screening and treatment–response prediction have been quite scarce. This situation may be attributed to (a) limited availability until recently of in-depth discovery technologies and limited understanding of the molecular features of cancers to guide the development of such biomarkers; (b) limited single investigator–based resources to accomplish objectives; (c) limited incentives for implementation of collaborative approaches to undertake the necessary studies; and (d) the lack of availability of biospecimens for discovery and validation that are highly relevant to the intended clinical application and that have been collected, processed, and stored using standard operating procedures (21), which is of crucial importance to the successful application of proteomics for developing blood-based tests for cancer.
Fig. 1. The potential circulating protein markers and their nature.
Depth and Breadth of Analysis of the Plasma Proteome with Current Technology
Innovations in MS technology and related informatics resources and increased availability of affinity capture agents that target particular proteins or protein subsets are likely to have a substantial impact on the prospects for identifying circulating cancer markers (21, 22). With each round of innovations in mass spectrometry resulting in increased resolving power and dynamic range, researchers have revisited the depth of analysis of the plasma proteome that can be achieved. With the use of a protein fractionation approach before trypsin digestion of individual fractions and mass spectrometry, more than 1400 proteins have been identified in a plasma sample, including isoforms that differ on the basis of their chromatographic mobility (23). The depth of analysis of plasma spans 7 orders of magnitude, allowing the detection of proteins with concentrations below 1 μg/L (24). More recently, the potential for increasing depth of analysis of the plasma proteome by using the currently available ultrahigh resolution linear ion trap Orbitrap TM Elite mass spectrometer (25) has been reexamined. With the current instrumentation available, some 2000 proteins have been identified with high confidence in a single plasma sample.
As an alternative to a systematic unbiased interrogation of the plasma proteome, selective approaches that target particular subsets of circulating proteins also have the potential to uncover cancer markers. A case in point is the selective targeting of glycoproteins. Glycan modifications of proteins are primarily Asn linked (N-linked glycans) or Ser or Thr linked (O-linked glycans). Glycoproteins with complex glycans are membrane bound or secreted. There is substantial evidence that cancer cells exhibit altered glycans relative to normal cells (26). The potential of targeting glycoproteins to identify biomarkers was investigated by enriching N-linked glycopeptides from tissues, cells, and plasma and identifying corresponding peptide sequences and proteins by mass spectrometry (27). A significant overlap was observed between glycoproteins identified in tissues and cells and glycoproteins identified in plasma, leading to the conclusion that extracellular glycoproteins originating from tissues and cells are released into the blood at concentrations that are detectable by mass spectrometry. The use of immunoaffinity depletion and multilectin chromatography integrated into an automated HPLC platform to remove high-abundance proteins followed by LC-MS analysis of protein digests has uncovered proteins with biological and disease significance for breast cancer (28). MALDI mass spectrometry has been applied to glycomic profile analyses of biological fluids (29). MALDI–mass spectrometry analysis of permethylated glycans in sera from breast cancer patients and disease-free controls identified several sialylated and fucosylated N-glycan structures as potential biomarkers (29). Increases in sialylation and fucosylation of glycan structures were associated with tumor progression.
Until recently, the study of glycoproteins by mass spectrometry has been based on the cleavage of glycans followed by separate analysis of glycans and deglycosylated proteins, which limits the ability to derive glycan compositions for individual glycoproteins. A methodology has been developed whereby a digital ion-trap mass spectrometer with a wide mass range is used for liquid chromatography–electrospray ionization/multistage mass spectrometry analysis of intact glycopeptides from fractionated and digested plasma (30). Both peptide and oligosaccharide compositions are elucidated by analysis of the ion fragmentation patterns of glycopeptides with an intact glycopeptide analysis pipeline.
Strategies based on the use of affinity capture agents, notably antibodies immobilized on microbarrays or aptamers, provide alternatives to the use of mass spectrometry for protein quantification (2, 31, 32). In one study the abundance of 813 proteins in plasma was measured with a new aptamer-based proteomic technology, leading to the identification of potential biomarkers that discriminated cases from controls with 91% diagnostic sensitivity and 84% diagnostic specificity in cross-validated training and 89% diagnostic sensitivity and 83% diagnostic specificity in a separate verification set, with similar performance for early and late-stage non–small cell lung cancer (32). A recombinant antibody microarray platform was used to identify serum biomarker signatures associated with pancreatic cancer. A resulting 25-serum biomarker signature discriminated pancreatic cancer patients from healthy controls and chronic pancreatitis patients with an area under the curve (AUC) of 0.88. Although affinity capture–based approaches provide an efficient method to search for biomarkers, success is dependent on the diversity of capture agents available, whereas mass spectrometry allows an unbiased approach to biomarker discovery.
Proteomic Profiling of the Humoral Immune Response to Tumor Antigens for Biomarker Discovery
The concept of testing for seropositivity to tumor antigens to determine the presence of cancer is quite appealing. This humoral response occurs early during tumor development and engenders an amplified signal detectable in the blood in the form of autoantibodies. Proteomic technologies are well suited for the identification of tumor antigens that induce autoantibodies. Available technologies target distinct repertoires of antigens and autoantibodies and thus complement each other (Table 1). A protein microarray approach was applied to discover and validate lung cancer tumor antigens associated with autoantibodies (33–36). Blinded validation of the autoantibody panel using prediagnostic sera yielded statistically significant reactivity among cases relative to controls, thus supporting the utility of the proteomic approach and of autoantibody tests for seropositivity as a means for detecting lung cancer at the presymptomatic stage (36).
Table 1. Proteomic technologies for the identification of tumor antigens that elicit autoantibodies.
| Advantages | Limitations | |
|---|---|---|
| Recombinant protein arrays | Content spans nearly the entire complement of proteins encoded in the genome | Not suited for epitopes associated with posttranslational modifications |
| Natural protein arrays | Well suited for epitopes associated with posttranslational modifications | Require isolation from tumor sources and verification of antigen identity |
| Peptide arrays | Scope covers the entire complement of peptides encoded in the genome | Limited to linear epitopes |
| Mass spectrometry identification of antigen bound to Ig or soluble HLA molecules | Allows detection of bound antigen | Complexes may be cleared from the circulation and thus not detectable |
Integrative Proteomic Approaches to Identify Blood-Based Cancer Markers
Given the substantial diversity of proteins released into the circulation both in health and in disease, clearly no single proteomic approach can capture the diversity and heterogeneity of proteins stemming from their chemical modifications, their cleavages, and their occurrences as complexes with other proteins and with antibodies. Therefore, efforts to fully elucidate cancer biomarkers would benefit from approaches that integrate multiple profiling technologies and biospecimen sources.
A case in point is an integrative study of lung cancer to identify plasma-based biomarkers (37, 38). In-depth quantitative proteomic analysis was applied to plasmas from 3 mouse models of lung adenocarcinoma driven by mutant epidermal growth factor receptor (EGFR) or Kras or induced by urethane exposure, and a mouse model of small-cell lung cancer driven by loss of Trp53 and Rb. To further refine lung cancer–specific and broad carcinoma signatures, the lung cancer proteome profiles were intersected with those from other well-established mouse models of pancreatic, ovarian, colon, prostate, and breast cancer, as well as 2 mouse models of inflammation. A set of proteins regulated by Nkx2–1 [NK2 homeobox 1 (Titf1)], a master transcription factor in cells from the peripheral airways and a known lineage-survival oncogene in lung cancer, was identified in plasmas of mouse models of lung adenocarcinoma. An EGFR network of upregulated proteins was discerned in the plasma of mice with lung tumors driven by a mutant human EGFR. The concentrations of these proteins returned toward baseline upon treatment with a tyrosine kinase inhibitor. To determine the potential relevance of findings from mouse models to human lung cancer, a set of protein markers identified in lung tumor–bearing mice for which ELISA assays were available were tested in human plasma samples. ROC analysis yielded an AUC of 0.88 with the combined panel for the newly diagnosed lung cancer cases relative to controls and an AUC of 0.799 for plasmas collected before the onset of symptoms and the diagnosis of lung cancer relative to controls. Given the success of proteomics approaches used to identify auto antibodies to tumor antigens in lung cancer, the findings from mass spectrometry profiling of lung cancer plasmas were integrated with autoantibody findings obtained by using micro array technology. The autoantibody panel yielded an AUC of 0.828 for the prediagnostic set, whereas the combined autoantibody and circulating protein marker panels together yielded an AUC of 0.896. These findings provide support for the merit of integrating data from multiple proteomic profiling technologies for the identification of blood-based cancer biomarkers. If additional proteomic profiling technologies were to be integrated into such a study, for example by including glycoproteomic profiling with analysis of glycan structures on individual proteins, the prospect of developing a lung cancer marker panel with high diagnostic sensitivity and diagnostic specificity would likely be further enhanced.
Realizing the Full Potential of Proteomics for the Development of Circulating Cancer Markers
The development of clinical applications based on the use of proteomics technologies for discovery of circulating cancer markers, although highly promising, represents a substantial undertaking. There are currently no gold standards. Thus there is a need for an organized collaborative effort. Some pilot projects have been initiated that support the feasibility of engaging in large-scale proteomic initiatives. The first proteome project to be conceived through the Human Proteome Organization (HUPO) was a plasma proteome project that resulted in completion of a pilot phase that had as objectives the comparison of a broad range of technology platforms for the characterization of proteins in human plasma and serum and assessment of the influence of various technical variables in sample collection, handling, and storage. Standardized samples were distributed to 18 participating laboratories, and an integrated analysis of resulting data was carried out (39). An initial integration of data resulted in 3020 proteins identified with 2 or more peptides. However, the application of rigorous statistical methodology, taking into account multiple hypothesis testing, resulted in a reduced set of 889 proteins identified with high confidence (40).
This HUPO plasma pilot project has represented a landmark in the burgeoning field of proteomics, demonstrating the potential of a collaborative approach to yield robust and insightful data, which were made publicly available. A collaborative cancer plasma proteome project would be far reaching. The project would contribute to the broader objective of defining variation in the plasma proteome in health in relation to age, sex, diet, energy expenditure, exercise, lifestyle, and common exposures such as tobacco, which would be relevant across disease applications. Objectives that are specific to cancer (Table 2) would consist of identifying potential markers with diagnostic specificity toward each of the common cancer types. A plasma proteome project with such a broad objective would benefit from close interactions with other related initiatives in proteomics underway at a number of NIH institutes and elsewhere.
Table 2. Objectives of a plasma proteome project to develop circulating cancer markers.
| 1. Comprehensive quantitative analysis of plasma protein constituents among individuals later diagnosed with cancer who were part of large study population cohorts for identifying proteins predictive of the onset or risk for major common cancers. Aliquots are distributed to participating investigator for analyses that encompass protein modifications and autoantibody responses. |
| 2. Comprehensive quantitative analysis of protein constituents in plasmas collected at diagnosis and postdiagnosis to identify signatures informative about the tissue of origin, molecular subtypes, prognosis, prediction of treatment response, and tumor recurrence. |
| 3. Development of affinity capture agents for plasma proteins to be used in assays that have platforms with the prerequisite analytical sensitivity and specificity. |
| 4. Development of proteotypic peptides applicable to proteins with cleavages and other modifications associated with cancer. |
| 5. Development of a publicly available cancer plasma proteome database with links to experimental data. |
Informatics would be an important component of the cancer plasma proteome project. Currently, however, there are few publicly available proteomics data related to cancer that can be interrogated. Databases have been developed for depositing and retrieving proteomics datasets, including PRIDE (Proteomics Identifications Database) (41), Peptide Atlas (42), UniPep (43), the Global Proteome Machine (44), Proteopedia (45), and Proteome Commons and its Tranche file-sharing system (https://proteomecommons.org/tranche/). The development of a plasma oncoproteome database containing detailed characteristics of samples and experimental conditions associated with the data, which are readily retrievable, would allow collective mining of the cancer plasma proteome. Such an oncoproteome database may also serve as a repository for other cancer proteomic data derived from cells, tissues, and biological fluids other than plasma.
Beyond Discovery: The Need for Validation
It should be emphasized that the strategies based on unbiased comprehensive quantitative proteomics, although suited for discovery, may not be suited for clinical implementation. Moreover, the greater the depth of analysis achieved in discovery, the more challenging it is to develop assays that are applicable in the clinic and that may be used to validate findings from unbiased comprehensive discovery studies. Given the ability to interrogate the plasma proteome down to protein concentrations well below 1 μg/L, there is a need to develop assays for targeted proteins that achieve the same level of analytical sensitivity. Such analytical sensitivity may not be routinely achieved with ELISA and may require several rounds of assay optimization. The current effort to increase the depth of analysis for discovery is paralleled with an effort to develop and implement assay technologies that meet requirements for analytical sensitivity, specificity, and throughput (Table 3). Strategies include sandwich assays, multiple-reaction monitoring (MRM), and nanotechnology-based methods such as magneto-nanosensors that achieve attomolar detection of protein biomarkers and 6 logs of dynamic range (46, 47). It should also be noted that the greater the rigor in the design of discovery studies, the greater the likelihood that candidate biomarkers will be successfully validated, provided that both discovery and validation studies aim at the same intended clinical application of the biomarkers being sought.
Table 3. Partial list of technologies for assays of circulating proteins.
| Assays | Characteristics |
|---|---|
| Affinity-based assays | |
| Sandwich ELISA [Zangar et al. (52 )] | Analytical sensitivity may be limited, requiring several rounds of antibody selection for optimization |
| Bead-based assays [Kim et al. (53)] | Increased throughput and reduced sample requirements compared with standard ELISA |
| Proximity ligation [Gu et al. (54)] | Utilizes oligonucleotides attached to affinity reagents as reporter molecules |
| Rolling circle amplification [Xue et al. (55)] | Utilizes DNA for amplification of signal, may be combined with DNA nanotags |
| Aptamers [Ostroff et al. (32); Ilyas et al. (56))] | May require several rounds to achieve desired analytical sensitivity and specificity |
| Nanosensors [Gaster et al. (46); Gaster et al. (47)] | Increased analytical sensitivity compared with standard sandwich ELISA using nanosensor technology |
| Surface plasmon resonance [Vaisocherova et al. (57)] | Label-free method with similar analytical sensitivity to sandwich ELISA |
| Mass spectrometry–based assays | |
| MRM [Lemoine et al. (58)] | Bypasses the need for affinity capture. Analytical sensitivity and specificity may be an issue |
| Immunoaffinity + MRM [Anderson et al. (59)] | Combines the use of antibody for capture and mass spectrometry for quantification |
Perspective
Numerous types of molecules and cell populations that occur in plasma may be potential sources of cancer biomarkers. These include mutated DNA, DNA fragments with altered methylation, microRNAs, metabolites, circulating tumor cells, and other cell populations. Massively parallel DNA sequencing technologies have undergone remarkable development in recent years, resulting in an exponential increase in the number of base pairs that can be sequenced and a dramatic reduction of sequencing costs per base pair. The reduced cost has also energized major international cancer genome projects, such as the International Cancer Genome Consortium. Also, epigenomic and transcriptomic sequencing are being accomplished in parallel. Likewise, advances in global metabolomic profiling allow capture of disease-specific signatures of metabolism by measuring the intermediary and end products of intercellular pathways that are responsive to genetic and environmental influences (48). Current methodologies involve mass spectrometry and high-resolution nuclear magnetic spectroscopy, which have the ability to detect hundreds of metabolites simultaneously (49). Given the dynamic nature of the metabolome, there exists an opportunity for biomarker discovery.
Although proteomics technologies remain largely labor-intensive and have a relatively high cost structure, the value that may be derived from proteomic data justifies the investment. Yet little consideration has been given to the implementation of proteomics at the necessary scale to derive clinical benefit. Genomic analysis requires for the most part the availability of tissue, which is usually obtained at the time of diagnosis, whereas proteomics is equally applicable to profiling biological fluids in addition to tissue profiling. Thus, a major advantage of proteomics is the possibility of profiling an individual's plasma at the prediagnostic stage to determine the impact of various exposures and detect disease at an early stage, as well as at the time of diagnosis and posttreatment to assess response to therapy and to monitor for disease regression, progression, or recurrence through noninvasive blood sampling. It may be argued that successes have been reported on the use of nucleic acid–based methods for detecting cancer or providing a means for patient follow-up through blood sampling (50, 51), thus reducing the need for proteomics. However, most such demonstrations have been based on analysis of advanced-stage disease (35, 36), and the utility of nucleic acid–based approaches for early detection remains to be determined. At the least, there is a need for side-by-side comparison of the merits of proteomics relative to other approaches based on blood sampling for risk assessment, early detection, and disease classification and monitoring, and for determining how these approaches may complement each other. Thus, there is a need to integrate various multiomic data to provide a better context for developing and evaluating blood biomarkers.
Acknowledgments
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled participants, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Nonstandard abbreviations: CT, computerized axial tomography; EGFR, epidermal growth factor receptor; AUC, area under the curve; HUPO, Human Proteome Organisation; MRM, multiple-reaction monitoring
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors' Disclosures or Potential Conflicts of Interest: No authors declared any potential conflicts of interest.
References
- 1.Hanash SM, Pitteri SJ, Faca VM. Mining the plasma proteome for cancer biomarkers. Nature. 2008;452:571–9. doi: 10.1038/nature06916. [DOI] [PubMed] [Google Scholar]
- 2.Hanash S, Taguchi A. The grand challenge to decipher the cancer proteome. Nat Rev Cancer. 2010;10:652–60. doi: 10.1038/nrc2918. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson T, Mann M, Aebersold R, Yates JR, 3rd, Bairoch A, Bergeron JJ. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat Methods. 2010;7:681–5. doi: 10.1038/nmeth0910-681. [DOI] [PubMed] [Google Scholar]
- 4.Croswell JM, Baker JM, Marcus PM, Clapp JD, Kramer BS. Cumulative incidence of false-positive test results in lung cancer screening: a randomized trial. Ann Intern Med. 2010;152:505–12. doi: 10.7326/0003-4819-152-8-201004200-00007. [DOI] [PubMed] [Google Scholar]
- 5.Chubak J, Boudreau DM, Fishman PA, Elmore JG. Cost of breast-related care in the year following false positive screening mammograms. Med Care. 2010;48:815–20. doi: 10.1097/MLR.0b013e3181e57918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Lung Screening Trial Research Team. Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gold L, Walker JJ, Wilcox SK, Williams S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N Biotechnol. 2012;29:543–9. doi: 10.1016/j.nbt.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Henschke CI, McCauley DI, Yankelevitz DF, Naidich DP, McGuinness G, Miettinen OS, et al. Early lung cancer action project: overall design and findings from baseline screening. Lancet. 1999;354:99–105. doi: 10.1016/S0140-6736(99)06093-6. [DOI] [PubMed] [Google Scholar]
- 9.Henschke C, Naidich D, Yankelevitz D, McGuinness G, McCauley D, Smith J, et al. Early lung cancer action project: initial findings on repeat screening. Cancer. 2001;92:153–9. doi: 10.1002/1097-0142(20010701)92:1<153::aid-cncr1303>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 10.Kaneko M, Eguchi K, Ohmatsu H, Kakunuma R, Naruke T, Suemasu K, Moriyama N. Peripheral lung cancer: screening and detection with low-dose spiral CT versus radiography. Radiology. 1996;201:798–802. doi: 10.1148/radiology.201.3.8939234. [DOI] [PubMed] [Google Scholar]
- 11.Sobue T, Moriyama N, Kaneko M, Kusumoso M, Kobayashi T, Tsuchiya R, et al. Screening for lung cancer with low-dose helical computed tomography: anti-lung cancer association project. J Clin Oncol. 2002;20:911–20. doi: 10.1200/JCO.2002.20.4.911. [DOI] [PubMed] [Google Scholar]
- 12.Sone S, Li F, Yang ZG, Honda T, Maruyama Y, Takashima S, et al. Results of three-year mass screening programme for lung cancer using mobile low-dose spiral computed tomography scanner. Br J Cancer. 2001;84:25–32. doi: 10.1054/bjoc.2000.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Sone S, Abe H, Macmahon H, Doi K. Malignant versus benign nodules at CT screenings for lung cancer: comparison of thin-section CT findings. Radiology. 2004;223:793–8. doi: 10.1148/radiol.2333031018. [DOI] [PubMed] [Google Scholar]
- 14.Swensen SJ, Jett JR, Sloan JA, Midthun DE, Hartman TE, Sykes AM, et al. Screening for lung cancer with low-dose spiral computed tomography. Am J Respir Crit Care Med. 2002;165:508–13. doi: 10.1164/ajrccm.165.4.2107006. [DOI] [PubMed] [Google Scholar]
- 15.Swensen SJ, Jett JR, Hartman JE, Midthun DE, Mandrekar S, Hillman SL, et al. CT screening for lung cancer: five-year prospective experience. Radiology. 2005;235:259–65. doi: 10.1148/radiol.2351041662. [DOI] [PubMed] [Google Scholar]
- 16.Diederich S, Wormanns D, Semik M, Thomas M, Lenzen H, Roos N, Heindel W. Screening for early lung cancer with low-dose spiral CT: prevalence in 817 asymptomatic smokers. Radiology. 2002;222:773–81. doi: 10.1148/radiol.2223010490. [DOI] [PubMed] [Google Scholar]
- 17.Nawa T, Nakagawa T, Kusano S, Kawasaki Y, Sugawara Y, Nakata H. Lung cancer screening using low-dose spiral CT: Results of baseline and 1-year follow-up studies. Chest. 2002;122:15–20. doi: 10.1378/chest.122.1.15. [DOI] [PubMed] [Google Scholar]
- 18.McWilliams AM, Mayo JR, Ahn MI, MacDonald SL, Lam SC. Lung cancer screening using multislice thin-section computed tomography and autofluorescence bronchoscopy. J Thorac Oncol. 2006;1:61–8. [PubMed] [Google Scholar]
- 19.Pastorino U, Bellomi M, Landoni C, De Flori E, Arnaldi P, Picchio M, et al. Early lung-cancer detection with spiral CT and positron emission tomography in heavy smokers: 2-year results. Lancet. 2003;362:593–7. doi: 10.1016/S0140-6736(03)14188-8. [DOI] [PubMed] [Google Scholar]
- 20.Roberts HC, Patsios D, Paul NS, McGregor M, Weisbrod G, Chung T, et al. Lung cancer screening with low dose computed tomography: Canadian experience. Can Assoc Radiol J. 2007;58:225–35. [PubMed] [Google Scholar]
- 21.Gold L, Walker JJ, Wilcox SK, Williams S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N Biotechnol. 2011;29:543–9. doi: 10.1016/j.nbt.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Holm A, Wu W, Lund-Johansen F. Antibody array analysis of labeled proteomes: how should we control specificity? N Biotechnol. 2012;29:578–85. doi: 10.1016/j.nbt.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 23.Faca V, Pitteri SJ, Newcomb L, Glukhova V, Phanstiel D, Krasnoselsky A, et al. Contribution of protein fractionation to depth of analysis of the serum and plasma proteomes. J Proteome Res. 2007;6:3558–65. doi: 10.1021/pr070233q. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Faca V, Hanash S. Mining the plasma proteome for disease applications across seven logs of protein abundance. J Proteome Res. 2011;10:46–50. doi: 10.1021/pr101052y. [DOI] [PubMed] [Google Scholar]
- 25.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11:O111.013698. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–88. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Liu AY, Loriaux P, Wollscheid B, Zhou Y, Watts JD, Aebersold R. Mass spectrometric detection of tissue proteins in plasma. Mol Cell Proteomics. 2007;6:64–71. doi: 10.1074/mcp.M600160-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Zeng Z, Hincapie M, Pitteri SJ, Hanash S, Schalkwijk J, Hogan JM, et al. A proteomics platform combining depletion, multi-lectin affinity chromatography (M-LAC), and isoelectric focusing to study the breast cancer proteome. Anal Chem. 2011;83:4845–54. doi: 10.1021/ac2002802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyselova Z, Mechref Y, Kang P, Goetz JA, Dobrolecki LE, Sledge GW, et al. Breast cancer diagnosis and prognosis through quantitative measurements of serum glycan profiles. Clin Chem. 2008;54:1166–75. doi: 10.1373/clinchem.2007.087148. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Wong CH, Chin A, Taguchi A, Taylor A, Hanash S, et al. Integrated mass spectrometry based analysis of plasma glycoproteins and their glycan modifications. Nat Protoc. 2011;6:253–69. doi: 10.1038/nprot.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez AB, Lampe PD. Discovery and validation of ovarian cancer biomarkers utilizing high density antibody microarrays. Cancer Biomark. 2010;8:293–307. doi: 10.3233/CBM-2011-0215. [DOI] [PubMed] [Google Scholar]
- 32.Ostroff RM, Bigbee WL, Franklin W, Gold L, Mehan M, Miller YE, et al. Unlocking biomarker discovery: large scale application of aptamer proteomic technology for early detection of lung cancer. PLOS One. 2010;5:e15003. doi: 10.1371/journal.pone.0015003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brichory F, Beer D, Le Naour F, Giordano T, Hanash S. Proteomics-based identification of protein gene product 9.5 as a tumor antigen that induces a humoral immune response in lung cancer. Cancer Res. 2001;61:7908–12. [PubMed] [Google Scholar]
- 34.Brichory FM, Misek DE, Yim AM, Krause MC, Giordano TJ, Beer DG, Hanash SM. An immune response manisfested by the common occurrence of annexins I and II autoantibodies and high circulating levels of IL-6 in lung cancer. Proc Natl Acad Sci USA. 2001;98:9824–9. doi: 10.1073/pnas.171320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira-Faca SR, Kuick R, Puravs E, Zhang Q, Krasnoselsky AL, Phanstiel D. Identification of 14–3-3 theta as an antigen that induces a humoral response in lung cancer. Cancer Res. 2007;67:12000–6. doi: 10.1158/0008-5472.CAN-07-2913. [DOI] [PubMed] [Google Scholar]
- 36.Qiu J, Choi G, Li L, Wang H, Pitteri SJ, Pereira-Faca SR, et al. Occurrence of autoantibodies to annexin I, 14–3-3 theta and LAMR1 in prediagnostic lung cancer sera. J Clin Oncol. 2008;26:5060–6. doi: 10.1200/JCO.2008.16.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi A, Politi K, Pitteri SJ, Lockwood WW, Faca VM, Kelly-Spratt K, et al. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer Cell. 2011;20:289–99. doi: 10.1016/j.ccr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liotta LA, Petricoin E. Cancer biomarkers: closer to delivering on their promise. Cancer Cell. 2011;20:279–80. doi: 10.1016/j.ccr.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omenn GS, States DJ, Adamski M, Blackwell T, Menon R, Hermjakob H, et al. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–45. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 40.States DJ, Omenn GS, Blackwell TW, Fermin D, Eng J, Speicher DW, Hanash SM. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol. 2006;24:333–8. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 41.Jones P, Cote RG, Martens L, Quinn AF, Taylor CF, Derache W, et al. PRIDE: A public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Res. 2006;34:D659–63. doi: 10.1093/nar/gkj138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deutsch EW, Eng JK, Zhang H, King NL, Nesvizhskii AI, Lin B, et al. Human plasma peptideatlas. Proteomics. 2005;5:3497–500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 43.Zhang H, Loriaux P, Eng J, Campbell D, Keller A, Moss P, et al. UniPep–a database for human N-linked glycosites: a resource for biomarker discovery. Genome Biol. 2006;7:R73. doi: 10.1186/gb-2006-7-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–42. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 45.Mathivanan S, Ahmed M, Ahn NG, Alexandre H, Amanchy R, Andrews PC, et al. Human proteinpedia enables sharing of human protein data. Nat Biotechnol. 2008;26:164–7. doi: 10.1038/nbt0208-164. [DOI] [PubMed] [Google Scholar]
- 46.Gaster RS, Hall DA, Nielsen CH, Osterfeld SJ, Yu H, Mach KE, et al. Matrix-insensitive protein assays push the limits of biosensors in medicine. Nat Med. 2009 doi: 10.1038/nm.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaster RS, Hall DA, Wang SX. Autoassembly protein arrays for analyzing antibody cross-reactivity. Nano Lett. 2010 doi: 10.1021/nl1026056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claudino WM, Quattrone A, Biganzoli L, Pestrin M, Bertini I, Di Leo A. Metabolomics: available results, current research projects in breast cancer, and future applications. J Clin Oncol. 2007;25:2840–6. doi: 10.1200/JCO.2006.09.7550. [DOI] [PubMed] [Google Scholar]
- 49.Serkova NJ, Glunde K. Metabolomics of cancer. Methods Mol Biol. 2009;520:273–95. doi: 10.1007/978-1-60327-811-9_20. [DOI] [PubMed] [Google Scholar]
- 50.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Rolf-Dieter H, Knippers R. DNA fragments in the blood plasma of cancer patients: quantifications and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–65. [PubMed] [Google Scholar]
- 51.Pathak AK, Bhutani M, Kumar S, Mohan A, Guleria R. Circulating cell-free DNA in plasma/serum of lung cancer patients as a potential screening and prognostic tool. Clin Chem. 2006;52:1833–42. doi: 10.1373/clinchem.2005.062893. [DOI] [PubMed] [Google Scholar]
- 52.Zangar RC, Daly DS, White AM. ELISA microarray technology as a high-throughput system for cancer biomarker validation. Expert Rev Proteomics. 2006;3:37–44. doi: 10.1586/14789450.3.1.37. [DOI] [PubMed] [Google Scholar]
- 53.Kim YW, Bae SM, Kim IW, Liu HB, Bang HJ, Chaturvedi PK, et al. Multiplexed bead-based immunoassay of four serum biomarkers for diagnosis of ovarian cancer. Oncol Rep. 2012;28:585–91. doi: 10.3892/or.2012.1829. [DOI] [PubMed] [Google Scholar]
- 54.Gu GJ, Friedman M, Jost C, Johnsson K, Kamali-Moghaddam M, Pluckthun A, et al. Protein tagmediated conjugation of oligonucleotides to recombinant affinity binders for proximity ligation. N Biotechnol. doi: 10.1016/j.nbt.2012.05.005. Epub ahead of print 2012 Jun 2. [DOI] [PubMed] [Google Scholar]
- 55.Xue Q, Wang Z, Wang L, Jiang W. Sensitive detection of proteins using assembled cascade fluorescent DNA nanotags based on rolling circle amplification. Bioconjug Chem. 2012;23:734–9. doi: 10.1021/bc200537g. [DOI] [PubMed] [Google Scholar]
- 56.Ilyas A, Asghar W, Allen PB, Duhon H, Ellington AD, Iqbal SM. Electrical detection of cancer biomarker using aptamers with nanogap breakjunctions. Nanotechnology. 2012;23:275502. doi: 10.1088/0957-4484/23/27/275502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaisocherova H, Faca VM, Taylor AD, Hanash S, Jiang S. Comparative study of SPR and ELISA methods based on analysis of CD166/ALCAM levels in cancer and control human sera. Biosens Bioelectron. 2009;24:2143–8. doi: 10.1016/j.bios.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 58.Lemoine J, Fortin T, Salvador A, Jaffuel A, Charrier JP, Choquet-Kastylevsky G. The current status of clinical proteomics and the use of MRM and MRM(3) for biomarker validation. Expert Rev Mol Diagn. 2012;12:333–42. doi: 10.1586/erm.12.32. [DOI] [PubMed] [Google Scholar]
- 59.Anderson NL, Jackson A, Smith D, Hardie D, Borchers C, Pearson TW. SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol Cell Proteomics. 2009;8:995–1005. doi: 10.1074/mcp.M800446-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]