

Abstract
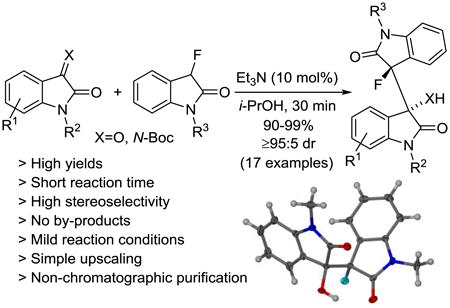
A highly diastereoselective organocatalytic method that produces 3-fluoro-3′-hydroxy-3,3′-bisoxindoles and the corresponding 3-fluoro-3′-amino derivatives having two adjacent chirality centers from fluorooxindoles and isatins in high yields is described. The reaction occurs in protic solvents at room temperature, it can be upscaled without compromising yield and stereoselectivity, and chromatographic product purification is not required.
Graphical abstract
The unique physicochemical properties and widespread use of fluorinated organic compounds in the health sciences continues to attract considerable attention. Numerous studies have shown that incorporation of fluorine can improve the therapeutic index of biologically active compounds.1 The introduction of synthetic methods that produce fluorinated derivatives of natural compounds and future drug candidates therefore remains of considerable interest. The construction of carbon-carbon bonds with reactive organofluorine intermediates, however, is often limited by undesirable side reactions and decomposition pathways. Various synthetic strategies that address these issues, for example by mild in situ production of fluoroenolates, have emerged in recent years.2 The 3,3-disubstituted oxindole scaffold is a privileged structural motif and a challenging synthetic target,3 especially if multiple stereocenters are present.4 The medicinal utility and potential of 3-fluorooxindoles, including the potassium ion channel modulator Maxipost (Figure 1),5 has inspired the development of several methods that accomplish direct fluorination of 3-alkyl and 3-aryloxindoles.6 More recently, synthetic alternatives that accomplish C-C bond formation with 3-fluorooxindoles have emerged.7
Figure 1.

Structures of Maxipost and biologically active 3,3′-bisoxindoles.
A remaining drawback of fluorooxindole transformations is that the use of inert reaction conditions and elaborate work-up procedures generating substantial amounts of chemical waste are required in most cases. Because environmental and sustainability aspects together with operational safety, time efficiency and overall cost considerations play an increasingly important role in industrial and academic laboratories we decided to develop a practical method that addresses these issues using 3-fluorooxindoles as starting material. The reaction with isatins was of particular interest to us as it produces a challenging dimeric oxindole scaffold exhibiting a 3,3′-linkage with two adjacent chirality centers.
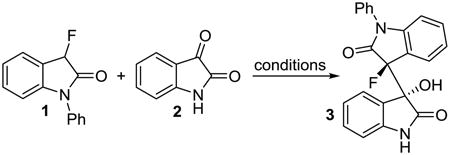
We began our search for an environmentally benign, economically attractive method by screening the reaction of N-phenyl-3-fluorooxindole, 1, and isatin, 2, in water and alcoholic solvents in the presence of catalytic amounts of inexpensive triethylamine at room temperature (Table 1). We found that the reaction proceeds smoothly in the presence of 20 mol% of base in water and is almost complete after stirring for 2 hours at room temperature (entry 1). The formation of the bisoxindole 3 was almost quantitative and occurred with high stereoselectivity. We did not detect formation of by-products and determined the diastereomeric ratio, dr, of 3 as 24:1. As expected, catalytic amounts of the base are required for this reaction and 3 was not formed in the absence of Et3N (entry 2). Screening of other protic solvents revealed that the conversion, reaction time and diastereoselectivity can be further improved (entries 3-6). In addition to optimization of the reaction conditions, we examined the possibility of non-chromatographic product isolation to minimize the overall solvent consumption and labor. Using 10 mol% of triethylamine in isopropyl alcohol we observed that 3 is produced quantitatively from 1 and 2 with 49:1 dr in just 30 minutes (entry 6). Under these conditions, the bisoxindole precipitated quantitatively which greatly facilitates product isolation and renders chromatographic work-up unnecessary.
Table 1.
Optimization of the organocatalytic C-C bond formation with N-phenyl-3-fluorooxindole, 1, and isatin, 2.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Solvent | Base (mol%) | Time (min) | Conversiona (%) | Diastereomeric ratioa |
| 1 | H2O | Et3N (20) | 120 | 93 | 24:1 |
| 2 | H2O | none | 120 | 0 | n/a |
| 3 | MeOH | Et3N (20) | 60 | 99 | 49:1 |
| 4 | MeOH/H2O (1:1) | Et3N (20) | 60 | 95 | 24:1 |
| 5 | EtOH | Et3N (20) | 60 | 96 | 24:1 |
| 6 | i-PrOH | Et3N (20) | 30 | 99 | 49:1 |
| 7 | i-PrOH | Et3N (10) | 30 | 99 | 49:1 |
General reaction conditions: Et3N (10-20 mol%) was added to a mixture of oxindole 1 (45.4 mg, 0.2 mmol) and isatin 2 (30.0 mg, 0.2 mmol) in 1.0 mL of the solvent at room temperature.
Based on 19F and 1H NMR analysis.
Having optimized reaction conditions and a work-up procedure that are both practical and environmentally benign, we continued to determine the reaction scope of the organocatalytic bisoxindole formation. 3-Fluoro-3′-hydroxy-1-phenyl-3,3′-bisoxindole, 3, was isolated in 96% yield and 99:1 dr (Scheme 1). The reaction between fluorooxindole 1 and isatins carrying a halide or a methyl group at position 5 in the fused benzene ring gave the corresponding bisoxindoles 4-7 in 90-96% yield and with at least 95:5 dr. The reaction tolerates substituents at all positions in the isatin electrophile. The chlorinated bisoxindoles 8-10 were produced with very similar results compared to 4. When we employed other brominated and fluorinated isatins in this reaction we obtained 11-13 in 91-92% yields and very high dr's. The isatin compound can also be substituted at the nitrogen. 3-Fluoro-3′-hydroxy-1,1′-diphenyl-3,3′-bisoxindole, 14, and the N-methyl analogue 15 were isolated in almost quantitative amounts and in excellent diastereomeric ratio. The reaction with N-methylisatin and N-methyl-3-fluorooxindole was also conducted in the presence of 20 mol% of triethylamine using either THF or dichloromethane as solvent. In both cases, the reaction occurs under homogeneous conditions and without precipitation of the product 15 which was obtained in quantitative yield and with >99:1 dr. This suggests that the high diastereoselectivity is achieved in solution and not a result of preferential crystallization of one diastereomer from a mixture of rapidly interconverting isomers of 15 (asymmetric transformation of the second kind). Finally the introduction of a strong electron-withdrawing nitro group and an electron-donating methoxy into the isatin ring showed little effects on the chemical and stereochemical outcome. We obtained 16 and 17 in 92-94% yield and very high dr. The triethylamine catalyzed reaction thus affords a variety of 3,3′-bridged bisoxindoles exhibiting two adjacent quaternary chiral centers in almost quantitative yields and with remarkable diastereoselectivity. Our organocatalytic method is operationally simple and leads to multifunctional bisoxindole alkaloid scaffolds. The protocol has several attractive features in addition to the high yields and dr values that are noteworthy. The C-C bond formation is accomplished within 30 minutes using mild reaction conditions, i.e. at room temperature and under air, and we did not observe by-product formation. All products 3-17 were isolated by precipitation and purified by careful washing with isopropyl alcohol-petroleum ether mixtures. The work-up does not require any chromatography which typically is time-consuming and increases both cost and waste production.8
Scheme 1.

Synthesis of 3,3′-bridged bisoxindoles 3-17 (only one enantiomer is shown).
To reveal the stereochemical outcome of this reaction we resorted to X-ray crystallography. We were able to grow a single crystal of racemic 3-fluoro-3′-hydroxy-1,1′-dimethyl-3,3′-bisoxindole, 15, by slow evaporation of a solution containing small amounts of ethyl acetate in hexanes.9 Crystallographic analysis confirmed that the reaction favors formation of the homochiral diastereomer (Figure 2).
Figure 2.
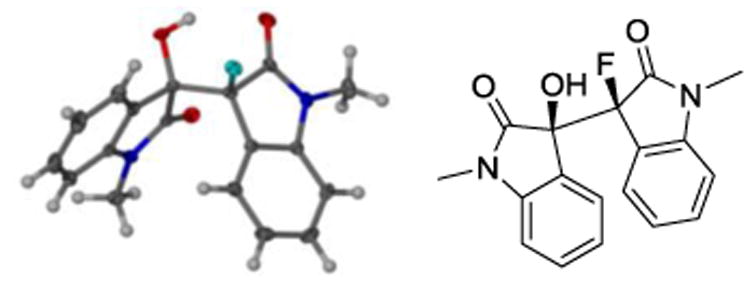
X-ray structure of 3,3′-bisoxindole 15 (only one enantiomer is shown).
To the best of our knowledge, the synthesis of 3-fluoro-3′-hydroxy-3,3′-bisoxindoles has not been reported to date and 3-17 are new compounds. Few examples of palladium catalyzed carbon-carbon bond formation with fluorooxindole 1 in dichloromethane and toluene, respectively, are known.7d,10 These methods accomplish asymmetric allylic alkylations and arylations with high yields and stereoselectivities but long reaction times and chromatographic work-up are required. Han and Soloshonok introduced a noncatalytic diastereoselective Mannich reaction via detrifluoroacetylative generation of intermediate 3-fluorooxindole enolates which achieves carbon-carbon bond formation with yields and dr's very similar to our method.7c,11 This reaction is fast and proceeds in etheral solvents or acetonitrile but large excess of base and LiBr additives are required in addition to chromatographic product purification. A very similar copper catalyzed asymmetric aldol-type reaction that utilizes the same detrifluoroacetylation concept was recently reported.12 An inherent drawback of the detrifluoroacetylative enolate generation, however, is the production of stoichiometric amounts of trifluoroacetic acid waste. In comparison to these methods our protocol establishes a significant green chemistry advance as waste resulting from by-products or additives, chromatographic work-up and the use of transition metals are avoided. Thakur and Meshram reported an interesting diastereoselective formation of 3-hydroxy-3,3′-bisoxindoles through catalyst-free on-water synthesis.13 We found, however, that this protocol cannot be generally used for the synthesis of the fluorinated bisoxindoles 3-17. While we successfully reproduced their results with oxindole and isatin, the reaction between N-phenyl-3-fluorooxindole and either isatin or N-phenylisatin using the on-water protocol gave 3 and 14, respectively, in only 3-5% yield after 24 hours.
We decided to run the reaction between 1 and 2 at the gram scale to determine if the overall efficiency and the environmentally attractive features of our method can be maintained without compromising yield and diastereoselectivity (see Experimental Section). We found that even at the increased reaction scale the product formation is complete within 30 minutes and 3 was isolated in 95% yield and with 99:1 dr. More than one gram of the bisoxindole 3 was thus obtained with an E-factor of 22 and without the use of expensive catalysts and additives or hazardous solvents.14
Our method is not limited to isatin electrophiles. When we applied the fluorooxindoles 1 and 20 in the reaction with N-Boc imine 18 we were pleased to find that the correspnding amines 19 and 21 were produced in 90-92% yield and with very high dr using the same method (Scheme 2). The reactivity of 3-fluorooxindoles in protic solvents and the utility of our environmentally benign C-C bond formation procedure also extends to Michael additions.15 Employing 1 and 1,1-bis(phenylsulfonyl)ethane, 22, in essentially the same protocol used above we were able to prepare 23 in 99% yield.7a,16 This reaction occurs in isopropyl alcohol in the presence of 10 mol% of triethylamine and is complete within 30 minutes. Again, chromatographic product purification was not necessary.
Scheme 2.

Additions to N-Boc imine 18 and the Michael acceptor 22.
In summary, we have introduced an organocatalytic method that produces 3-fluoro-3′-hydroxy-3,3′-bisoxindoles or the corresponding 3-fluoro-3′-amines carrying two vicinal chirality centers in high yields and stereoselectivities. The reaction occurs in non-hazardous isopropyl alcohol or other protic solvents at room temperature within 30 minutes in the presence 10 mol% of triethylamine as catalyst and the bisoxindole formation can be upscaled without compromising yields and diastereoselectivity. Furthermore, the formation of by-products was not observed and chromatographic product purification is not necessary.
Experimental Section
Commercially available isatins, reagents and solvents were used as purchased without further purification. 3-Fluorooxindole was synthesized by following literature procedures.7d NMR spectra were obtained at 400 MHz (1H NMR), 376 MHz (19F NMR) and 100 MHz (13C NMR) in deuterated dimethylsulfoxide, acetone or deuterated chloroform. Proton chemical shifts are reported in ppm relative to the solvent peak or TMS.
General Procedure
A mixture of 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and an isatin (0.20 mmol) were added to 1.0 mL of isopropyl or methyl alcohol. Triethylamine (2.8 μL, 0.020 mmol) was added and the solution was stirred for 30 minutes. The resulting solid was isolated after addition of 1.0 mL of petroleum ether and decanting off the liquid. The crude product was purified by washing the solid three times with 1.0 mL of petroleum ether-isopropyl alcohol (1:1).
3-Fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 3 was obtained as a white crystalline solid in 96% yield (72 mg, 0.19 mmol) and >99:1 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and isatin (30 mg, 0.20 mmol) after sonication in isopropyl alcohol for 30 minutes as described above. Decomp. 204 °C. 1H NMR (399 MHz, DMSO-d6): δ = 10.44 (s, 1H), 7.51 – 7.33 (m, 4H), 7.28 (dd, J = 7.6, 7.6 Hz, 1H), 7.15 (dd, J = 7.5, 7.5 Hz, 1H), 6.98 (s, 1H), 6.95 – 6.90 (m, 2H), 6.84 – 6.77 (m, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.37 (d, J = 6.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 173.8 (d, JC-F = 2.4 Hz), 168.5 (d, JC-F = 21.6 Hz), 144.4 (d, JC-F = 5.1 Hz), 142.9, 132.9, 132.0 (d, JC-F = 2.6 Hz), 130.7, 129.7, 128.6, 126.9, 126.4, 125.7 (d, JC-F = 4.1 Hz), 124.8, 123.2 (d, JC-F = 2.5 Hz), 121.6 (d, JC-F = 18.8 Hz), 121.3, 109.8, 109.2, 94.0 (d, JC-F = 205.3 Hz), 77.0 (d, JC-F = 23.8 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.2. Anal. Calcd. for C22H15FN2O3: C, 70.58; H, 4.04; N, 7.48. Found: C, 70.39; H, 4.20; N, 7.40.
Large Scale Synthesis of 3
A mixture of 3-fluoro-1-phenylindolin-2-one (911 mg, 4.0 mmol) and isatin (602 mg, 4.0 mmol) were added to 4.0 mL of isopropyl alcohol. Triethylamine (56.5 μL, 0.40 mmol) was added and the solution was stirred for 30 minutes. The resulting product 3 was obtained as a white solid in 95% yield (1.43 g, 3.8 mmol) and >99:1 dr after adding 6.0 mL of isopropyl alcohol, filtration and washing the solid with a total of 20 mL of petroleum ether-isopropyl alcohol (1:1).
5′-Chloro-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 4 was obtained as a white solid in 94% yield (77 mg, 0.19 mmol) and 96:4 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5-chloroisatin (37 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 10.62 (s, 1H), 7.54 - 7.38 (m, 5H), 7.35 (dd, J = 8.3, 2.1 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.01 – 6.93 (m, 2H), 6.82 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 8.0 Hz, 1H), 6.26 (bs, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 173.4 (d, JC-F = 2.2 Hz), 168.2 (d, JC-F = 21.6 Hz), 144.4 (d, JC-F = 5.1 Hz), 141.8, 132.9, 132.3 (d, JC-F = 2.6 Hz), 130.5, 129.8, 128.7, 127.6 (d, JC-F = 4.1 Hz), 127.0, 126.3, 125.3, 124.8, 123.4 (d, JC-F = 2.4 Hz), 121.2 (d, JC-F = 19.0 Hz), 111.4, 109.2, 93.9 (d, JC-F = 206.4 Hz), 77.1 (d, JC-F = 24.2 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.9. Anal. Calcd. for C22H14ClFN2O3: C, 64.64; H, 3.45; N, 6.85. Found: C, 64.46; H, 3.50; N, 6.71.
5′-Bromo-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 5 was obtained as a white solid in 91% yield (83 mg, 0.18 mmol) and 95:5 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5-bromoisatin (48 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, acetone-d6): δ = 9.40 (s, 1H), 7.48 – 7.41 (m, 3H), 7.40 – 7.26 (m, 2H), 7.24 – 7.16 (m, 2H), 7.10 (s, 1H), 6.95 (dd, J = 7.3, 7.3 Hz, 1H), 6.82 – 6.73 (m, 2H), 6.63 (d, J = 7.7 Hz, 1H), 5.85 (s, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 173.3 (d, JC-F = 2.0 Hz), 168.2 (d, JC-F = 21.6 Hz), 144.4 (d, JC-F = 5.0 Hz), 142.1, 133.3, 132.8, 132.3, 129.8, 128.7, 128.0 (d, JC-F = 4.4 Hz), 127.6, 127.0, 126.3, 123.4, 121.2 (d, JC-F = 19.1 Hz), 112.8, 111.9, 109.2, 94.0 (d, JC-F = 206.9 Hz), 77.0 (d, JC-F = 24.1 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.4. Anal. Calcd. for C22H14BrFN2O3: C, 58.30; H, 3.11; N, 6.18. Found: C, 58.30; H, 3.19; N, 6.08.
3,5′-Difluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 6 was obtained as a white solid in 96% yield (75 mg, 0.19 mmol) and 96:4 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5-fluoroisatin (34 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 10.50 (s, 1H), 7.54 – 7.47 (m, 2H), 7.46 – 7.40 (m, 2H), 7.34 (m, 1H), 7.21 – 7.12 (m, 3H), 7.04 – 6.96 (m, 2H), 6.81 (dd, J = 8.6, 4.2 Hz, 1H), 6.66 (d, J = 8.0 Hz, 1H), 6.14 (m, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 173.7 (d, JC-F = 2.4 Hz), 168.2 (d, JC-F = 21.6 Hz), 157.4 (d, JC-F = 237.8 Hz), 144.4 (d, JC-F = 5.2 Hz), 139.1 (d, JC-F = 1.9 Hz), 132.9, 132.3 (d, JC-F = 2.5 Hz), 129.8, 128.7, 127.2 (dd, JC-F = 7.8, 4.0 Hz), 126.9, 126.3, 123.3 (d, JC-F = 2.4 Hz), 121.2 (d, JC-F = 19.0 Hz), 117.1 (d, JC-F = 23.1 Hz), 112.4 (d, JC-F = 25.0 Hz), 110.8 (d, JC-F = 7.8 Hz), 109.3, 93.8 (d, JC-F = 205.7 Hz), 77.4 (d, JC-F = 24.1 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -122.5 (m), -177.51. Anal. Calcd. for C22H14F2N2O3: C, 67.35; H, 3.60; N, 7.14. Found: C, 67.05; H, 3.56; N, 7.03.
3-Fluoro-3′-hydroxy-5′-methyl-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 7 was obtained as a white solid in 90% yield (70 mg, 0.18 mmol) and 97:3 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5-methylisatin (33 mg, 0.20 mmol) after sonication in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 10.31 (s, 1H), 7.54 – 7.37 (m, 4H), 7.27 (m, 1H), 7.17 – 7.07 (m, 2H), 6.99 – 6.93 (m, 2H), 6.87 (s, 1H), 6.67 (d, J = 8.0 Hz, 1H), 6.61 (d, J = 8.0 Hz, 1H), 6.23 (bs, 1H), 2.06 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ = 174.2 (d, JC-F = 2.8 Hz), 168.9 (d, JC-F = 21.7 Hz), 144.9 (d, JC-F = 5.1 Hz), 140.8, 133.5, 132.4 (d, JC-F = 2.7 Hz), 131.1, 130.6, 130.2, 129.0, 127.3, 126.9, 126.2 (d, JC-F = 3.8 Hz), 126.1, 123.5 (d, JC-F = 2.4 Hz), 122.1, 121.9, 109.7 (d, JC-F = 48.5 Hz), 94.4 (d, JC-F = 205.1 Hz), 77.8 (d, JC-F = 23.8 Hz), 21.0. 19F NMR (376 MHz, DMSO-d6): δ = -177.6. Anal. Calcd. for C23H17FN2O3: C, 71.13; H, 4.41; N, 7.21. Found: C, 70.76; H, 4.51; N, 7.13.
7′-Chloro-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 8 was obtained as a white solid in 91% yield (74 mg, 0.18 mmol) and >99.1 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 7-chloroisatin (37 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 10.94 (s, 1H), 7.54 – 7.40 (m, 4H), 7.40 – 7.32 (m, 2H), 7.23 – 7.14 (m, 2H), 7.01 – 6.95 (m, 2H), 6.87 (dd, J = 7.9, 7.9 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.36 (m, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 173.7 (d, JC-F = 2.3 Hz), 168.2 (d, JC-F = 21.6 Hz), 144.4 (d, JC-F = 5.1 Hz), 140.6, 132.9, 132.3 (d, JC-F = 2.7 Hz), 130.6, 129.8, 128.7, 127.6 (d, JC-F = 4.1 Hz), 126.9, 126.4, 123.4, 123.3 (d, JC-F = 2.5 Hz), 122.7, 121.3 (d, JC-F = 18.8 Hz), 114.0, 109.3, 93.8 (d, JC-F = 205.7 Hz), 77.5 (d, JC-F = 24.1 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.0. Anal. Calcd. for C22H14ClFN2O3: C, 64.64; H, 3.45; N, 6.85. Found: C, 64.40; H, 3.47; N, 6.78.
4′-Chloro-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 9 was obtained as a white solid in 94% yield (77 mg, 0.19 mmol) and 99:1 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 4-chloroisatin (37 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 10.64 (s, 1H), 7.58 – 7.52 (m, 2H), 7.47 (m, 1H), 7.38 – 7.30 (m, 2H), 7.25 – 7.20 (m, 2H), 7.02 (d, J = 8.1 Hz, 1H), 6.97 (dd, 7.6, 7.6 Hz, 1H), 6.83 (m, 1H), 6.79 (s, 1H), 6.72 (d, J = 7.7 Hz, 1H), 6.66 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 173.6 (d, JC-F = 6.0 Hz), 168.6 (d, JC-F = 22.1 Hz), 144.8, 144.4 (d, JC-F = 5.2 Hz), 138.9, 133.2, 132.2 (d, JC-F = 2.8 Hz), 132.1, 131.6, 129.7, 128.5, 126.5, 126.3, 123.6, 122.9 (dd, JC-F = 6.3, 2.1 Hz), 121.9 (d, JC-F = 18.7 Hz), 109.4, 108.7, 93.5 (d, JC-F = 206.1 Hz), 80.0 (d, JC-F = 24.9 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -169.5. Anal. Calcd. for C22H14ClFN2O3: C, 64.64; H, 3.45; N, 6.85. Found: C, 64.38; H, 3.48; N, 6.75.
6′-Chloro-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 10 was obtained as a white solid in 96% yield (79 mg, 0.19 mmol) and 98:2 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 6-chloroisatin (37 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 10.64 (s, 1H), 7.53 – 7.38 (m, 5H), 7.21 – 7.14 (m, 2H), 7.02 – 6.95 (m, 2H), 6.89 (d, J = 8.0, 1H), 6.83 (s, 1H), 6.66 (d, J = 7.9 Hz, 1H), 6.35 (m, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 173.7 (d, JC-F = 2.2 Hz), 168.3 (d, JC-F = 21.6 Hz), 144.4, 144.3 (d, JC-F = 5.1 Hz), 135.1, 132.9, 132.3 (d, JC-F = 2.6 Hz), 129.8, 128.7, 127.0, 126.3, 124.6 (d, JC-F = 4.3 Hz), 123.4 (d, JC-F = 2.5 Hz), 121.4, 121.2, 121.2, 109.9, 109.3, 93.9 (d, JC-F = 205.8 Hz), 76.7 (d, JC-F = 24.1 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.1. Anal. Calcd. for C22H14ClFN2O3: C, 64.64; H, 3.45; N, 6.85. Found: C, 64.49; H, 3.63; N, 6.69.
3,6′-Difluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 11 was obtained as a white solid in 92% yield (72 mg, 0.18 mmol) and 98:2 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 6-fluoroisatin (34 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 10.63 (bs, 1H), 7.54 – 7.37 (m, 5H), 7.18 (dd, J = 7.5, 7.5 Hz, 1H), 7.08 (s, 1H), 7.03 – 6.96 (m, 2H), 6.69 – 6.59 (m, 3H), 6.36 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 174.0 (d, JC-F = 2.2 Hz), 168.4 (d, JC-F= 21.7 Hz), 163.6 (d, JC-F = 245.3 Hz), 144.8 (d, JC-F = 12.7 Hz), 144.3 (d, JC-F = 5.1 Hz), 132.9, 132.2 (d, JC-F = 2.7 Hz), 129.8, 128.6, 127.0, 126.5 (m), 126.3, 123.3 (d, JC-F = 2.4 Hz), 121.7 (dd, JC-F = 4.4, 2.8 Hz), 121.4 (d, JC-F = 18.9 Hz), 109.3, 107.6 (d, JC-F = 22.5 Hz), 98.0 (d, JC-F = 27.0 Hz), 93.9 (d, JC-F = 205.5 Hz), 76.5 (d, JC-F = 24.1 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -109.8 (m), -176.7. Anal. Calcd. for C22H14F2N2O3: C, 67.35; H, 3.60; N, 7.14. Found: C, 66.96; H, 3.76; N, 7.08.
6′-Bromo-3-fluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 12 was obtained as a white solid in 91% yield (83 mg, 0.18 mmol) and 97:3 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 6-bromoisatin (46 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 10.62 (s, 1H), 7.54 – 7.36 (m, 5H), 7.22 – 7.12 (m, 2H), 7.06 – 6.93 (m, 4H), 6.65 (d, J = 7.9 Hz, 1H), 6.29 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 173.5 (d, JC-F = 2.3 Hz), 168.3 (d, JC-F = 21.4 Hz), 144.5, 144.3 (d, JC-F = 5.2 Hz), 132.9, 132.2 (d, JC-F = 2.7 Hz), 129.7, 128.7, 126.9, 126.5, 126.3, 125.0 (d, JC-F = 4.2 Hz), 124.1, 123.5, 123.3 (d, JC-F = 2.5 Hz), 121.3 (d, JC-F = 18.9 Hz), 112.6, 109.3, 93.8 (d, JC-F = 206.0 Hz), 76.8 (d, JC-F = 24.2 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.2. Anal. Calcd. for C22H14BrFN2O3: C, 58.30; H, 3.11; N, 6.18. Found: C, 58.04; H, 3.29; N, 6.07.
3,5′,6′-Trifluoro-3′-hydroxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 13 was obtained as a white solid in 91% yield (75 mg, 0.18 mmol) and 98:2 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5,6-difluoroisatin (38 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 10.66 (bs, 1H), 7.55 – 7.51 (m, 2H), 7.47 – 7.43 (m, 2H), 7.33 (m, 1H), 7.28 – 7.14 (m, 3H), 7.09 – 7.04 (m, 2H), 6.89 (dd, J = 10.3, 6.8 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.36 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 173.7 (d, JC-F = 2.6 Hz), 168.1 (d, JC-F = 21.8 Hz), 150.9 (dd, JC-F = 247.8, 13.9 Hz), 144.9 (dd, JC-F = 240.0, 13.5 Hz), 144.3 (d, JC-F = 5.3 Hz), 139.8 (d, JC-F = 10.2 Hz), 132.9, 132.3, 129.8, 128.7, 126.8, 126.2, 123.4, 121.7, 121.0 (d, JC-F = 18.9 Hz), 114.3 (d, JC-F = 20.2 Hz), 109.3, 99.9 (d, JC-F = 22.4 Hz), 93.6 (d, JC-F = 205.6 Hz), 77.0 (d, JC-F = 24.5 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -135.0 (m), -148.5 (m), -177.0. Anal. Calcd. for C22H13F3N2O3: C, 64.39; H, 3.19; N, 6.83. Found: C, 64.30; H, 3.24; N, 6.81.
3-Fluoro-3′-hydroxy-1,1′-diphenyl-[3,3′-biindoline]-2,2′-dione
Compound 14 was obtained as a white solid in 97% yield (87 mg, 0.19 mmol) and 98:2 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 1-phenylisatin (46 mg, 0.20 mmol) after sonication in methyl alcohol for 30 minutes as described above. 1H NMR (400 MHz, DMSO-d6): δ = 7.58 – 7.37 (m, 9H), 7.36 – 7.19 (m, 4H), 7.01 – 6.83 (m, 3H), 6.66 (dd, J = 12.6, 8.0 Hz, 2H), 6.42 (bs, 1H). 13C NMR (101 MHz, DMSO-d6): δ = 171.7 (d, JC-F = 1.4 Hz), 168.5 (d, JC-F = 21.6 Hz), 144.4 (d, JC-F = 5.2 Hz), 144.0, 133.7, 132.8, 132.3 (d, JC-F = 2.5 Hz), 130.9, 129.8, 129.7, 128.7, 128.3, 127.2, 126.6, 126.4, 124.9, 124.7 (d, JC-F = 4.4 Hz), 123.4 (d, JC-F = 2.4 Hz), 122.6, 121.3 (d, JC-F = 18.9 Hz), 109.3, 109.0, 94.5 (d, JC-F = 207.4 Hz), 76.7 (d, JC-F = 23.7 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -176.8. Anal. Calcd. for C28H19FN2O3: C, 74.66; H, 4.25; N, 6.22. Found: C, 74.55; H, 4.27; N, 6.21.
3-Fluoro-3′-hydroxy-1,1′-dimethyl-[3,3′-biindoline]-2,2′-dione
Compound 15 was obtained as a white crystalline solid in 99% yield (65 mg, 0.20 mmol) and >99:1 dr from 3-fluoro-1-methylindolin-2-one (33 mg, 0.20 mmol) and 1-methylisatin (33 mg, 0.20 mmol) after sonication in methanol for 30 minutes by as described above. Decomp. 196 °C. 1H NMR (399 MHz, CDCl3): δ = 7.72 (d, J = 7.1 Hz, 1H), 7.46 (dd, J = 7.8, 7.8 Hz, 1H), 7.31 (dd, J = 7.8, 7.8 Hz, 1H), 7.22 (dd, J = 7.6, 7.6 Hz, 1H), 6.81 (d, J = 7.8 Hz, 1H), 6.79 – 6.73 (m, 2H), 6.10 (d, J = 7.5 Hz, 1H), 5.68 (s, 1H), 3.26 (s, 3H), 2.85 (s, 3H). 13C NMR (100 MHz, CDCl3): δ = 173.4 (d, JC-F = 8.3 Hz), 171.4 (d, JC-F = 21.2 Hz), 144.9 (d, JC-F = 4.8 Hz), 144.6, 132.3 (d, JC-F = 3.0 Hz), 131.3, 127.0 (d, JC-F = 3.0 Hz), 125.2, 124.3, 123.7, 123.2 (d, JC-F = 2.6 Hz), 121.5 (d, JC-F = 18.2 Hz), 109.3, 108.4, 90.4 (d, JC-F = 200.8 Hz), 79.2 (d, JC-F = 24.6 Hz), 26.4, 26.1. 19F NMR (376 MHz, CDCl3): δ = -174.4. Anal. Calcd. for C18H15FN2O3: C, 66.25; H, 4.63; N, 8.58. Found: C, 66.39; H, 4.68; N, 8.71.
3-Fluoro-3′-hydroxy-5′-nitro-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 16 was obtained as a white solid in 92% yield (77 mg, 0.18 mmol) and 97:3 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 5-nitroisatin (36 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 11.22 (bs, 1H), 8.25 (dd, J = 8.7, 2.3 Hz, 1H), 7.56 – 7.41 (m, 6H), 7.25 (dd, J = 7.6, 7.6 Hz, 1H), 7.11 (s, 1H), 7.01 (d, J = 8.6 Hz, 1H), 6.97 – 6.87 (m, 2H), 6.67 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 174.0 (d, JC-F = 1.7 Hz), 168.0 (d, JC-F = 21.5 Hz), 149.2, 144.2 (d, JC-F = 5.1 Hz), 141.7, 132.7, 132.5 (d, JC-F = 2.7 Hz), 129.8, 128.7, 127.8, 127.2, 126.4, 126.1, 123.6 (d, JC-F = 2.2 Hz), 120.8 (d, JC-F = 19.0 Hz), 120.4, 110.2, 109.5, 93.8 (d, JC-F = 207.0 Hz), 76.5 (d, JC-F = 24.5 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -177.5. Anal. Calcd. for C22H14FN3O5: C, 63.01; H, 3.37; N, 10.02. Found: C, 63.10; H, 3.57; N, 9.90.
3-Fluoro-3′-hydroxy-6′-methoxy-1-phenyl-[3,3′-biindoline]-2,2′-dione
Compound 17 was obtained as a white solid in 94% yield (76 mg, 0.19 mmol) and 99:1 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 6-methoxyisatin (36 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. 1H NMR (399 MHz, DMSO-d6): δ = 10.35 (s, 1H), 7.49 – 7.30 (m, 5H), 7.11 (dd, J = 7.2, 6.7 Hz, 1H), 6.96 – 6.90 (m, 2H), 6.80 (s, 1H), 6.58 (d, J = 7.9 Hz, 1H), 6.33 – 6.27 (m, 2H), 6.25 – 6.16 (m, 1H), 3.67 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ = 174.2 (d, JC-F = 2.2 Hz), 168.6 (d, JC-F = 21.7 Hz), 161.4, 144.4 (d, JC-F = 6.9 Hz), 133.0, 132.0 (d, JC-F = 2.7 Hz), 129.7, 128.6, 126.9, 126.4, 125.8, 123.2 (d, JC-F = 2.4 Hz), 121.8, 121.7, 117.4 (d, JC-F = 4.4 Hz), 109.2, 106.3, 96.4, 94.0 (d, JC-F = 205.4 Hz), 76.7 (d, JC-F = 23.9 Hz), 55.4. 19F NMR (376 MHz, DMSO-d6): δ = -176.4. Anal. Calcd. for C23H17FN2O4: C, 68.31; H, 4.24; N, 6.93. Found: C, 68.26; H, 4.29; N, 6.91.
tert-Butyl (1′-benzyl-3-fluoro-2,2′-dioxo-1-phenyl-[3,3′-biindolin]-3′-yl)carbamate
Compound 19 was obtained as a white crystalline solid in 92% yield (104 mg, 0.18 mmol) and >99:1 dr from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and tert-butyl (1-benzyl-2-oxoindolin-3-ylidene)carbamate (67 mg, 0.20 mmol) after stirring in isopropyl alcohol for 24 hours as described above. Decomp. 178 °C. 1H NMR (399 MHz, CDCl3): δ = 7.70 (d, J = 7.5 Hz, 1H), 7.59 – 7.40 (m, 6H), 7.32 (dd, J = 7.8, 7.8 Hz, 1H), 7.24 – 7.02 (m, 5H), 6.74 – 6.56 (m, 5H), 5.83 (d, J = 7.5 Hz, 1H), 4.89 (d, J = 15.7 Hz, 1H), 4.45 – 4.28 (m, 1H), 1.28 (s, 9H). 13C NMR (100 MHz, CDCl3): δ = 172.4 (d, JC-F = 8.0 Hz), 170.0 (d, JC-F = 21.8 Hz), 154.09, 145.1 (d, JC-F = 5.1 Hz), 144.0, 135.2, 133.1, 132.1 (d, JC-F = 2.4 Hz), 130.1, 129.8, 128.8, 128.6, 127.2, 126.9, 126.8, 126.0, 125.4 (d, JC-F = 2.8 Hz), 123.2 (d, JC-F = 2.4 Hz), 123.1, 120.5 (d, JC-F = 18.0 Hz), 110.0, 109.1, 90.5 (d, JC-F = 204.3 Hz), 80.3, 66.6 (d, JC-F = 22.9 Hz), 44.3, 28.1. 19F NMR (376 MHz, CDCl3): δ = -170.5. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C34H30FN3O4Na 586.2118; Found 586.2111.
tert-Butyl (1,1′-dibenzyl-3-fluoro-2,2′-dioxo-[3,3′-biindolin]-3′-yl)carbamate
Compound 21 was obtained as a white solid in 91% yield (105 mg, 0.18 mmol) and 96:4 dr from 1-benzyl-3-fluoroindolin-2-one (48 mg, 0.20 mmol) and tert-butyl (1-benzyl-2-oxoindolin-3-ylidene)carbamate (67 mg, 0.20 mmol) after stirring in isopropyl alcohol for 24 hours as described above. 1H NMR (399 MHz, CDCl3): δ = 7.67 (d, J = 7.4 Hz, 1H), 7.54 (s, 1H), 7.44 – 7.37 (m, 2H), 7.33 – 7.24 (m, 4H), 7.18 (dd, J = 7.5, 7.5 Hz, 1H), 7.15 – 7.06 (m, 2H), 6.99 (dd, J = 7.5, 7.5 Hz, 2H), 6.68 – 6.59 (m, 3H), 6.57 – 6.45 (m, 2H), 5.69 (d, J = 7.5 Hz, 1H), 5.13 (d, J = 15.9 Hz, 1H), 4.93 (d, J = 15.7 Hz, 1H), 4.79 (d, J = 15.8 Hz, 1H), 4.47 – 4.25 (m, 1H), 1.29 (s, 9H). 13C NMR (100 MHz, CDCl3): δ = 172.2 (d, JC-F = 7.5 Hz), 170.7 (d, JC-F = 23.1 Hz), 154.1, 144.1 (d, JC-F = 5.0 Hz), 143.9, 135.2, 134.5, 132.1 (d, JC-F = 3.1 Hz), 130.0, 128.9, 128.6, 127.8, 127.4, 127.1, 126.9, 125.9, 125.5 (d, JC-F = 3.0 Hz), 123.1, 122.9 (d, JC-F = 2.7 Hz), 120.9 (d, JC-F = 17.8 Hz), 110.2, 109.1, 91.4 (d, JC-F = 201.9 Hz), 80.3, 66.2 (d, JC-F = 23.1 Hz), 44.5, 44.3, 28.2. 19F NMR (376 MHz, CDCl3): δ = -167.1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C35H32FN3O4Na 600.2275; Found 600.2267.
3-(2,2-Bis(phenylsulfonyl)ethyl)-3-fluoro-1-phenylindolin-2-one
Compound 23 was obtained as a white crystalline solid in 99% yield (106 mg, 0.20 mmol) from 3-fluoro-1-phenylindolin-2-one (45 mg, 0.20 mmol) and 1,1-bis(phenylsulfonyl)ethene (65 mg, 0.20 mmol) after stirring in isopropyl alcohol for 30 minutes as described above. Mp. 127-128 °C. 1H NMR (399 MHz, DMSO-d6): δ = 7.99 – 7.87 (m, 4H), 7.84 – 7.76 (m, 2H), 7.72 -7.56 (m, 7H), 7.54 – 7.41 (m, 4H), 7.23 (dd, J = 7.5, 7.5 Hz, 1H), 6.82 (d, J = 7.9 Hz, 1H), 5.69 (dd, J = 4.4, 4.4 Hz, 1H), 3.42 (ddd, J = 17.9, 14.1, 4.3 Hz, 1H), 3.02 (ddd, J = 32.4, 17.3, 4.5 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 170.4 (d, JC-F = 23.0 Hz), 143.1 (d, JC-F = 5.2 Hz), 137.8, 136.9, 135.1, 134.9, 132.9, 132.2 (d, JC-F = 2.9 Hz), 129.8, 129.5, 129.4, 129.4, 129.0, 128.7, 126.4, 125.3, 124.1, 123.9 (d, JC-F = 2.1 Hz), 110.1, 89.3 (d, JC-F = 188.9 Hz), 76.1, 30.3 (d, JC-F = 30.6 Hz). 19F NMR (376 MHz, DMSO-d6): δ = -155.2 (dd, J = 32.4, 13.9 Hz). HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C28H22FNO5S2Na 558.0821; Found 558.0816.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from NIH (GM106260) and the Georgetown Environment Initiative (GEI).
Footnotes
Supporting Information: Copies of NMR spectra and crystallographic details.
References
- 1.Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Acena JL, Soloshonok VA, Izawa K, Liu H. Chem Rev. 2016;116:422–518. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- 2.a) Han C, Kim EH, Colby DA. J Am Chem Soc. 2011;133:5802–5805. doi: 10.1021/ja202213f. [DOI] [PubMed] [Google Scholar]; b) Zhang P, Wolf C. J Org Chem. 2012;77:8840–8844. doi: 10.1021/jo3017583. [DOI] [PubMed] [Google Scholar]; c) Saidalimu I, Fang X, He XP, Liang J, Yang XY, Wu FH. Angew Chem Int Ed. 2013;52:5566–5570. doi: 10.1002/anie.201301443. [DOI] [PubMed] [Google Scholar]; d) Zhang P, Wolf C. Angew Chem Int Ed. 2013;52:7869–7873. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]; e) Wu C, Li G, Sun W, Zhang M, Hong L, Wang R. Org Lett. 2014;16:1960–1963. doi: 10.1021/ol500517d. [DOI] [PubMed] [Google Scholar]; f) Xie C, Wu L, Han J, Soloshonok VA, Pan Y. Angew Chem Int Ed. 2015;54:6019–6023. doi: 10.1002/anie.201500908. [DOI] [PubMed] [Google Scholar]; g) Xie C, Dai Y, Mei H, Han J, Soloshonok VA, Pan Y. Chem Commun. 2015;51:9149–9152. doi: 10.1039/c5cc02256h. [DOI] [PubMed] [Google Scholar]; h) Saadi J, Wennemers H. Nat Chem. 2016;8:276–280. doi: 10.1038/nchem.2437. [DOI] [PubMed] [Google Scholar]; i) Ding R, Wolf C. Chem Commun. 2016;52:3576–3579. doi: 10.1039/c5cc09753c. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Balaraman K, Moskowitz M, Liu Y, Wolf C. Synthesis. 2016;48:2376–2384. doi: 10.1055/s-0035-1561433. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Ding R, Bakhshi PR, Wolf C. J Org Chem. 2017;82:1273–1278. doi: 10.1021/acs.joc.6b02704. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a general review : Mei H, Acena JL, Soloshonok VA, Roeschenthaler GV, Han J. Eur J Org Chem. 2015:6401–6412.
- 3.a) Ma S, Han X, Krishnan S, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:8037–8041. doi: 10.1002/anie.200902943. [DOI] [PubMed] [Google Scholar]; b) Antonchick AP, Gerding-Reimers C, Catarinella M, Schürmann M, Preut H, Ziegler S, Rauh D, Waldmann H. Nat Chem. 2010;2:735–740. doi: 10.1038/nchem.730. [DOI] [PubMed] [Google Scholar]; c) Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S. Angew Chem Int Ed. 2012;51:5217–5221. doi: 10.1002/anie.201201132. [DOI] [PubMed] [Google Scholar]; d) Biswas P, Paul S, Guin J. Angew Chem Int Ed. 2016;55:7756–7760. doi: 10.1002/anie.201603809. [DOI] [PubMed] [Google Scholar]
- 4.a) Wu MY, He WW, Liu XY, Tan B. Angew Chem Int Ed. 2015;54:9409–9413. doi: 10.1002/anie.201504640. [DOI] [PubMed] [Google Scholar]; b) Ohmatsu K, Ando Y, Ooi T. J Am Chem Soc. 2013;135:18706–18709. doi: 10.1021/ja411647x. [DOI] [PubMed] [Google Scholar]; c) Tan B, Candeias NR, Barbas CF. Nat Chem. 2011;3:473–477. doi: 10.1038/nchem.1039. [DOI] [PubMed] [Google Scholar]; d) Zhao J, Fang B, Luo W, Hao X, Liu X, Lin L, Feng X. Angew Chem Int Ed. 2015;54:241–244. doi: 10.1002/anie.201408730. [DOI] [PubMed] [Google Scholar]; e) Engl OD, Fritz SP, Wennemers H. Angew Chem Int Ed. 2015;54:8193–8197. doi: 10.1002/anie.201502976. [DOI] [PubMed] [Google Scholar]
- 5.a) Gribkoff VK, Starrett JE, Jr, Dworetzky SL, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. Nat Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]; b) Hewawasam P, Gribkoff VK, Pendri Y, Dworetzky SI, Meanwell NA, Martinez E, Boissard CG, Post-Munson DJ, Trojnacki JT, Yeleswaram K, Pajor LM, Knipe J, Gao Q, Perrone R, Starrett JE., Jr Bioorg Med Chem Lett. 2002;12:1023–1026. doi: 10.1016/s0960-894x(02)00101-4. [DOI] [PubMed] [Google Scholar]
- 6.Selected examples: Shibata N, Suzuki E, Asahi T, Shiro M. J Am Chem Soc. 2001;123:7001–7009. doi: 10.1021/ja010789t.Hamashima Y, Suzuki T, Takano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164–10165. doi: 10.1021/ja0513077.Shibata N, Kohno J, Takai K, Ishimaru T, Nakamura S, Toru T, Kanemasa S. Angew Chem, Int Ed. 2005;44:4204–4207. doi: 10.1002/anie.200501041.Ishimaru T, Shibata N, Horikawa T, Yasuda N, Nakamura S, Toru T, Shiro M. Angew Chem, Int Ed. 2008;47:4157–4161. doi: 10.1002/anie.200800717.Deng QH, Wadepohl H, Gade LH. Chem Eur J. 2011;17:14922–14928. doi: 10.1002/chem.201102375.Shen K, Liu XH, Lin LL, Feng XM. Chem Sci. 2012;3:327–334.Li J, Cai Y, Chen W, Liu X, Lin L, Feng X. J Org Chem. 2012;77:9148–9155. doi: 10.1021/jo301705t.Gu X, Zhang Y, Xu ZJ, Che CM. Chem Commun. 2014;50:7870–7873. doi: 10.1039/c4cc01631a.; For an orthogonal approach based on asymmetric ring construction, see Wu L, Falivene L, Drinkel E, Grant S, Linden A, Cavallo L, Dorta R. Angew Chem Int Ed. 2012;51:2870–2873. doi: 10.1002/anie.201200206.
- 7.a) Dou X, Lu Y. Org Biomol Chem. 2013;11:5217–5221. doi: 10.1039/c3ob41267a. [DOI] [PubMed] [Google Scholar]; b) Wang T, Hoon DL, Lu Y. Chem Commun. 2015;51:10186–10189. doi: 10.1039/c5cc03289j. [DOI] [PubMed] [Google Scholar]; c) Xie C, Zhang L, Sha W, Soloshonok VA, Han J, Pan Y. Org Lett. 2016;18:3270–3273. doi: 10.1021/acs.orglett.6b01516. [DOI] [PubMed] [Google Scholar]; d) Balaraman K, Wolf C. Angew Chem Int Ed. 2017;56:1390–1395. doi: 10.1002/anie.201608752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8 In some cases, sonication of the reaction mixture facilitates product precipitation and isolation (see Experimental Section).
- 9 CCDC 1557938 contains the supplementary crystallographic data for bisoxindole 15. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (44) 1223-336-033; or deposit@ccdc.cam.ac.uk)
- 10.Jin Y, Chen M, Ge S, Hartwig JF. Org Lett. 2017;19:1390–1393. doi: 10.1021/acs.orglett.7b00294. [DOI] [PubMed] [Google Scholar]
- 11.Xie C, Sha W, Zhu Y, Han J, Soloshonok VA, Pan Y. RSC Adv. 2017;7:5679–5683. [Google Scholar]
- 12.Zhang L, Zhang W, Mei H, Han J, Soloshonok VA, Pan Y. Org Biomol Chem. 2017;15:311–315. doi: 10.1039/c6ob02454h. [DOI] [PubMed] [Google Scholar]
- 13.Thakur PB, Meshram HM. RSC Adv. 2014;4:5343–5350. [Google Scholar]
- 14 We isolated 1.43 g of 3 using 0.911 g of 1, 0.602 g of 2, 56.5 μL of Et3N and a total solvent amount of 30 mL (reaction solvent and for product purification). For discussion of the E-factor: Sheldon RA. Chem Ind. 1997:12–15.Andraos J, Sayed M. J Chem Ed. 2007;84:1004–1010.Sheldon RA. Green Chem. 2007;9:1273–1283.
- 15.Balaraman K, Ding R, Wolf C. Adv Synth Catal. 2017;359 doi: 10.1002/adsc.201701107. 10.1002/adsc.201701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Zhang W, Mei H, Han J, Soloshonok VA, Pan Y. Chem Eur J. 2017;23:11221–11225. doi: 10.1002/chem.201702091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.