

Abstract
Human biology is regulated by a complex network of protein–protein interactions (PPIs), and disruption of this network has been implicated in many diseases. However, the targeting of PPIs remains a challenging area for chemical probe and drug discovery. Although many methodologies have been put forth to facilitate these efforts, new technologies are still needed. Current biochemical assays for PPIs are typically limited to motif–domain and domain–domain interactions, and assays that will enable the screening of full-length protein systems, which are more biologically relevant, are sparse. To overcome this barrier, we have developed a new assay technology, “PPI catalytic enzyme-linked click chemistry assay” or PPI cat-ELCCA, which utilizes click chemistry to afford catalytic signal amplification. To validate this approach, we have applied PPI cat-ELCCA to the eIF4E–4E-BP1 and eIF4E–eIF4G PPIs, key regulators of cap-dependent mRNA translation. Using these examples, we have demonstrated that PPI cat-ELCCA is amenable to full-length proteins, large (>200 kDa) and small (∼12 kDa), and is readily adaptable to automated high-throughput screening. Thus, PPI cat-ELCCA represents a powerful new tool in the toolbox of assays available to scientists interested in the targeting of disease-relevant PPIs.
Keywords: chemical probes, protein–protein interactions, click chemistry, high-throughput screening
Graphical abstract

INTRODUCTION
More than 80% of the cellular proteome is regulated by 40,000–200,000 protein–protein interactions (PPIs) that comprise the human protein interactome.1 Dysregulation of PPIs has been implicated in many human disease areas, including oncology, immunology, and cardiovascular disorders, making the targeting of PPIs of high therapeutic interest.2–4 In fact, the first PPI-targeted drug, venetoclax, a BH3 mimetic designed to inhibit the antiapoptotic Bcl-2 protein, recently received FDA approval for refractory chronic lymphocytic leukemia.5,6 Thus, the development of new technologies that will facilitate the discovery of PPI modulators is of growing importance in chemistry and biology.
In the present toolbox of high-throughput screening (HTS) assays for PPIs, a missing piece is the ability to screen against full-length protein systems in a noncellular format. The most commonly used biochemical assays for PPIs rely on methods such as fluorescence polarization (FP), fluorescence resonance energy transfer (FRET), time-resolved FRET (TR-FRET), and AlphaScreen.7,8 Although easy to implement (mix-and-measure), these assays are typically limited to the analysis of motif–domain or domain–domain interactions due to size and labeling requirements.8 Thus, by using these methods in an in vitro biochemical setting, probe discovery efforts are often focused solely on “hot spot” interactions while eliminating the possibility of targeting potentially more druggable allosteric binding sites. These approaches also require structural knowledge about the PPI to design appropriate peptide substrates (e.g., FP) or for proximity-matched labeling (e.g., FRET), which may be difficult, particularly for large or disordered proteins. Other disadvantages of these approaches include single-turnover readout, which limits the sensitivity of the measurement, and compound interference by assay-specific interferents (e.g., fluorescent molecules or fluorescence quenchers) yielding many false positive and negative hits.8–12
To efficiently assay and discover chemical modulators of full-length PPIs which are more biologically relevant, we have developed a new platform assay technology termed “PPI catalytic enzyme-linked click chemistry assay” or PPI cat-ELCCA. In our approach, a biotinylated protein is first immobilized in the wells of a streptavidin-coated microtiter plate (Figure 1). The wells are then incubated with a click chemistry-armed protein-binding partner to form the PPI. Detection occurs via an initial click reaction with labeled horseradish peroxidase (HRP), followed by addition of an HRP substrate and chemiluminescence measurement. Importantly, this catalytic system, similar to enzyme-linked immunosorbent assays (ELISA; Figure 1), allows for increased sensitivity due to signal amplification and a significantly reduced risk of compound interference from screening diverse libraries due to added washing steps. Unlike ELISA, however, it eliminates the requirement for antibodies, which is important for targets for which monoclonal antibodies may not already exist or are difficult to generate against. Herein, we describe our work toward the development of PPI cat-ELCCA for two interactions that play a crucial role in the initiation of cap-dependent mRNA translation: eIF4E–4E-BP1 and eIF4E–eIF4G (Figure 2A). Through these examples, we demonstrate the applicability of our method toward proteins of varying size (12–220 kDa), structural ordering, and stability, highlighting the advantages of PPI cat-ELCCA over existing methodologies.
Figure 1.

Schematic of PPI cat-ELCCA and its comparison to ELISA.
Figure 2.

eIF4E-initiated cap-dependent mRNA translation. (A) Regulation of eIF4E by its binding partners 4E-BP1 and eIF4G. mTORC1 = mechanistic target of rapamycin complex 1. (B) Consensus sequences of eIF4E “hot spot” binding to 4E-BP1 (G49–N64) and eIF4GI (E607–F622).
RESULTS AND DISCUSSION
As a proof-of-concept for PPI cat-ELCCA, we focused our efforts on a critical initiator of protein synthesis, eukaryotic translation initiation factor 4E (eIF4E) (Figure 2A). By binding to m7GpppX-cap, eIF4E selectively enhances the translation of transcripts undergoing cap-dependent mRNA translation, many of which encode for oncoproteins and growth and survival factors.13 Thus, overexpression of eIF4E has been identified as a biomarker in many human cancers.13,14 Because of the important nature of eIF4E, it is highly regulated, primarily through the work of two binding proteins, 4E-BP1 and eIF4G. The proteins interact with eIF4E using a conserved helical binding motif, YXXXFLϕ (Figure 2B), establishing a competitive binding model for the initiation of cap-dependent translation.15 4E-BP1 is an intrinsically disordered protein that serves as a gate-keeper of this process by sequestering eIF4E from eIF4G and the eIF4F translation initiation complex, whereas eIF4G binding stimulates translation initiation.16 Previous work has demonstrated the potential for targeting eIF4E PPIs in cancer drug discovery;13,14 however, selective chemical probes for interrogating eIF4E biology have yet to be reported.17,18 Thus, we developed and applied PPI cat-ELCCA for eIF4E–4E-BP1 and eIF4E–eIF4G to address this unmet need.
To test the applicability of PPI cat-ELCCA, full-length eIF4E, 4E-BP1, and eIF4G were first expressed as N-terminal HaloTag fusion proteins for selective labeling with either biotin for immobilization or methyltetrazine (mTet) for click chemistry (Figure 3).19 N-Terminal labeling was chosen due to the known geometries of the eIF4E PPIs to inhibit possible disruption of the interactions due to immobilization or the click chemistry detection step. However, C-terminal HaloTag vectors are also available, and it is anticipated that labeling sites will need to be optimized for application to other PPI systems. For the click chemistry detection step, we chose our second-generation approach, which utilizes inverse-electron demand Diels–Alder (IEDDA) chemistry, and HRP was labeled with trans-cyclooctene (TCO).20 We have previously demonstrated that, because of its kinetic superiority, replacing our first-generation copper-catalyzed alkyne–azide cycloaddition (CuAAC) click chemistry detection step21,22 with IEDDA allowed us to develop a technology with improved sensitivity and reproducibility, enabling automated HTS.20 Similar results were observed using CuAAC with PPI cat-ELCCA (Figure S3).
Figure 3.

PPI cat-ELCCA for eIF4E PPIs. mTet = methyltetrazine; TCO = trans-cyclooctene. For the eIF4E–4E-BP1 assay, eIF4E was immobilized, and 4E-BP1 was mTet labeled. For the eIF4E–eIF4G assay, eIF4G was immobilized, and eIF4E was mTet labeled.
With our assay components in hand and verified (Figures S1 and S2), we tested PPI cat-ELCCA. Biotinylated eIF4E or eIF4G was first immobilized in the wells of a 384-well streptavidin plate and incubated with mTet-4E-BP1 or -eIF4E, respectively. Of note, for the eIF4E–eIF4G PPI, eIF4G was immobilized due to its large size (220 kDa) and crude preparation from overexpressing HEK293T cells (Figure S1), as eIF4G cannot be purified to homogeneity. Following click reaction with HRP-TCO, the wells were treated with SuperSignal West Pico, and chemiluminescence signal was measured. As shown in Figure 4A, our preliminary experiments were successful, and >500-fold chemiluminescence signal increases were observed for both PPIs, whereas controls without either protein yielded no signal as expected. Importantly, this is the first time that full-length eIF4G protein has been used in a biochemical assay of eIF4E binding as previous reports focused solely on eIF4G peptide or protein fragments due to its size and instability and limitations of the assay formats used (FP and TR-FRET).17,18,23,24 The interactions were also found to be dose-dependent, yielding apparent Kd values of 3.8 ± 0.7 and 8.3 ± 0.5 nM for 4E-BP1 and eIF4G binding, respectively (Figure 4B). These values are in line with previous biophysical affinity measurements of 4E-BP1 protein and eIF4G fragments for eIF4E.15,25,26 Of note, a complete loss of signal (eIF4E–eIF4G) or drastic increase in the apparent Kd (eIF4E–4E-BP1) was observed when eIF4E was exposed to a freeze/thaw cycle (Figure S5A). Interestingly, this phenonmenon was observed only when the protein was free in solution as the mTet-labeled substrate (Figure S5B). Because eIF4E instability is well-documented in the literature,27 we hypothesize that immobilization enhances its stability during the assay. A similar result was observed with eIF4G, and the assay using mTet-eIF4G in solution failed (data not shown). Thus, our immobilization-based PPI cat-ELCCA may enable the analysis of full-length proteins that exhibit stability issues. On the basis of these results, we propose that protein stability be a guiding principle in determining which protein is to be immobilized for use in PPI cat-ELCCA.
Figure 4.

PPI cat-ELCCA for eIF4E PPIs. (A) Proof-of-concept data; X refers to protein. (B) Kd,app measurement. (C) Comparison to ELISA.
To provide a direct comparison of PPI cat-ELCCA to ELISA, we measured the Kd of the eIF4E–4E-BP1 PPI. As shown in Figure 4C, a comparable affinity of 19 ± 1 nM was observed, showing that our results with PPI cat-ELCCA correlate with this standard methodology. Although the robustness of the assays were similar, distinct advantages of our approach were noted. With respect to sensitivity, PPI cat-ELCCA exhibited a superior limit of detection (0.014 ng for PPI cat-ELCCA and 0.15 ng for ELISA) and comparable limit of quantitation (0.43 and 0.47 ng, respectively) (Figure S6). ELISA was also slower by 2 h due to the required additional incubation step with the enzyme-linked secondary antibody and subsequent washing steps. Finally, a similar ELISA for the eIF4E–eIF4G PPI could not be performed due to the necessity for using crude eIF4G protein, which was contaminated with endogenous eIF4E and would yield false results as the primary antibody would detect both endogenous and exogenous eIF4E.
Because our goal is to use PPI cat-ELCCA to discover inhibitory chemical probes, we examined the competitive effect of 4E-BP1 proteins and previously reported small molecule modulators of the eIF4E–eIF4G PPI.17,18 As shown in Figure 5A, unlabeled 4E-BP1 protein was able to readily compete with the mTet-labeled protein (IC50 value of 11.0 ± 0.1 nM), whereas null-binding 4E-BP1 mutants28 exhibited reduced inhibition of the PPI-dependent signal (IC50 values of 67, 370, 618 and 41,000 nM for the M60A, L59A, Y54A, and L59A/M60A mutants, respectively). For the small molecules 4EGI-1 and 4E1RCat, apparent IC50 values of 12 ± 1 and 3.1 ± 0.4 μM for eIF4E–eIF4G and 5.1 ± 0.1 and 1.8 ± 0.6 μM for eIF4E–4E-BP1 were measured, respectively (Figure 5B, C). Importantly, this is the first study demonstrating that these molecules can directly disrupt eIF4E–4E-BP1 binding.17,18 For 4EGI-1, this is in contrast to its initial report, which indicated that it stabilized 4E-BP1 binding to eIF4E in cells.17 However, this is likely due to its complex cellular activity, which may involve inhibition of mTOR,29 and inhibition or stabilization of 4E-BP1 binding by this molecule has never been analyzed in a biochemical or biophysical assay, only binding to eIF4E or eIF4E–4E-BP1 fragment complexes.17,30,31 4EGI-1 was recently found to bind allosterically to eIF4E at its lateral surface, which is distinct from that of eIF4G and 4E-BP1 that competitively bind at the dorsal surface.32 Because both eIF4G33,34 and 4E-BP130,35–37 contain second, yet weaker, binding sites at this lateral surface of eIF4E, it is not surprising that 4EGI-1 would disrupt the binding of both. It is important to note, however, that both 4EGI-1 and 4E1RCat exhibit potentially nonspecific inhibitory mechanisms for the PPIs, as indicated by their Hill slopes < −2. On the other hand, a 4E-BP1 peptide (Gly49-Asn64) exhibited specific inhibition of both PPIs with apparent IC50 values of 27 ± 4 and 74 ± 5 nM for the eIF4E–eIF4G and eIF4E–4E-BP1 PPIs, respectively (Figure 5D), and Hill slopes of −1.
Figure 5.
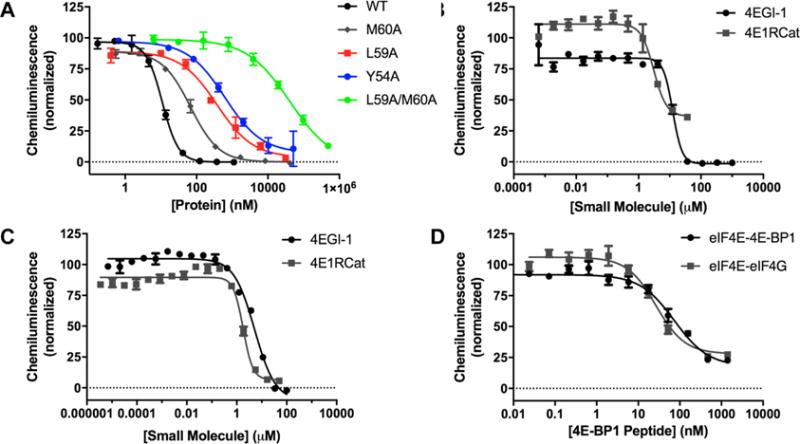
Characterization of PPI cat-ELCCA for chemical probe discovery. (A) Inhibition by 4E-BP1 proteins. (B, C) Inhibition of eIF4E–eIF4G and eIF4E–4E-BP1 by 4EGI-1 and 4E1RCat, respectively. (D) Inhibition by 4E-BP1 peptide.
To support the use of PPI cat-ELCCA in high-throughput inhibitor discovery, we further characterized the eIF4E–4E-BP1 assay. We first found that it was tolerant to up to 10% DMSO (Figure S7). Subsequently, we analyzed the effect of potential compound interferents. We had previously demonstrated the compatibility of cat-ELCCA with fluorescent molecules and fluorescence quenchers;21 however, we had not examined the impact of known aggregators, which are littered within screening libraries.38,39 Well-known aggregate-forming molecules quercetin, benzyl benzoate, and Congo Red were tested at 12.5, 25, and 50 μM; of these, only Congo Red was found to inhibit the assay (Figure S8A). Because our assay buffer contains 0.01% Tween-20, we were interested in determining if this mechanism of inhibition was still due to colloid formation. Upon examining the activity of this molecule in the presence and absence of detergent (Figure S8B, C), we found that dose-dependent inhibition was observed in the presence of Tween-20, whereas an extreme increase in chemiluminescence signal was observed in its absence. This indicates that Congo Red is likely a real yet nonspecific inhibitor of the PPI, as evidenced by its Hill slope < −2 similar to 4EGI-1 and 4E1RCat.
Finally, to demonstrate the utility of PPI cat-ELCCA for HTS, we adapted it to automated liquid handling. Importantly, the assay performed excellently with a measured Z′ factor of 0.66, signal-to-background (S/B) ratio of 23, and signal-to-noise (S/N) ratio of >10,000 (Figure S8D). For HTS potential, the Z′ factor is the most important of these statistical parameters, and assays exhibiting Z′ factors of ≥0.5 are regarded as suitable assays.40 As additional characterization, we screened a small collection of ∼3,000 fragment molecules. Fragments were chosen to further analyze the impact of a high compound concentration (400 μM to 1 mM) on our assay and the fact that fragment screening is typically performed using low-throughput biophysical methods due to compound interference in biochemical assays.41,42 Although no hit compounds were identified (Figure 6), the assay performed well with Z′ factors ranging between 0.44 and 0.67. Plate-to-plate high signal deviations were observed, however, as gain values for chemiluminescence measurements were normalized to the first plate read. With respect to compound interference, we did observe signals higher than the negative control in a few of the sample-containing wells. Based on our results with Congo Red in the absence of detergent, we attribute this to aggregation, or compounds that have crashed out of solution, due to insolubility. Nonetheless, from the presented promising preliminary data, we are confident that PPI cat-ELCCA can be employed in HTS to discover chemical probes for eIF4E PPIs.
Figure 6.

HTS data using PPI cat-ELCCA against the eIF4E–4E-BP1 PPI. Black = negative controls; red = positive controls; blue = samples.
In summary, we have described a new assay technology that readily facilitates the analysis of full-length PPIs. Although obvious advantages of PPI cat-ELCCA over ELISA have been noted, we anticipate that based on its similarity to this assay format that our new technology will be useful for PPIs with Kd values ≤1 μM.8 As PPIs are of great interest in both basic science and drug discovery, we hope that PPI cat-ELCCA will provide a key methodology in advancing PPI-targeted investigations and chemical probe discovery. An expanded HTS campaign against eIF4E PPIs is ongoing in our laboratory, the details of which will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported through a generous start-up package from the University of Michigan College of Pharmacy, a discovery grant from the American Brain Tumor Association (A.L.G.), a pilot grant from the University of Michigan Center for the Discovery of New Medicines, the NIH (R01 CA202018 to A.L.G., T32 GM008353 to J.M.S., T32 CA140044 to D.C.M., T32 GM008597 to O.T.J.), and the NSF (O.T.J.). We thank Martha Larsen and Steve Vander Roest for assistance with HTS equipment setup.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombs-ci.7b00128.
General synthetic, protein expression/purification, and bioconjugation methods, PPI cat-ELCCA protocol, and supplemental figures (PDF)
ORCID
Amanda L. Garner: 0000-0002-0870-3347
Notes
The authors declare no competing financial interest.
References
- 1.Whitty A. Small-molecule inhibitors of protein-protein interactions: challenges and prospects. In: Lackey KE, editor. Gene Family Targeted Molecular Designs. John Wiley & Sons, Inc; Hoboken, NJ: 2009. pp. 199–233. [Google Scholar]
- 2.Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discovery. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 3.Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garner AL, Janda KD. Protein-protein interactions and cancer: targeting the central dogma. Curr Top Med Chem. 2011;10:258–280. doi: 10.2174/156802611794072614. [DOI] [PubMed] [Google Scholar]
- 5.Mullard A. Pioneering apoptosis-targeted cancer drug poised for FDA approval. Nat Rev Drug Discovery. 2016;15:147–149. doi: 10.1038/nrd.2016.23. [DOI] [PubMed] [Google Scholar]
- 6.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advances selective BCL-2 family inhibitors. Nat Rev Drug Discovery. 2017;16:273–284. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 7.Zhou M, Li Q, Wang R. Current experimental methods for characterizing protein-protein interactions. ChemMedChem. 2016;11:738–756. doi: 10.1002/cmdc.201500495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkin MR, Glicksman MA, Fu H, Havel JJ, Du Y. Assay Guidance Manual. Eli Lilly and Company, National Center for Advancing Translational Sciences; Bethesda, MD: 2004. Inhibition of protein-protein interactions: non-cellular assay formats. [PubMed] [Google Scholar]
- 9.Janzen WP. Screening technologies for small molecule discovery: the state of the art. Chem Biol. 2014;21:1162–1170. doi: 10.1016/j.chembiol.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 10.Schorpp K, Rothenaigner I, Salmina E, Reinshagen J, Low T, Brenke JK, Gopalakrishnan J, Tetko IV, Gul S, Hadian K. Identification of small-molecule frequent hitters from AlphaScreen high-throughput screens. J Biomol Screening. 2014;19:715–726. doi: 10.1177/1087057113516861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imbert P-E, Unterreiner V, Siebert D, Gubler H, Parker C, Gabriel D. Recommendations for the reduction of compound artifacts in time-resolved fluorescence resonance energy transfer assays. Assay Drug Dev Technol. 2007;5:363–372. doi: 10.1089/adt.2007.073. [DOI] [PubMed] [Google Scholar]
- 12.Comley J. Assay interference a limiting factor in HTS? Drug Disc World. 2003;4:91–98. [Google Scholar]
- 13.Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75:250–263. doi: 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discovery. 2015;14:261–278. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- 15.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell. 1999;3:707–716. doi: 10.1016/s1097-2765(01)80003-4. [DOI] [PubMed] [Google Scholar]
- 16.Martineau Y, Azar R, Bousquet C, Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013;32:671–677. doi: 10.1038/onc.2012.116. [DOI] [PubMed] [Google Scholar]
- 17.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh M, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 18.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011;108:1046–1051. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Friedman Ohana R, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugirls G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz DA, Garner AL. A click chemistry-based microRNA maturation assay optimized for high-throughput screening. Chem Commun. 2016;52:8267–8270. doi: 10.1039/c6cc02894b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenz DA, Song JM, Garner AL. High-throughput platform assay technology for the discovery of pre-microRNA-selective small molecule probes. Bioconjugate Chem. 2015;26:19–23. doi: 10.1021/bc500544v. [DOI] [PubMed] [Google Scholar]
- 22.Garner AL, Janda KD. cat-ELCCA: a robust method to monitor the fatty acid acyltransferase activity of ghrelin O-acyltransferase (GOAT) Angew Chem, Int Ed. 2010;49:9630–9634. doi: 10.1002/anie.201003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lama D, Quah ST, Verma CS, Lakshminarayanan R, Beuerman RW, Lane DP, Brown CJ. Rational optimization of conformational effects induced by hydrocarbon staples in peptides and their binding interfaces. Sci Rep. 2013;3:3451. doi: 10.1038/srep03451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morino S, Imataka H, Svitkin YV, Pestova TV, Sonenberg N. Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Mol Cell Biol. 2000;20:468–477. doi: 10.1128/mcb.20.2.468-477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross JD, Moerke NJ, von der Haar T, Lugovskoy AA, Sachs AB, McCarthy JEG, Wagner G. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell. 2003;115:739–750. doi: 10.1016/s0092-8674(03)00975-9. [DOI] [PubMed] [Google Scholar]
- 26.Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2014;519:106–109. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 27.Volpon L, Osborne MJ, Topisirovic I, Siddiqui N, Borden KLB. Cap-free structure of eIF4E suggests a basis for conformational regulation by its ligands. EMBO J. 2006;25:5138–5149. doi: 10.1038/sj.emboj.7601380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grosso S, Pesce E, Brina D, Beugnet A, Loreni F, Biffo S. Sensitivity of global translation to mTOR inhibition in REN cells depends on the equilibrium between eIF4E and 4E-BP1. PLoS One. 2011;6:e29136. doi: 10.1371/journal.pone.0029136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yefidoff-Freedman R, Chen T, Sahoo R, Chen L, Wagner G, Halperin JA, Aktas BH, Chorev M. 3-Substituted indazoles as configurationally locked 4EGI-1 mimetics and inhibitors of the eIF4E/eIF4G interaction. ChemBioChem. 2014;15:595–611. doi: 10.1002/cbic.201300723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, Leger-Abraham M. Molecular mechanism of the dual activity of 4EGI-1: dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci U S A. 2015;112:E4036–E4045. doi: 10.1073/pnas.1512118112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Yefidoff-Freedman R, Supko JG, Chorev M, Wagner G, Halperin JA. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012;3:869–881. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadopoulos E, Jenni S, Kabha E, Takrouri KJ, Yi T, Salvi N, Luna RE, Gavathiotis E, Mahalingam P, Arthanari H, Rodriguez-Mias R, Yefidoff-Freedman R, Aktas BH, Chorev M, Halperin JA, Wagner G. Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proc Natl Acad Sci U S A. 2014;111:E3187–E3195. doi: 10.1073/pnas.1410250111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umenaga Y, Paku KS, In Y, Ishida T, Tomoo K. Identification and function of the second eIF4E-binding region in N-terminal domain of eIF4G: comparison with eIF4E-binding protein. Biochem Biophys Res Commun. 2011;414:462–467. doi: 10.1016/j.bbrc.2011.09.084. [DOI] [PubMed] [Google Scholar]
- 34.Gruner S, Peter D, Weber R, Wohlbold L, Chung M-Y, Weichenrieder O, Valkov E, Igreja C, Izaurralde E. The structures of eIF4E-eIF4G complexes reveal an extended interface to regulate translation initiation. Mol Cell. 2016;64:467–479. doi: 10.1016/j.molcel.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 35.Paku KS, Umenaga Y, Usui T, Fukuyo A, Mizuno A, In Y, Ishida T, Tomoo K. A conserved motif with the flexible C-terminus of the translational regulator 4E-BP is required for tight binding to the mRNA cap-binding protein eIF4E. Biochem J. 2012;441:237–245. doi: 10.1042/BJ20101481. [DOI] [PubMed] [Google Scholar]
- 36.Lukhele S, Bah A, Lin H, Sonenberg N, Forman-Kay JD. Interaction of the eukaryotic initiation factor 4E with 4E-BP2 at a dynamic bipartite interface. Structure. 2013;21:2186–2196. doi: 10.1016/j.str.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 37.Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Weichenrieder O, Izaurralde E. Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol Cell. 2015;57:1074–1087. doi: 10.1016/j.molcel.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 38.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 39.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J-H, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 41.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discovery. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- 42.Barker J, Courtney S, Hesterkamp T, Ullmann D, Whittaker M. Fragment screening by biochemical assay. Expert Opin Drug Discovery. 2006;1:225–236. doi: 10.1517/17460441.1.3.225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.