

Abstract
A complex cell envelope, composed of a mixture of lipid types including complex lipopolysaccharides, protects bacteria from the external environment. Clearly, the proteins embedded within the various components of the cell envelope have an intricate relationship with their local environment. Therefore, to obtain meaningful results, molecular simulations need to mimic as far as possible this chemically heterogeneous system. However, setting up such systems for computational studies is far from trivial, and consequently the vast majority of simulations of outer membrane proteins still rely on oversimplified phospholipid membrane models. This work presents an update of CHARMM-GUI Martini Maker for coarse-grained modeling and simulation of complex bacterial membranes with lipopolysaccharides. The qualities of the outer membrane systems generated by Martini Maker are validated by simulating them in bilayer, vesicle, nanodisc, and micelle environments (with and without outer membrane proteins) using the Martini force field. We expect this new feature in Martini Maker to be a useful tool for modeling large, complicated bacterial outer membrane systems in a user-friendly manner.
Keywords: coarse-grained simulation, outer membrane vesicles, bilayers, nanodiscs, micelles
GRAPHICAL ABSTRACT
An outer membrane vesicle generated by CHARMM-GUI Martini Maker with lipopolysaccharides in the outer leaflet and phospholipids in the inner leaflet.
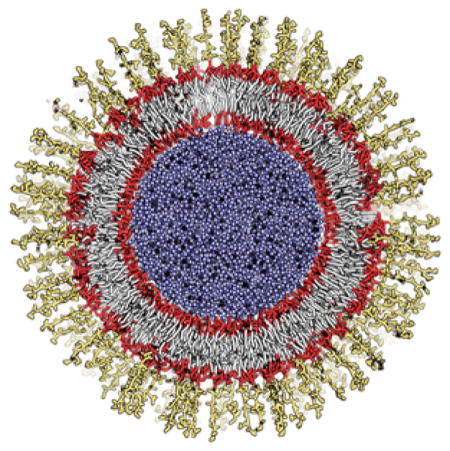
INTRODUCTION
Computational studies and in particular molecular dynamics (MD) simulations are now a firmly established technique for the study of biological membranes. At the all-atom or united-atom level, molecular simulations have been successfully used to study various membrane properties, the permeation of small molecules directly through model membranes, transport of ions and small molecules through specific channels, and the dynamics of membrane protein structures solved in different environments.1–4
Over the past decade, the biological simulation community has seen a widespread embracing of coarse-grained (CG) models and methods, which have widened the scope of simulations by allowing access to longer time and length scales. The Martini force field developed by Marrink and co-workers5,6 is perhaps the most widely used model for CG simulations of biological membranes. Some examples of recent successes are combination of the force field with the metadynamics method of enhanced sampling to study the energetics of conformational rearrangements in the epidermal growth factor receptor;7 high throughput simulations showing the effects of alcohol on a mechanosensitive protein;8 studies of local phase transitions in bacterial membranes induced by an antimicrobial peptide;3 and unraveling of the plastoquinone exchange pathways of the photosystem II complex.9 A key advantage of CG models is the ability to self-assemble the lipid component whether this be a flat bilayer, a micelle, or a small spherical vesicle in the presence or absence of the proteins of interest, thereby eliminating the initial ‘guesswork’ of determining how proteins are oriented and packed by the lipids and/or detergents in each environment. However, such a spontaneous self-assembly becomes rather more complex and questionable when the membranes are asymmetric or composed of lipids with large, complex carbohydrate components such as lipopolysaccharides (LPS) as in the case of bacterial membranes.10
Bacterial membranes are complex in terms of their chemical composition. In particular, the cell envelope surrounding the cytoplasm of Gram-negative bacteria is composed of the inner membrane, the periplasm, and the outer membrane (OM). The OM is a unique asymmetrical bilayer composed of LPS molecules in the outer leaflet and a mixture of zwitterionic and anionic phospholipids in the inner leaflet.11,12 The inner membrane is more or less symmetric, and both leaflets closely resemble the inner leaflet of the OM in terms of the phospholipid composition.13 Given that modification of these membranes is one of the ways that bacteria achieve resistance to our current arsenal of antibiotics,14,15 studying the relationship between the membrane/membrane proteins and drugs is imperative for the rational design of novel antibiotics.4,16–18 However, simulation studies of biologically relevant bacterial OM models are complicated by the chemical complexity of the membrane constituents, in particular, the LPS molecules.13,19–22 Detailed atomistic simulations of the OM have only recently become widespread,23–27 and consequently the first CG models of these membranes are only just being reported in the last couple of years.28,29
CHARMM-GUI (http://www.charmm-gui.org) is a web-based graphical user interface (GUI) to prepare complex molecular simulation systems and input files to facilitate the usage of common and advanced simulation techniques.30 Recently, taking advantage of the frameworks in all-atom CHARMM-GUI modules,31–33 CHARMM-GUI Martini Maker (http://www.charmm-gui.org/input/martini) was developed to prepare various CG simulation systems in solution, bilayer, micelle, nanodisc, and vesicle environments using the Martini force field.34 Although more than 200 lipid types are available in Martini Maker, incorporating the CG LPS molecules in the system building process poses new challenges and thus requires specific methods for system size determination and counterion placement. To address the practical difficulties of setting up simulation systems with LPS molecules, we have updated Martini Maker to automate the building process of LPS-containing complex bilayer, micelle, nanodisc, and vesicle systems, as well as systems with randomly distributed LPS molecules. In this work, the update in Martini Maker is described and its robustness is tested by building and simulating various LPS-containing systems to illustrate the simulation contents that one can perform with Martini Maker.
METHODS
CHARMM-GUI Implementation
Martini models of two different LPS, Ra LPS (RAMP) and Re LPS (REMP), were added in CHARMM-GUI Martini Maker (Figure 1). The LPS models follow a 4-to-1 mapping scheme of the Martini force field and the parameters were optimized based on united-atom LPS simulations to improve accuracy.35,36
Figure 1.

Structural comparison between (A) E. coli R3-core type LPS and corresponding Martini (B) Ra and (C) Re LPS models. The carbohydrate names are: Kdo for 2-keto-3-deoxyoctulosonate, Hep for L-glycero-D-manno heptose, Glc for D-glucose, Gal for D-galactose, and GalNAc for N-acetyl-D-galactosamine. The lipid A molecule in this study consists of two D-glucosamine (GlcN) residues joined by a β-(1→6)-linkage, two monophosphoester groups at O1 and O4′, and six amide/ester-linked fatty acids. Lipid A head and tail groups are colored in red and light blue, respectively. Two Kdo (Re glycans) are colored in dark gray, and the rest (Ra) glycans are colored in light gray. Phosphates and Kdo carboxyl groups are colored in tan and dark blue, respectively.
The overall building procedures of all LPS-containing Martini Maker modules (Bilayer/Nanodisc/Vesicle/Micelle/Random Builders) are identical from the original implementation.34 Briefly, in STEP 1, a user-specified protein structure is read-in through PDB Reader. In STEP 2, the protein orientation is changed based on the user-specific input; by definition, the Z axis is the membrane normal and Z = 0 is the membrane center. In STEP 3, the system size is determined, and the pseudo spheres are placed for assigning lipid head group positions. Note that this is the first step when the membrane-only generation option is selected. In STEP 4, the system components (lipids, water, and ions) are generated. Finally, all the components are assembled in STEP 5. During STEP 5, the CHARMM structure (PSF) and coordinate (CRD/PDB) files of each component generated in STEP 4 are merged into single PSF and CRD/PDB files, and water beads in close proximity to the solutes are removed.
Some LPS-specific changes were introduced in the system size calculation and ion placement steps. As described above, the system size was previously determined in STEP 3. However, as the LPS molecule has a long carbohydrate chain, a portion of the LPS molecule can be stretched out beyond the system box determined in STEP 3 based on phospholipids. To resolve this issue, if the system contains LPS, the system size is recalculated by taking the LPS height into account in STEP 4, and the updated system size information is used for further steps (building water box and placing ions).
As divalent cations play an important role in stabilizing the bacterial OM by interacting with the LPS,22,37–40 the ion placement procedure in STEP 4 was modified to use Ca2+ as the counterions for LPS lipid A and core oligosaccharides. By default, CHARMM-GUI adds Ca2+ ions to neutralize lipid A, but for the LPS core, CHARMM-GUI provides an option to select an ion type (Na+ or Ca2+).
Martini Force Fields Used in This Study
The standard lipid parameters for palmitoyl-oleoyl-phosphatidylethanolamine (POPE) and palmitoyl-oleoyl-phosphatidylglycerol (POPG) were taken from Wassenaar et al.41 and those for CDL2 (a cardiolipin with a net charge of −2e) from Dahlberg and Maliniak.42 The parameters for Na+ and Cl– were taken from Marrink et al.5,6 For Ca2+, a well-tested Martini model is not available yet. Here, Ca2+ was simply modeled as Na+ with +2e, as this has been used before in other published studies.43 As in most applications, the standard water model was taken from Marrink et al.44 The parameters for LPS are those defined in Hsu et al.35 Note that a few bonds with large force constants in the original LPS models were replaced with constraints to improve stability and allow larger integration time steps.6 For the proteins, the Martini 2.1 protein force field was used45 in combination with an elastic network.46 The common settings associated with the Martini model were used to perform the simulations, including a 12-Å cutoff for the non-bonded interactions using shifted potentials.47 In this study, unless specified explicitly, all NPT (constant particle number, pressure and temperature) simulations were performed at 310 K, atmospheric pressure of 1 bar, and physiological salt concentration (150 mM NaCl for bulk solution with additional Na+ or Ca2+ ions to neutralize the LPS core and Lipid A, respectively). The systems generated by Martini Maker using default options (unless specified explicitly) were energy-minimized and equilibrated using the default settings of the README output file and the GROMACS 5.1 molecular dynamics package.48
Bilayer Systems
Three CG OM systems were constructed to test Bilayer Builder in Martini Maker; Table S1 provides a summary of the system information. The membrane composition mimics the E. coli OM and contains Ra LPS in the outer leaflet and a composition of POPE, POPG, and CDL2 at a ratio of 18:1:1 at the inner leaflet.17,41,42,49 Two of the OM systems contained an inserted β-barrel outer membrane protein (OMP), the monomeric OmpA (PDB:1QJP)50 or the trimeric OmpF (PDB:3POX);51 see Figure 2 for their structures. Both are major OMPs in E. coli as identified by proteomics studies.52,53 The proteins were inserted into the OM using the orientation predefined in the OPM database.54 The OM-only and the OM-OmpA systems were set to be 100 × 100 Å2 in XY, the OM-OmpF system was bigger (150 × 150 Å2) due to the relatively large size of the OmpF trimer. All systems initially had a layer of 45 Å of Martini water between the periodic images of the OM. After equilibration, each system was simulated for 5- to 9-μs production with a time step of 20 fs.
Figure 2.

Top view structures of outer membrane proteins OmpA (PDB ID: 1QJP) and trimeric OmpF porins (PDB ID: 3POX). Four and Eight loops of OmpA and OmpF are also depicted in different colors. Red and blue spheres in the OmpF porins are acidic and basic residues in the constriction zone.
In this work, the membrane thickness was defined as the distance between the average Z positions of phosphate atoms in each leaflet and calculated using MDAnalysis.55 For Ra LPS (containing phosphates in Hep sugars), only the Lipid A phosphates were used for the thickness calculation. The area per lipid of each lipid type was determined by two-dimensional Voronoi analysis using the phosphate group of each lipid and an in-house adaptation of the pyvoro library (created by Joe Jordan) and MDAnalysis; for LPS, PO1 and PO2 in lipid A were used. All protein beads within a distance of 10 Å from any selected phosphate groups of neighboring lipids were used for the protein-lipid interface. Only the last microsecond of the production run was used to measure this value. In all cases the leaflets were analyzed separately. All means and standard deviations were calculated using GROMACS 5.1 gmx analyze48. Visual inspection, image rendering, and calculation of the root-mean-square deviation (RMSD) of the protein from their initial structure were performed with VMD,56 and in case of the RMSD, the full production run of 5-μs was used.
Nanodisc Systems
Two symmetric discoidal LPS nanodiscs with neutralizing Na+ or Ca2+ ions were constructed with Nanodisc Builder in Martini Maker; see Table S2 for the system information. In each system, the discoidal LPS bilayers were encased by two membrane scaffold protein MSP1D1. Given the nanodisc area by MSP1D1 (a diameter of ~95 Å), the first system was made with 37 Re LPS molecules per leaflet, and the second system was built with 33 Ra LPS molecules per leaflet. After the equilibration, we performed 1-μs production runs with a time step of 20 fs. The membrane thickness analysis was done on a per-phosphate basis through the use of two-dimensional Voronoi diagrams and Delaunay triangulations as a function of distance from the nanodisc center.
Vesicle Systems
Two outer membrane vesicle (OMV)-only and two OMV-OmpF systems of different diameters (150 Å or 100 Å) were constructed with Vesicle Builder in Martini Maker; see Table S3 for the system information. The outer leaflet of the vesicles contained either Re or Ra LPS molecules, while the inner leaflets contained POPE, POPG, and CDL2 lipids at a ratio of 18:1:1. Following energy minimization and NPT equilibration, each vesicle system was simulated for several μs with a time step of 20 fs (Table S3). Before the production run, the vesicle was built with two pores on the surface to allow free movement of water particles to equilibrate the interior and exterior water.57 The vesicle properties were determined using the last 500 ns of simulation time. The bilayer thickness was interpreted using radial distribution function (RDF) of the selected phosphate groups in the inner and outer leaflets of the OMV with respect to the center of mass of vesicle, and the area per lipid was estimated by calculating Voronoi cells for each of these isolated lipid phosphate groups.58 Vesicle radii were estimated using an in-house script by calculating the distance between the center of mass of the OM and the vesicle center.59 The membrane density profiles were measured as the position of the lipid phosphate groups along the bilayer normal using a bin width of 1 Å.
Micelle and Random Systems
To demonstrate the robustness and flexibility of Micelle and Random Builders with LPS CG models, we constructed and simulated various systems with different numbers of Ra or Re LPS molecules and with/without OmpF; see Table S4 for the system information. The LPS-only systems contained 5, 10, 15, and 20 LPS molecules in a micelle form or randomly distributed in 0.15 M NaCl solution with neutralizing Ca2+ ions added near to the Lipid A phosphate groups. The OmpF micelle and random systems were composed of a monomer of OmpF and 40 Ra LPS molecules at the same salt concentration. After the conventional CHARMM-GUI protocol for minimization and equilibration of the systems, we performed 10-μs production runs with a time step of 20 fs. As simulations with 20 Ra LPS (starting from a preformed micelle or a random distribution) showed some discrepancies regarding the shape of aggregates formed, we also performed additional simulations: i) simulations without Ca2+ ions, but replaced by Na+; ii) simulations with polarizable Martini water model;60 iii) simulations at higher temperature (400 K) and NVT (constant particle number, volume, and temperature) ensemble (with the volume equal to the average values obtained in the NPT simulations at 310 K and 1 bar). All average properties were calculated over the last 2 μs of the simulations.
RESULTS AND DISCUSSION
Martini (Re and Ra) LPS models (Figure 1) have been added to the following Martini Maker modules: Bilayer, Nanodisc, Vesicle, Micelle, and Random Builder. In this section, the robustness and applications of these modules are tested and illustrated by a wide content of practical simulation studies and their analyses.
Bilayer Systems
Martini Maker has shown its capability to construct complex plasma membranes (with or without a membrane protein) using Bilayer Builder.34 Here, to illustrate its ability to build complex, asymmetric OMs, three representations of the E. coli OMs were constructed, constituted of pure Ra LPS in the outer leaflet and a mixture of POPE, POPG, and CDL2 in the inner leaflet (see METHODS). The OM-only, OM-OmpA, and OM-OmpF systems (Fig. 3A, B) contained a total of 216, 205, and 402 lipids fully hydrated in 150 mM NaCl solution. The negative charges of Ra LPS were neutralized by a mixture of Ca2+ and Na+. All bilayers were rather rigid in the outer leaflet, with little to no diffusion of Ra LPS, in the course of the entire simulation. This caused the inserted protein to stay trapped between its initial neighboring Ra LPS molecules throughout the production run. In the lower leaflet, lipids diffuse freely and do not phase separate. None of the proteins left the OM during any of the runs.
Figure 3.

(A, B) The side and top view of the final snapshot of 5 μs simulations (at 310 K and 1 bar) of different Ra LPS systems. The unit cells are depicted by a blue dotted wireframe. (C) The OM thickness varied only little over the span of the simulation for each system (the red line shows the running average; n=20).
The objective of the LPS Bilayer Builder extension was to generate an initial structure that is close to an equilibrated state of an LPS-containing bilayer. Therefore, we investigated three bilayer properties. First, the OM thickness was defined as the distance between the phosphate groups of the inner leaflet and the phosphate groups of the lipid A in the outer leaflet. The average bilayer thickness and standard error over the last μs of each system are 37.61 ± 0.15 (OM-only), 37.25 ± 0.20 (OM-OmpA), and 37.18 ± 0.12 Å (OM-OmpF). For all three systems, the change in bilayer thickness over 5 μs is very small (less than 1 %). Interestingly, even though the change is less than 0.5 Å compared to the OM-only simulation, the insertion of OmpA and OmpF appears to slightly reduce the overall bilayer thickness. This is probably due to hydrophobic mismatch at the protein-lipid interface.
The second property we tested is the area per lipid (APL). The change in APL is slightly positive (<5%) for all lipids in all systems over 5 μs. POPG and CDL2 show the highest change probably due to their low copy number (Fig. S1). A table containing the average APL over the last μs for each lipid in each system can be found in Table S5. The slight increase in APL corresponds well with the observed slight decrease in the OM thickness. As for the OM thickness, the initial APL appears to be close to its equilibrium value.
The third property is the protein integrity. Since the Martini ElNeDyn elastic network used for OmpA and OmpF should preserve the global protein structure, the protein integrity was examined by calculating the backbone RMSD with respect to the CG crystal structure. The calculated RMSD for OmpA (1.60 ± 0.05 Å) and OmpF (1.88 ± 0.07 Å) was in the same order of magnitude reported in the original CG Martini ElNeDyn publication.46 Therefore, the insertion process of the protein in Bilayer Builder does not alter the protein to any relevant extent.
All of the bilayers tested are stable. The membrane thickness and APL after 5 μs are close to their initial values, which normally indicates that the generated initial configuration is close to an equilibrium state of an LPS-containing bilayer. However, due to the extremely slow dynamics of Ra LPS and the measured small drift in the bilayer thickness and APL, it is difficult to prove that we are indeed close to equilibrium. This is a general difficulty of working with Ra LPS or any model with extremely slow dynamics and has little to do with the capacities of Bilayer Builder. In addition, through Bilayer Builder, users can change the number of lipids or each lipid’s initial area in each leaflet, so that one can examine various properties of asymmetric bilayers of their own interest.
Nanodisc Systems
Discoidal lipid/protein particles, termed nanodiscs, are synthetic model membrane systems which are useful in the study of membrane proteins and native membrane environments.61,62 Each nanodisc generally contains two amphipathic membrane scaffold proteins (MSPs) that encase and thereby support a cylindrical lipid bilayer. Nanodiscs are increasingly being used as platforms for investigating integral membrane proteins such as bacteriorhodopsin, cytochrome P450, and G-protein coupled receptors.63 While the application of these discoidal lipid/protein particles is largely limited to simple phospholipids, the incorporation of LPS molecules into nanodiscs would enable the fabrication and investigation of more realistic Gram-negative OM environments. To provide insight into the dynamic properties of LPS nanodiscs, and to test the capabilities of Nanodisc Builder in Martini Maker, we constructed nanodiscs of Re or Ra LPS that were encased in two MPS1D1 proteins (see METHODS).
The RMSD of the MSPs reached asymptotic values within the simulation time, indicating that the nanodisc properties had converged (Figure S2). The equilibrium RMSD values are similar in the Ra LPS nanodisc systems; 4.3 ± 0.2 and 4.3 ± 0.3 Å (for each MSP with neutralizing Ca2+ ions) and 4.6 ± 0.2 and 4.1 ± 0.2 Å (with neutralizing Na+ ions) over the last 100-ns of simulation time. For comparison, the RMSD values in the Re LPS nanodisc systems are 4.7 ± 0.3 and 4.2 ± 0.4 Å (Ca2+ ions) and 5.2 ± 0.4 and 5.0 ± 0.3 Å (Na+ ions). The data reveal a slight reduction in the MSP stability when they encase LPS molecules of smaller head groups, and likewise reveal a slight decrease in protein stability when divalent cations are not present to stabilize the repulsive electrostatic interactions between LPS phosphate and carboxylate groups.
Figure 4 shows the bilayer thickness as a function of radial distance (R) from the nanodisc center for each nanodisc system. While the Ra LPS nanodisc bilayers generally preserve their thickness as a function of R, the Re LPS nanodisc bilayers taper towards the nanodisc edge. This deformation of lamellar structure is rationalized in terms of the incomplete coverage afforded by the MSPs that encase LPS acyl tails, and the relatively small size of the Re LPS lipid head groups. The Re LPS lipid head groups are able to perforate small gaps in the amphipathic protein belts leading to deformation of bilayer structure and reduction of the membrane thickness towards the disc edge (Figure 4E, F). This insight has important implications for experiments involving LPS nanodiscs, as it is evident that realistic Gram-negative OM environments are more easily achieved using LPS variants with bulkier core oligosaccharide sections, which are less able to perforate small holes in the encasing protein belts. Note that the symmetric LPS nanodisc bilayers are thinner than the planar OM thickness (Figure 3) because the OM contains phospholipids with longer acyl chains in the inner leaflet. The average thickness of the nanodisc bilayers within R = 20 Å are 28.46 ± 0.74 Å (Ra LPS with Ca2+), 28.32 ± 0.82 Å (Ra LPS with Na+), 34.65 ± 0.34 Å (Re LPS with Ca2+), and 35.89 ± 1.32 Å (Re LPS with Na+).
Figure 4.

Nanodisc bilayer thickness as a function of radial distance from the nanodisc center for the Ra LPS systems with (A) Na+ or (B) Ca2+ neutralizing ions and for the Re LPS systems with (C) Na+ or (D) Ca2+ neutralizing ions. The thickness values are calculated for the last 100 ns of the simulations. The error bars represent one standard deviation. (E) Side view of the symmetric Re LPS nanodisc after 1-μs of simulation time. The component Re LPS molecules are colored white, and the encasing MSP1D1 helices are represented as red and green isosurfaces, extracted from a volumetric Gaussian density map. (F) The symmetric Ra LPS nanodisc with the MSPs omitted to accentuate the Re LPS molecules that have perforated gaps in the encasing MSP helices. These perforating Re LPS lipids are colored yellow, grey, mauve and purple.
Vesicle Systems
Martini Maker has previously been used to manufacture multifaceted membrane systems that incorporate different membrane proteins34. Here, we built outer membrane vesicles (OMVs) that were made of lipids alone (OMV-only), and complex vesicles that incorporate lipids and embedded OmpF (OMV-OmpF). Vesicle radii of 100 and 150 Å were studied for each OMV composition (Figure 5A, B;). The outer leaflet of the vesicles contained Re or Ra LPS lipids, and the inner leaflet contained a combination of POPE, POPG, and CDL2 (see METHODS). To assess simulation convergence, we monitored the radius of each OMV (ROMV), which was defined as the radial distance between the OMV center and the center of mass of the encapsulating membranes. The ROMV from the last 500-ns simulations are 142.36 ± 0.31 (150-Å Re OMV-only), 141.96 ± 0.03 (150-Å Re OMV-OmpF), 140.07 ± 0.02 (150-Å Ra OMV-only), and 96.93 ± 0.04 Å (100-Å Ra OMV-OmpF), respectively (Figure S3).
Figure 5.

(A, B) OMVs structures and (C, D) Re and Ra LPS phosphate particle positions. The color scheme in (A) and (B) is the same as in Figure 3, and water beads are omitted for clarity. The phosphate particle positions are shown relative to the center of mass of the vesicle bilayers, and the average ROMV is at position 0. The black line shows data for (C) 150-Å Re OMV-only and (D) 150-Å Ra OMV-only, while the magenta line shows data for (C) 150-Å Re OMV-OmpF and (D) 100-Å Ra OMV-OmpF. Note that there are two phosphate density profiles in (D) Ra OMVs in the outer leaflet, one from lipid A and the other from the core region.
Seeking to investigate if the embedded protein influenced vesicle morphology, we measured the positions of the lipid phosphate groups relative to ROMV (Figure 5C, D). The incorporation of the OmpF protein does not significantly affect the distribution of LPS phosphate groups in either Re or Ra OMV. The OMV thickness was measured using the RDF of the outer and inner leaflets with respect to the center of mass of the vesicles. The OMV thicknesses from the last 500 ns are 35.88 ± 2.51 (150-Å Re OMV-only), 36.18 ± 2.15 (150-Å Re OMV-OmpF), 34.97 ± 4.16 (150-Å Ra OMV-only), and 34.45 ± 6.26 Å (100-Å Ra OMV-OmpF), respectively. The average LPS APL in the OMV systems over the last 500-ns simulations were 179.2 ± 0.014 (150-Å Re OMV-only), 179.4 ± 0.015 (150-Å Re OMV-OmpF), 200.5 ± 0.018 (150-Å Ra OMV-only), and 200.8 ± 0.029 Å2 (100-Å Ra OMV-OmpF), respectively, while the APL for the phospholipids in the OMV systems can be found in Table S6. The OMV thickness was smaller than the bilayer system (Figure 3) and the area per lipid of LPS was larger than the bilayer system (Table S5), indicating that our OMVs were less packed. Given the complexity of the system building and simulation, this could be acceptable, but also suggest that users may need to try a few different set of the number of LPS in the outer leaflet, as it would be difficult for us to come up with a set of general parameters applicable to various systems.
Micelle and Random Systems
Regarding their immune response, LPS aggregates are considered to be biologically active as monomers.64,65 However, little is known about their supramolecular organization. Experimental evidences indicate that LPS molecules can form micelles, lamellar, tubular or even cubic assemblies.65–68 In this section, we tested Random Builder and Micelle Builder as tools to get the first estimates of how the LPS Martini CG models predict the type of aggregates formed by small numbers of Re and Ra LPS molecules. MD simulations of 5 to 20 LPS molecules at 310 K and 1 bar were performed in order to predict their self-assemblies (see METHODS). The results are presented in Figure 6, giving emphasis to the systems containing 20 Ra LPS molecules. Other results, including those obtained with Re LPS molecules, are detailed in Figures S4–S6.
Figure 6.

(A–C) Initial structure and last snapshot of 10-μs MD simulations (at 310 K and 1 bar) performed with different Ra LPS systems. (A) 20 Ra LPS molecules were randomly placed and self-assembled during the simulations. In this case, an auxiliary 5-μs MD simulation was also performed at higher temperature (400 K) and fixed density (NVT ensemble). (B) 20 Ra LPS molecules were preassembled in a micelle configuration. (C) 40 Ra LPS molecules and one monomer of OmpF in a micelle configuration. For sake of clarity, only half of the LPS molecules are shown. (D) Time-series of the radius of gyration (Rg) of 20 Ra LPS systems. (E) Time-series of the tail-tail/core-core contacts ratio of 20 Ra LPS systems. Tail-tail and core-core contacts were defined using a distance cutoff of 6 Å between the beads. (F) Average radius of gyration of 20 Ra LPS molecules in four different conditions: (i) with Ca2+ ions added near to the phosphate groups of lipid A; (ii) with Na+ ions replacing Ca2+ ions; (iii) with Ca2+ ions and polarizable water model; (iv) with Ca2+ ions and higher temperature (400K). (G) Average radius of gyration of 5, 10, 15, and 20 Ra and Re LPS molecules in MD simulations performed with random and micelle initial configurations.
The 10-μs simulations of 20 Ra LPS molecules randomly placed in a water cubic box at 310 K did not yield any form of micelle structure or small bilayer patches. Instead, after 6 μs, Ra LPS aggregates appear to be trapped in stable elongated structures with an average radius of gyration (Rg) of 30.73 ± 0.03 Å (Fig. 6A, D). This unusual aggregate indicates more favorable interactions between the oligosaccharide cores than the aliphatic tails (as indicated by the tail-tail/core-core contacts ratio in Figure 6E). Different counterions (Na+ or Ca2+) or improvements in the electrostatic interactions (through the usage of polarizable models) were also not able to produce micelle or bilayer-like structures (Fig. 6F). Only an auxiliary 5-μs MD simulation performed at higher temperature (400 K but the same density of systems simulated at 310 K) was converged to bilayer-like aggregates with an average Rg of 25.60 ± 0.01 Å (Fig. 6A), showing a highly compact sugar head, as already exemplified in the previous bilayer sections.
The usage of Micelle Builder clearly showed the importance of the initial configuration for MD simulations with LPS molecules. The pre-assembled micelle structures at physiological conditions (310 K and 1 bar) and with less simulation time (Fig. 6B) allowed the formation of aggregates similar to those predicted by MD simulations of random Ra LPS initial configuration at high temperatures. About 1 μs was sufficient for the convergence of the bilayer-like structure. For MD simulations performed with less than 15 LPS molecules, the aggregates obtained from the simulations of Ra and Re LPS systems from both Micelle and Random Builders are structurally very similar with similar Rg, as shown in Figure 6G (and also in snapshots of Figures S5–6). For the creation of aggregates with 15 to 44 LPS molecules, we do not recommend the usage of Random Builder. In these cases, Micelle builder proved to be a more suitable tool, including its application for MD simulations of LPS-protein aggregates, as exemplified by the pre-assembled Ra LPS–OmpF micelle (Fig. 6C). In pure LPS systems containing 44 or more LPS molecules, Vesicle Builder or Bilayers Builder should be a reasonable choice, depending on the application. It should be stressed that the LPS aggregates presented here are not necessarily the supramolecular structures expected for specific LPS:water ratio, but an easy route to generate aggregates in specific sizes. Further MD simulations are necessary to understand if the current CG model is able to predict the most thermodynamically stable structure in specific LPS concentration.
CONCLUSIONS
In conclusion, we have demonstrated the utility of the CHARMM GUI Martini-Maker for the automated construction of systems that are of relevance to bacterial membranes containing LPS that are much more complex than the typical phospholipids. While phospholipids are often used to mimic bacterial membranes in computational studies, for molecular dynamics simulations of bacterial membranes to be biologically relevant, models should incorporate the diverse chemical moieties that are present in the native bacterial membranes, as well as accessing timescales that are longer than those practically possible using atomistic-level models. This argues for CG systems that can be easily set up. To this end, web-based user-friendly Martini-Maker is developed to construct flat bilayers, vesicles, micelles and nanodiscs with minimal user inputs. We have shown that the resulting structures are robust and similar compared to their atomistic counterparts, although there is room for improvement (i) by optimizing the Martini parameters for Ca2+ and by reducing the overall stickiness of sugar-based molecules as was recently performed for gangliosides69 and (ii) the Martini-Maker system building parameters to better estimate the number of lipid molecules in Vesicle Builder. Martini-Maker is expected to replace the laborious process of manual system setup of these complex LPS-containing systems, with an easy to use, freely available, online procedure that requires minimal manual intervention. Finally, the current work provides a framework to include all LPS molecules (including O-antigen polysaccharides) available in LPS Modeler in CHARMM-GUI (http://www.charmm-gui.org/input/lps).70
Supplementary Material
Acknowledgments
The work was supported in part by grants from NSF MCB-1727508, NIH GM087519 and GM103695, XSEDE MCB-070009 (to WI) and an ERC Advanced grant “COMP-MICR-CROW-MEM” (to SJM).
References
- 1.Mori T, Miyashita N, Im W, Feig M, Sugita Y. Biochim Biophys Acta. 2016;1858(7 Pt B):1635–1651. doi: 10.1016/j.bbamem.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingólfsson HI, Arnarez C, Periole X, Marrink SJ. J Cell Sci. 2016;129:257–268. doi: 10.1242/jcs.176040. [DOI] [PubMed] [Google Scholar]
- 3.Jefferies D, Hsu PC, Khalid S. Biochemistry. 2017;56(11):1672–1679. doi: 10.1021/acs.biochem.6b01063. [DOI] [PubMed] [Google Scholar]
- 4.Pothula KR, Solano CJ, Kleinekathöfer U. Biochim Biophys Acta. 2016;1858(7 Pt B):1760–1771. doi: 10.1016/j.bbamem.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 5.Marrink SJ, Tieleman DP. Chem Soc Rev. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- 6.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. J Phys Chem B. 2007;111(27):7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 7.Lelimousin M, Limongelli V, Sansom MS. J Am Chem Soc. 2016;138(33):10611–10622. doi: 10.1021/jacs.6b05602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melo MN, Arnarez C, Sikkema H, Kumar N, Walko M, Berendsen HJ, Kocer A, Marrink SJ, Ingólfsson HI. J Am Chem Soc. 2017;139(7):2664–2671. doi: 10.1021/jacs.6b11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Eerden FJ, Melo MN, Frederix PWJM, Periole X, Marrink SJ. Nature Communication. 2017;8:15214. doi: 10.1038/ncomms15214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knirel YA, Valvano MA. Bacterial Lipopolysaccharides. Springer; Vienna: 2011. [Google Scholar]
- 11.Bos MP, Robert V, Tommassen J. Annu Rev Microbiol. 2007;61:191–214. doi: 10.1146/annurev.micro.61.080706.093245. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz N, Kahne D, Silhavy TJ. Nat Rev Microbiol. 2006;4(1):57–66. doi: 10.1038/nrmicro1322. [DOI] [PubMed] [Google Scholar]
- 13.Parkin J, Chavent M, Khalid S. Biophys J. 2015;109(3):461–468. doi: 10.1016/j.bpj.2015.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikaido H. Pharmacol Ther. 1985;27(2):197–231. doi: 10.1016/0163-7258(85)90069-5. [DOI] [PubMed] [Google Scholar]
- 15.Nikaido H. Microbiol Mol Biol Rev. 2003;67(4):593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel DS, Re S, Wu EL, Qi Y, Klebba PE, Widmalm G, Yeom MS, Sugita Y, Im W. Biophys J. 2016;110(4):930–938. doi: 10.1016/j.bpj.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piggot TJ, Holdbrook DA, Khalid S. J Phys Chem B. 2011;115(45):13381–13388. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- 18.Wu EL, Fleming PJ, Yeom MS, Widmalm G, Klauda JB, Fleming KG, Im W. Biophys J. 2014;106(11):2493–2502. doi: 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khalid S, Berglund NA, Holdbrook DA, Leung YM, Parkin J. Biochem Soc Trans. 2015;43(2):162–167. doi: 10.1042/BST20140262. [DOI] [PubMed] [Google Scholar]
- 20.Patel DS, Qi Y, Im W. Curr Opin Struct Biol. 2017;43:131–140. doi: 10.1016/j.sbi.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Pavlova A, Hwang H, Lundquist K, Balusek C, Gumbart JC. Biochim Biophys Acta. 2016;1858(7 Pt B):1753–1759. doi: 10.1016/j.bbamem.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Kim S, Patel DS, Park S, Slusky J, Klauda JB, Widmalm G, Im W. Biophys J. 2016;111(8):1750–1760. doi: 10.1016/j.bpj.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleming PJ, Patel DS, Wu EL, Qi Y, Yeom MS, Sousa MC, Fleming KG, Im W. Biophys J. 2016;110(12):2698–2709. doi: 10.1016/j.bpj.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holdbrook DA, Piggot TJ, Sansom MS, Khalid S. Biochim Biophys Acta. 2013;1828(2):715–723. doi: 10.1016/j.bbamem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Patel DS, Kucharska I, Tamm LK, Im W. Biophys J. 2017;112(2):346–355. doi: 10.1016/j.bpj.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piggot TJ, Holdbrook DA, Khalid S. Biochim Biophys Acta. 2013;1828(2):284–293. doi: 10.1016/j.bbamem.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 27.Jo S, Wu EL, Stuhlsatz D, Klauda JB, MacKerell AD, Jr, Widmalm G, Im W. Methods Mol Biol. 2015;1273:391–406. doi: 10.1007/978-1-4939-2343-4_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma H, Irudayanathan FJ, Jiang W, Nangia S. J Phys Chem B. 2015;119(46):14668–14682. doi: 10.1021/acs.jpcb.5b07122. [DOI] [PubMed] [Google Scholar]
- 29.Van Oosten B, Harroun TA. J Mol Graph Model. 2016;63:125–133. doi: 10.1016/j.jmgm.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Jo S, Kim T, Iyer VG, Im W. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 31.Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W. J Comput Chem. 2014;35(27):1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jo S, Lim JB, Klauda JB, Im W. Biophys J. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng X, Jo S, Lee HS, Klauda JB, Im W. J Chem Inf Model. 2013;53(8):2171–2180. doi: 10.1021/ci4002684. [DOI] [PubMed] [Google Scholar]
- 34.Qi Y, Ingólfsson HI, Cheng X, Lee J, Marrink SJ, Im W. J Chem Theory Comput. 2015;11(9):4486–4494. doi: 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
- 35.Hsu PC, Jefferies D, Khalid S. J Phys Chem B. 2016;120(43):11170–11179. doi: 10.1021/acs.jpcb.6b06615. [DOI] [PubMed] [Google Scholar]
- 36.Graham JA, Essex JW, Khalid S. J Chem Inf Model. 2017;57(4):650–656. doi: 10.1021/acs.jcim.7b00096. [DOI] [PubMed] [Google Scholar]
- 37.Coughlin RT, Tonsager S, McGroarty EJ. Biochemistry. 1983;22(8):2002–2007. doi: 10.1021/bi00277a041. [DOI] [PubMed] [Google Scholar]
- 38.Schindler M, Osborn MJ. Biochemistry. 1979;18(20):4425–4430. doi: 10.1021/bi00587a024. [DOI] [PubMed] [Google Scholar]
- 39.van Alphen L, Verkleij A, Leunissen-Bijvelt J, Lugtenberg B. J Bacteriol. 1978;134(3):1089–1098. doi: 10.1128/jb.134.3.1089-1098.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu EL, Engstrom O, Jo S, Stuhlsatz D, Yeom MS, Klauda JB, Widmalm G, Im W. Biophys J. 2013;105(6):1444–1455. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wassenaar TA, Ingólfsson HI, Bockmann RA, Tieleman DP, Marrink SJ. J Chem Theory Comput. 2015;11(5):2144–2155. doi: 10.1021/acs.jctc.5b00209. [DOI] [PubMed] [Google Scholar]
- 42.Dahlberg M, Maliniak A. J Chem Theory Comput. 2010;6(5):1638–1649. doi: 10.1021/ct900654e. [DOI] [PubMed] [Google Scholar]
- 43.Pannuzzo M, de Jong DH, Raudino A, Marrink SJ. The Journal of Chemical Physics. 2014;140:124905. doi: 10.1063/1.4869176. [DOI] [PubMed] [Google Scholar]
- 44.Marrink SJ, de Vries AH, Mark AE. J Phys Chem B. 2004;108(2):750–760. [Google Scholar]
- 45.Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. J Chem Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 46.Periole X, Cavalli M, Marrink SJ, Ceruso MA. J Chem Theory Comput. 2009;5(9):2531–2543. doi: 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- 47.de Jong DH, Baoukina S, Ingólfsson HI, Marrink S. J Comput Phys Commun. 2016;199:1–7. [Google Scholar]
- 48.Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, Lindahl E. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 49.Lugtenberg EJ, Peters R. Biochim Biophys Acta. 1976;441(1):38–47. doi: 10.1016/0005-2760(76)90279-4. [DOI] [PubMed] [Google Scholar]
- 50.Pautsch A, Schulz GE. J Mol Biol. 2000;298(2):273–282. doi: 10.1006/jmbi.2000.3671. [DOI] [PubMed] [Google Scholar]
- 51.Efremov RG, Sazanov LA. J Struct Biol. 2012;178(3):311–318. doi: 10.1016/j.jsb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Molloy MP, Herbert BR, Slade MB, Rabilloud T, Nouwens AS, Williams KL, Gooley AA. Eur J Biochem. 2000;267(10):2871–2881. doi: 10.1046/j.1432-1327.2000.01296.x. [DOI] [PubMed] [Google Scholar]
- 53.Neidhardt FC, Ingraham JL, Schaechter M. A molecular approach. Sinauer associates; Sunderland, MA: 1990. Physiology of the bacterial cell. [Google Scholar]
- 54.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 55.Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. J Comput Chem. 2011;32(10):2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14(1):33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 57.Risselada HJ, Mark AE, Marrink SJ. J Phys Chem B. 2008;112(25):7438–7447. doi: 10.1021/jp0758519. [DOI] [PubMed] [Google Scholar]
- 58.Buchoux S. Bioinformatics. 2017;33(1):133–134. doi: 10.1093/bioinformatics/btw563. [DOI] [PubMed] [Google Scholar]
- 59.Holdbrook DA, Huber RG, Piggot TJ, Bond PJ, Khalid S. PLoS One. 2016;11(6):e0156963. doi: 10.1371/journal.pone.0156963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yesylevskyy SO, Schafer LV, Sengupta D, Marrink SJ. PLoS Comput Biol. 2010;6(6):e1000810. doi: 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bibow S, Polyhach Y, Eichmann C, Chi CN, Kowal J, Albiez S, McLeod RA, Stahlberg H, Jeschke G, Guntert P, Riek R. Nat Struct Mol Biol. 2017;24(2):187–193. doi: 10.1038/nsmb.3345. [DOI] [PubMed] [Google Scholar]
- 62.Denisov IG, Sligar SG. Chem Rev. 2017;117(6):4669–4713. doi: 10.1021/acs.chemrev.6b00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denisov IG, Sligar SG. Nat Struct Mol Biol. 2016;23(6):481–486. doi: 10.1038/nsmb.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mueller M, Lindner B, Kusumoto S, Fukase K, Schromm AB, Seydel U. J Biol Chem. 2004;279(25):26307–26313. doi: 10.1074/jbc.M401231200. [DOI] [PubMed] [Google Scholar]
- 65.Sasaki H, White SH. Biophys J. 2008;95(2):986–993. doi: 10.1529/biophysj.108.129197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brandenburg K, Seydel U. Eur J Biochem. 1990;191(1):229–236. doi: 10.1111/j.1432-1033.1990.tb19114.x. [DOI] [PubMed] [Google Scholar]
- 67.Santos NC, Silva AC, Castanho MA, Martins-Silva J, Saldanha C. ChemBioChem. 2003;4(1):96–100. doi: 10.1002/cbic.200390020. [DOI] [PubMed] [Google Scholar]
- 68.Wilkinson SG. Prog Lipid Res. 1996;35(3):283–343. doi: 10.1016/s0163-7827(96)00004-5. [DOI] [PubMed] [Google Scholar]
- 69.Gu RX, Ingólfsson HI, de Vries AH, Marrink SJ, Tieleman DP. J Phys Chem B. 2017;121:3262–3275. doi: 10.1021/acs.jpcb.6b07142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo S, Cheng X, Lee J, Kim S, Park SJ, Patel DS, Beaven AH, Lee KI, Rui H, Park S, Lee HS, Roux B, MacKerell AD, Jr, Klauda JB, Qi Y, Im W. J Comput Chem. 2017;38(15):1114–1124. doi: 10.1002/jcc.24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.