

Abstract
Most G protein-coupled receptors (GPCRs) signal through both heterotrimeric G proteins and β–arrestins (βarr1 and βarr2). Although synthetic ligands can elicit biased signaling by G protein-vis-à-vis βarr-mediated transduction, endogenous mechanisms for biasing signaling remain elusive. Here we report that S-nitrosylation of a novel site within βarr1/2 provides a general mechanism to bias ligand-induced signaling through GPCRs by selectively inhibiting βarr-mediated transduction. Concomitantly, S-nitrosylation endows cytosolic βarrs with receptor-independent function. Enhanced βarr S-nitrosylation characterizes inflammation and aging, as well as human and murine heart failure. In genetically engineered mice lacking βarr2-Cys253 S-nitrosylation, heart failure is exacerbated in association with greatly compromised β-adrenergic chronotropy and inotropy, reflecting βarr-biased transduction and β-adrenergic receptor down-regulation. Thus, S-nitrosylation regulates βarr function and thereby biases transduction through GPCRs, demonstrating a novel role for nitric oxide in cellular signaling with potentially broad implications for patho/physiological GPCR function, including a previously unrecognized role in heart failure.
eTOC Blurb
S-nitrosylation, the ubiquitous nitric oxide-derived post-translational modification of proteins, inhibits the canonical function of β-arrestins to promote preferential G protein signaling (‘bias’) via G protein-coupled receptors. This general mechanism for biased signaling profoundly impacts the severity of heart failure, and provides a novel function for nitric oxide broadly.

Ligand-induced stimulation of G protein-coupled receptors (GPCRs) activates heterotrimeric G proteins to initiate a broad range of intracellular signaling cascades. G protein-mediated signaling through most GPCRs is truncated by recruitment of β-arrestins (βarr1 and βarr2; also designated arrestin-2 and arrestin-3), resulting in both receptor desensitization and down-regulation by internalization (Ahn et al., 2003). The functional purview of βarrs was greatly expanded by the discovery that βarrs serve to scaffold elements mediating signals that may be largely independent of G proteins, best characterized in the case of MAPK-based transduction (Lefkowitz and Shenoy, 2005; Shenoy et al., 2006). Furthermore, βarrs interact with cytoplasmic partners independent of G proteins, and thereby exhibit diverse functionality (Lefkowitz and Shenoy, 2005; Ma and Pei, 2007).
Analysis of GPCR-dependent transduction induced by synthetic ligands including multiple therapeutically relevant drugs has shown that man-made ligands can elicit “biased “signaling, i.e. differential activation of G protein-versus βarr-mediated pathways, and biased agonism remains the subject of intensive pharmacological analysis with broad potential clinical applicability (Wisler et al., 2014). The ability of synthetic ligands to elicit biased signaling by GPCRs points to the possibility that endogenous mechanisms might operate to bias signaling. Examples exist of endogenous allosteric modulators that interact directly with GPCRs to bias signaling through particular GPCRs (van der Westhuizen et al., 2015). However, the physiological relevance of receptor allostery that may impact biased signaling is not well established, and general mechanisms that might bias signaling through GPCRs have not been described.
We considered that signaling bias might be endogenously regulated by a mechanism common to multiple GPCRs, and in particular that post-translational regulation of the βarrs by the ubiquitous signaling molecule nitric oxide (NO) might provide such a mechanism. NO generation is coupled to stimulation of multiple GPCRs and regulates receptor-affiliated proteins through S-nitrosylation (Haldar and Stamler, 2013; Hess et al., 2005; Ozawa et al., 2008; Wang et al., 2006; Whalen et al., 2007). Prior analysis has shown that eNOS-mediated S-nitrosylation of βarr2 at the C-terminus Cys410 facilitates conformational changes in βarr2 that enhance receptor internalization (Ozawa et al., 2008). Here we report that S-nitrosylation by neuronal (nNOS) or inducible NOS (iNOS) of a newly discovered site that is conserved in βarr1/2, Cys251/253, provides a general mechanism to bias GPCR signaling by suppressing canonical βarr-based function but not G protein-mediated signaling. Moreover, S-nitrosylation of Cys251/253 alters cytoplasmic interactions of βarrs, enabling βarrs to function independently and providing an additional signature of bias. These results provide a novel role for NO in cellular signaling and new understanding of βarr-based function, with potential implications for physiology and disease, in particular heart failure, where β-adrenergic receptor (βAR) compromise may be newly understood in terms of βarr versus G protein bias.
Results
βarr1 and βarr2 Bind and Are S-nitrosylated by both nNOS and iNOS at a Single Common Cys
In human embryonic kidney (HEK293) cells stably transfected with rat nNOS (HEK-nNOS) or human iNOS (HEK-iNOS) and transiently transfected with rat βarr1 or βarr2, both βarr1 and βarr2 were S-nitrosylated by both nNOS (Figure 1A) and iNOS (Figure 1B), as assessed by the SNO-RAC method (Forrester et al., 2009). In addition, endogenous βarr2 was S-nitrosylated by endogenous iNOS in RAW264.7 cells following iNOS induction by bacterial lipopolysaccharide and interferon-γ (LPS/IFγ) (Figure 1C) (see Figures S1D and S1E for validation of antibodies).
Figure 1. S-nitrosylation by nNOS and by iNOS of Cys 251/253 within βarr1/2 and Association of n/iNOS with βarr1/2.
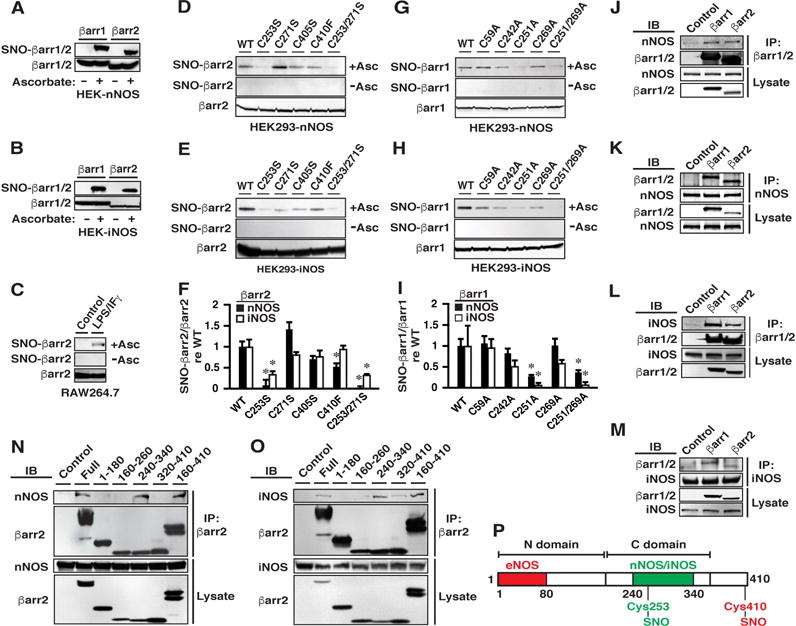
(A and B) Both βarr1 and βarr2 are S-nitrosylated by (A) nNOS and (B) iNOS in HEK cells stably transfected with rat/human n/iNOS and transiently transfected with rat βarr1/2. Assay by SNO-RAC followed by Western blotting; omission of ascorbate serves as a control for specificity.
(C) βarr2 is S-nitrosylated in RAW264.7 treated with LPS/IFγ to induce iNOS.
(D-F) Mutational analysis demonstrates that Cys253 within βarr2 is the predominant target of S-nitrosylation by n/iNOS. In F, n=3; *p<0.05 versus WT.
(G-I) As in D-F, Cys251 is the predominant target within βarr1 of S-nitrosylation by n/iNOS. In I, n=3; *p<0.05 versus WT.
(J-M) Co-immunoprecipitation demonstrates that both βarr1 and βarr2 associate with both nNOS (J and K) and iNOS (L and M). IB=immunoblot; IP=immunoprecipitation; Control=IP with non-specific IgG.
(N and O) Deletional analysis demonstrates that (N) nNOS and (O) iNOS interact principally with a region within βarr2 comprising residues 240-340. The examples shown are representative of three independent experiments. Control= transfection with empty vector.
(P) Diagrammatic representation of binding of n/iNOS and eNOS to the mapped regions within the C- and N-domains of βarr1/2, respectively, and alternative sites of S-nitrosylation by n/iNOS (Cys 253/251) and eNOS (βarr2-Cys 410). See also Figures S1 and S2.
S-nitrosylation of βarr2 by eNOS selectively targets Cys410 (Ozawa et al., 2008), the only Cys not conserved in βarr1 (numbering of Cys here and below is according to the rat sequence). Analysis of HEK-nNOS or HEK-iNOS cells transfected with βarr1 or βarr2 in which Cys were mutated to Ser, Phe or Ala demonstrated that S-nitrosylation by nNOS or iNOS targets selectively Cys253 of βarr2 (Figures 1D–1F) and the equivalent Cys251 of βarr1 (Figures 1G–1I). S-nitrosylation of βarr1 by the S-nitrosylating agent S-nitrosocysteine was also eliminated by Cys251 mutation, whereas S-nitrosylation of βarr2 was greatly diminished by Cys253 mutation; remaining S-nitrosylation of βarr2 was eliminated by mutation of Cys410 (Figures S1A-C). Cys253/251 within βarr2/1 (but not within the visual arrestins) is conserved across phylogeny (see Figure S2G).
Targeting of proteins modified by S-nitrosylation often reflects the direct interaction of substrates and NOSs (Hess et al., 2005). Binding of eNOS (Ozawa et al., 2008) and iNOS (Kuhr et al., 2010) with βarr2 has been reported. In HEK-nNOS and HEK-iNOS cells transiently transfected with βarr1 or βarr2, immunoprecipitation of either βarr1 or βarr2 (Figures 1J and 1L) or conversely of either nNOS or iNOS (Figures 1K and 1M) demonstrated that both nNOS (Figures 1J and 1K) and iNOS (Figures 1L and 1M) interact with both βarr1 and βarr2. Binding of βarr1 and of βarr2 by nNOS (Figures S2A and S2B) and by iNOS (Figures S2C and S2D) was unaffected by mutation of Cys251/253 and was therefore independent of S-nitrosylation. βarr1 also interacted with eNOS (Figure S2E), but eNOS does not S-nitrosylate βarr1 (Ozawa et al., 2008). The N-terminus of βarr2 is largely responsible for eNOS binding (Ozawa et al., 2008). Deletional analysis demonstrated that both nNOS (Figures 1N and 1P) and iNOS (Figures 1O and 1P) bound to a region comprising residues 240-340 within the C-domain of βarr2. Thus, direct interaction with nNOS and iNOS mediates selective S-nitrosylation of Cys251/253 within βarr1/βarr2, whereas the direct interaction of eNOS with βarr2 mediates selective S-nitrosylation of Cys410 in βarr2 (Figure S2F). Targeting of alternative Cys within a substrate by different NOS isoforms has not been reported previously.
S-nitrosylation of βarr1/βarr2 by n/iNOS Mediates Homodimerization
Homo/hetero-oligomerization (dimerization) of βarr1/2 has been previously observed in vitro and in situ (Boularan et al., 2007; Milano et al., 2006; Storez et al., 2005), and potentially suppresses ligand-induced βarr function (Milano et al., 2006), but the physiological relevance of dimerization remains unclear. Cys251/253 are located within the inositol 1,2,3,4,5,6-hexakisphosphate (IP6)-binding domain (residues 233-251 within βarr2), which has been implicated in βarr dimerization (Boularan et al., 2007; Milano et al., 2006), and which includes a short loop of residues (242-246) that interacts with the ligand-activated β2-adrenergic receptor (β2AR) (Shukla et al., 2014) (Figure S2G). Thus, we initially examined the effects of S-nitrosylation on βarr dimerization and interaction with the β2AR.
In HEK293 cells transiently co-transfected with separate βarr2 constructs bearing either a FLAG- or hemagglutinin (HA)-tag or separate βarr1 constructs bearing either a FLAG- or HA-tag, homo-dimerization as assessed by co-immunoprecipitation was not observed in the absence of nNOS or iNOS expression (Figures 2A–2D, HEK), but dimerization of WT and not of Cys253/251Ser βarr1/2 was induced by co-expression of either iNOS (Figures 2A, 2B and 2F) or nNOS (Figures 2C, 2D and 2F). Dimerization was not detected in HEK293 cells expressing eNOS (Figure S2H). Thus, S-nitrosylation of Cys253/251 by n/iNOS induces homodimerization of βarr1/2. We also observed dimerization of endogenous βarr2 in RAW264.7 cells in which iNOS was induced by treatment with LPS/IFγ (Figures 2E and 2F), and inhibition of iNOS with 1400W diminished βarr2 dimerization (Figures 2G and 2H) and S-nitrosylation (Figures 2I and 2J) in parallel. Note that detection of dimerization by Western blotting here and below does not rule out the presence of hetero-dimers.
Figure 2. Dimerization of βarr1 and of βarr2 Induced by Expression of nNOS or iNOS, Dependent upon Cys 251/253.
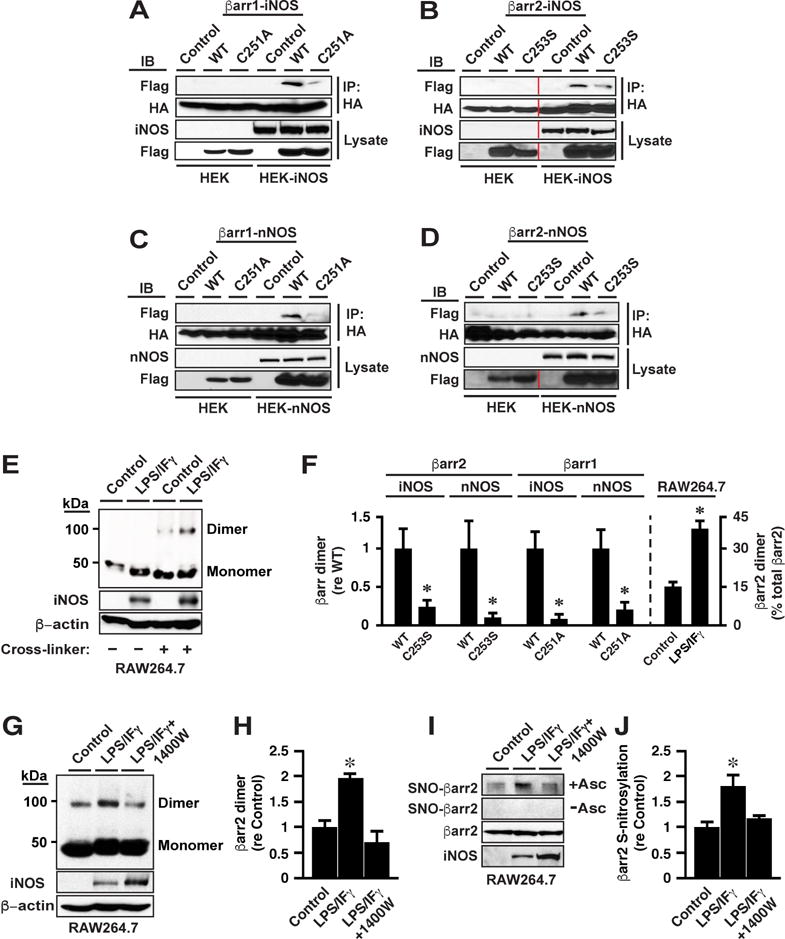
(A – D) Expression of (A and B) iNOS or (C and D) nNOS in HEK cells induces dimerization of βarr1 and of βarr2, dependent respectively upon Cys251 and Cys253 (co-IP following dual transfection with FLAG-βarr1 and HA-βarr1 or FLAG-βarr2 and HA-βarr2). Control =transfection with empty vector. Vertical red lines indicate within-blot crops. See also Figure S2H.
(E) Induction of endogenous iNOS in RAW264.7 cells induces dimerization of native βarr2. Control=non-induced.
(F) Quantification of results exemplified in A-E; n=3; *p<0.01.
(G-J) Treatment with 1400W reverses the enhanced dimerization (G and H) and S-nitrosylation (I and J) of βarr2 that is induced by LPS/IFγ treatment of RAW 264.7 cells. In H and J, n=3; *p<0.01 versus other groups.
S-nitrosylation of βarr1/βarr2 by n/iNOS Suppresses Ligand-induced Recruitment of βarr1/βarr2 to the β2AR and Receptor Internalization
To test whether S-nitrosylation by n/iNOS of Cys 251/253 might directly regulate the ligand-induced interaction of βarr1/2 with the β2AR, we focused on βarr2 because internalization of the β2AR is mediated with substantially greater efficacy by βarr2 versus βarr1 (Ahn et al., 2003; Kohout et al., 2001). We evaluated binding of βarr2 to ligand-activated β2AR by co-immunoprecipitation from extracts prepared from HEK293 cells stably overexpressing the β2AR (designated W9 cells) and transiently transfected with WT βarr2 alone (control cells) or WT or C253S βarr2 and either nNOS or iNOS. Whereas β2AR activation with ISO enhanced co-immunoprecipitation of β2AR with WT βarr2 in control W9 cells, co-immunoprecipitation was largely eliminated in W9-nNOS and W9-iNOS cells (Figures 3A and 3B). Co-immunoprecipitation was not observed in the absence of ISO (Figure 3A). Diminishment by n/iNOS of the ligand-induced interaction between βarr2 and the β2AR could be ascribed to S-nitrosylation of βarr2 because interaction was restored by mutation of Cys253 within βarr2 (Figures 3C and 3D). Mutation of Cys253 within βarr2 had no effect on the ISO-induced interaction between βarr2 and the β2AR in the absence of n/iNOS (Figures S3A and S3B).
Figure 3. S-nitrosylation of βarrs Suppresses ISO-induced βarr2 Recruitment to the β2AR and Receptor Internalization, and biases Signal Transduction toward G protein.
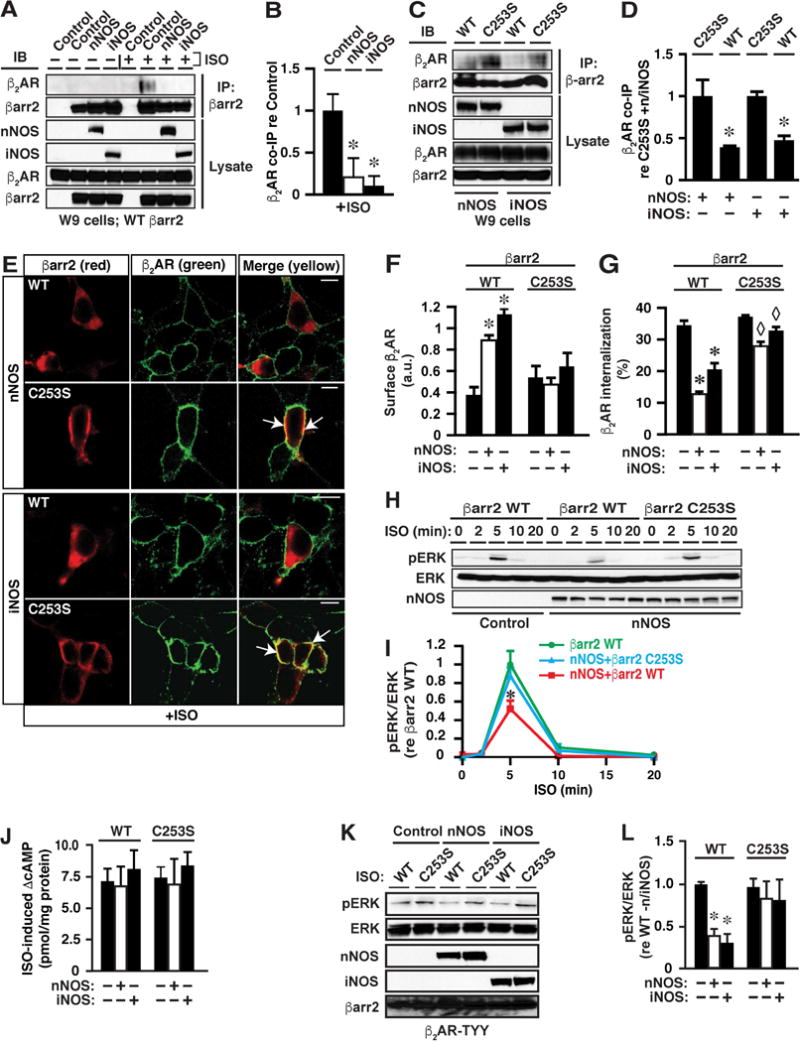
(A and B) ISO-induced (10 μM, 5 min) co-immunoprecipitation of βarr2 and the β2AR is greatly diminished by expression of either nNOS or iNOS. The first (left-hand) Control in each set of lanes (− or + ISO) = transfection with empty vector; the second (right-hand) Control = absence of i/nNOS. In B, n=3; *p<0.01 versus control.
(C and D) n/iNOS-dependent suppression of the interaction between βarr2 and the β2AR is rescued by mutation of Cys253 within βarr2. In D, n=3; *p<0.01 versus C253S.
(E) Confocal microscopy reveals that ISO-induced (10 μM, 5 min) recruitment of WT but not C253S βarr2 to the plasma membrane (as shown in Figure S3C) is largely prevented by expression of n/iNOS. Scale bars=10μm.
(F) Cell-surface biotinylation demonstrates that ISO-induced (10 μM, 10 min) internalization of the β2AR is greatly diminished in W9 cells expressing n/iNOS and WT βarr2, and that suppression is abrogated in the case of C253S βarr2; *p<0.01 versus all other groups; no other inter-group differences were significant; n=3. See Figure S3D for raw data.
(G) FACS analysis confirms inhibition of ISO-induced internalization of the β2AR by S-nitrosylation of βarr2; n=3-5; * and ◊ p<0.01 re internalization in the absence of n/iNOS and ◊ p<0.01 versus WT in the presence of n/iNOS (ANOVA).
(H and I) S-nitrosylation of Cys253 within βarr2 by nNOS suppresses ISO-induced ERK phosphorylation, a signature of βarr-mediated signaling (see Figures S4A and S4B for identical effects of iNOS expression). In I, n=3; *p<0.05 versus βarr2 WT.
(J) There are no significant differences between any conditions in ISO-induced (10 μM, 2 min) cAMP production, a signature of G protein-mediated β2AR signaling; n=4-6.
(K and L) Suppression of ISO-induced (10 μM, 5 min) ERK phosphorylation by n/iNOS is dependent upon C253 within βarr2 in HEK293 cells stably expressing β2AR-TYY, which can couple to βarrs but cannot activate G proteins (in L, n=3; *p<0.05 versus WT without n/iNOS and C253S).
Confocal immunofluorescence microscopy of W9 cells transiently transfected with either WT or C253S βarr2, and either nNOS or iNOS, or without transfection of NOS showed that both WT and C253S βarr2 were predominantly cytoplasmic in the absence of ISO (Figure S3C) and that stimulation of the β2AR with ISO resulted in the re-distribution of both WT and C253S βarr2 from cytoplasm to the plasma membrane and co-localization with the β2AR (Figure S3C). Co-expression of either nNOS or iNOS largely prevented ISO-induced membrane recruitment of WT but not of C253S βarr2 (Figure 3E) (whereas n/iNOS had no effect on localization of WT or C253S βarr2 in the absence of ISO; see Figure S6E). Recruitment of βarrs to GPCRs promotes internalization through the clathrin/AP2-based endocytotic machinery (Drake et al., 2006). Consistent with the suppression of membrane recruitment of βarr2 by S-nitrosylation, ISO-induced internalization of the β2AR was greatly diminished in W9 cells expressing either nNOS or iNOS and WT βarr2, whereas internalization was not significantly suppressed in cells expressing n/iNOS and βarr2-C253S, as assessed by cell-surface labeling (biotinylation of intact W9 cells followed by avidin pull-down) (Figures 3F and S3D). Expression of WT or C253S βarr2, with or without co-expression of nNOS or iNOS, had no effect on the membrane localization of the β2AR in the absence of stimulation (Figure S3E). Suppression of ISO-induced β2AR internalization by S-nitrosylation of Cys253 within βarr2 was confirmed by fluorescence-based cell sorting (FACS) (Figure 3G). We also determined that the expression of neither AP2 nor clathrin was altered by overexpression of nNOS or iNOS (Figures S3F and S3G). Note that analysis by FACS (Figure 3G) revealed a relatively small but significant suppression by n/iNOS of ISO-induced β2AR internalization in cells expressing βarr2-C253S. This observation would be consistent with a role for S-nitrosylation by n/iNOS of elements in addition to βarr2-C253S, which might include WT (endogenous) βarr2 and/or βarr1. However, the finding that the suppressive effect of n/iNOS on receptor internalization is largely eliminated by mutation of Cys253 within βarr2 indicates that S-nitrosylation of Cys253 within βarr2 is largely responsible for suppressed β2AR internalization.
S-nitrosylation of βarr1/βarr2 by n/iNOS Suppresses Ligand-induced Signaling by the β2AR via βarrs (MAPK) But Not G Protein (cAMP)
Recruitment of βarrs to ligand-activated β2ARs and other GPCRs initiates βarr-mediated signaling through MAPKs. Our findings suggest the possibility that inhibitory S-nitrosylation of βarrs might suppress βarr-dependent but not G protein-dependent signaling, thereby providing an endogenous mechanism for biasing signaling. We found that ERK activation consequent upon ISO stimulation of the β2AR as assessed by ERK phosphorylation was substantially (~50%) suppressed in cells expressing WT but not C253S βarr2 and either nNOS (Figures 3H and 3I) or iNOS (Figures S4A and S4B). In contrast, ISO-induced cAMP production was unaffected (assessed following ISO stimulation for 2 min) (Figure 3J). Co-immunoprecipitation demonstrated that βarr2 and ERK were complexed in the absence of agonist, and that complex formation was unaltered by C253S mutation of βarr2 or by expression of n/iNOS (Figures S4C and S4D). Suppression of ERK activity by ~50% when βarr2 recruitment to the β2AR is almost entirely eliminated is consistent with an independent role for Gαs/i in ERK activation (Daaka et al., 1997; Shenoy et al., 2006). Notably, the magnitude of suppression of ISO-induced ERK phosphorylation consequent upon S-nitrosylation of βarr2 (Figures 3I and S4B) is comparable to the magnitude of suppression of ERK phosphorylation resulting from siRNA-mediated knockdown of βarr(s) (Shenoy et al., 2006). Finally, we determined that C253- and NOS-dependent suppression of ISO-induced ERK phosphorylation did not reflect a contribution from altered G protein-mediated signaling (i.e., that potential G-protein/βarr2 cross-talk (Alvarez-Curto et al., 2016) was not involved): suppression of ERK phosphorylation by n/iNOS was completely dependent upon C253 in HEK293 cells stably expressing TYY mutant β2AR (TYY: T68F, Y132G, Y219A) (Shenoy et al., 2006) that can recruit βarrs but cannot activate G proteins (Figures 3K and 3L).
S-nitrosylation of βarr1/βarr2 by n/iNOS Suppresses Signaling by βarrs But Not G Protein Following Activation of Multiple GPCRs
To examine the possibility that S-nitrosylation of βarrs might provide a general mechanism to alter βarr-mediated signal transduction, we examined the effect of S-nitrosylation of βarr2 on recruitment of βarr2 to the AT1aR and on AT1aR signaling. The β2AR and the AT1aR are prototypes of GPCRs that can be differentiated by differences in their affinity for βarrs (designated class A and class B; Lefkowitz and Shenoy, 2005). We employed HEK cells stably expressing the AT1aR, and transiently transfected with either nNOS or iNOS and either WT or C253S βarr2. In the absence of n/iNOS, stimulation with angiotensin II (AngII; 0.1 μM) resulted in the recruitment of both WT and C253S βarr2 to the plasma membrane where they co-localized with the AT1aR, as assessed by confocal immunofluorescence microscopy (Figure S5A). In cells expressing either nNOS or iNOS, AngII-induced membrane recruitment of WT but not C253S βarr2 was suppressed (Figure 4A). Thus, S-nitrosylation suppresses ligand-induced membrane recruitment of βarr2 by at least two prototypic GPCRs that are distinguished by their modes of interaction with βarrs.
Figure 4. S-nitrosylation of βarr Biases Signaling through the AT1aR and the B1R.
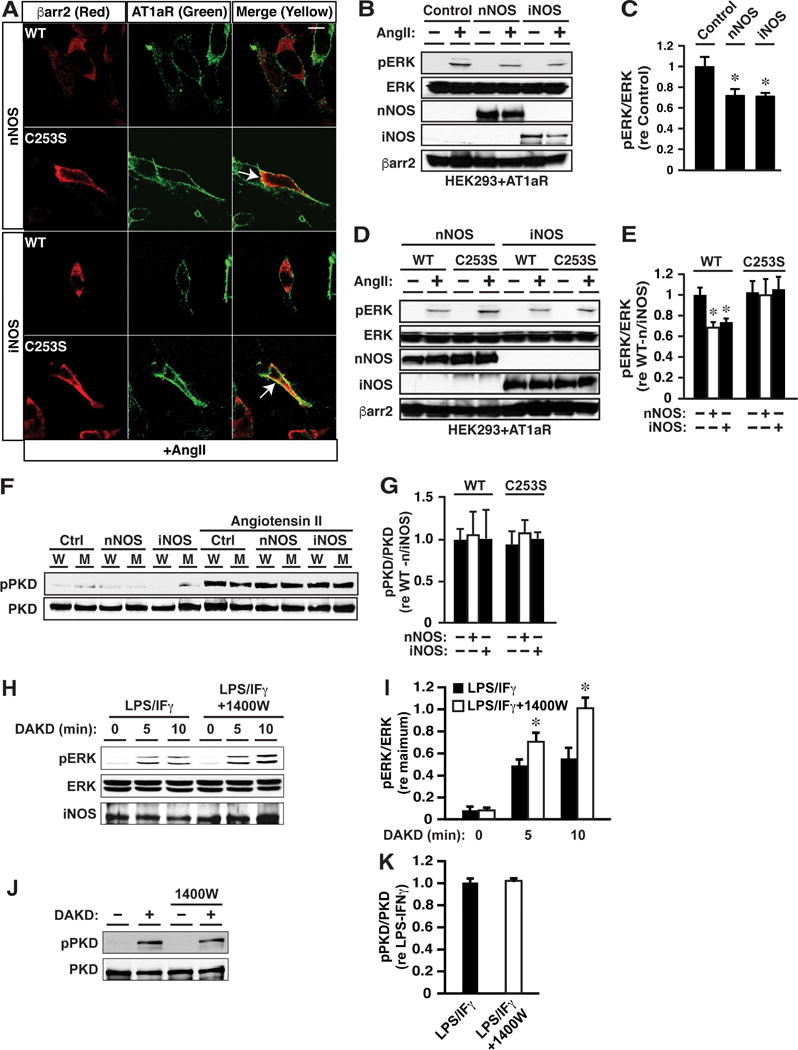
(A) In HEK cells expressing the AT1aR, confocal immunofluorescence microscopy reveals angiotensin II-induced (0.1 μM, 5 min) membrane recruitment of both WT and C253S βarr2 in control cells (without n/iNOS expression), and recruitment of WT but not C253S βarr2 is suppressed by expression of nNOS or iNOS. Scale bar (upper right)=10 μm.
(B and C) Angiotensin II-induced (0.1 μM, 5 min) ERK phosphorylation in cells expressing the AT1aR and WT βarr2 is suppressed by expression of n/iNOS versus control cells (without n/iNOS expression). In C, n=3; *p<0.05 versus Control.
(D and E) Suppression of angiotensin II-induced ERK phosphorylation by n/iNOS is dependent upon βarr2 Cys253. In E, n=3; *p<0.05 versus WT in the absence of n/iNOS and versus C253S.
(F and G) Angiotensin II-induced (0.1 μM, 2 min) phosphorylation of PKD, a signature of G protein-coupled signaling through the AT1aR, does not differ between control cells (without n/iNOS) and cells expressing n/iNOS, which also express either WT (W) or C253S βarr2 (M, mutant); n=3.
(H and I) In BAEC following induction of iNOS and the B1R by LPS/IFγ, ERK phosphorylation induced by selective stimulation of the B1R with DAKD (1 μM, 2 min) is enhanced by inhibiting iNOS with 1400W; n=3; *p<0.05. (J and K) In contrast, endogenous iNOS inhibition had no effect on DAKD-induced PKD phosphorylation (G protein activation); n=3.
We also determined that S-nitrosylation of Cys253 within βarr2 inhibits AngII-induced ERK activation (Figures 4B–4E); AngII-induced ERK activation did not differ in cells expressing WT versus C253S βarr2 in the absence of NOS (Figures 4E and S5B). In contrast, S-nitrosylation of Cys253 within βarr2 had no effect on Ang-II-induced activation of protein kinase C (PKC), a principal mediator of G protein (Gq/11)-coupled signaling (Hunyady and Catt, 2006), as assessed by assay of AngII-induced and PKC-specific phosphorylation (Figure S5C) of protein kinase D (PKD) (Tan et al., 2004) (Figures 4F and 4G). Thus, S-nitrosylation of βarrs provides an endogenous mechanism for biasing signaling through both the β2AR and the AT1aR, by selectively suppressing βarr–dependent versus G protein-coupled transduction.
Bradykinin-coupled GPCRs (B1R, B2R) mediate the effects of the kallikrein-kinin system through Gq/11 and βarrs (Leeb-Lundberg et al., 2005). Whereas the B2R is expressed constitutively, the B1R is induced by inflammatory stimuli that also induce iNOS (Leeb-Lundberg et al., 2005). Following stimulation of the B1R, βarr2 scaffolds iNOS to promote iNOS activation consequent upon phosphorylation by ERK (Kuhr et al., 2010). In native (untransfected) bovine aortic endothelial cells (BAEC) following induction of the B1R and iNOS by treatment with LPS/IFγ, selective activation of B1Rs with des-Arg(10)-kallidin (DAKD; 1 μM) resulted in phosphorylation of ERK (Figures 4H and 4I) and of PKD (Figures 4J and 4K), indicative of transduction via βarr and Gq/11. DAKD did not result in ERK phosphorylation in the absence of LPS/IFγ (Figure S6A), consistent with the requirement for inflammatory activation in B1R expression (Figure S6B). Inhibition of iNOS with 1400W enhanced ERK phosphorylation (Figures 4H and 4I) but had no effect on PKD phosphorylation (Figures 4J and 4K) (or on B1R induction; Figure S6B), consistent with selective suppression of βarr-versus Gq/11-mediated signaling by iNOS-derived NO through S-nitrosylation of βarr2. In addition, dimerization of βarr2 was significantly enhanced following induction of iNOS, and 1400W suppressed dimerization (Figures S6C and S6D). Thus (consistent with our findings in RAW264.7 cells expressing endogenous iNOS (Figures 2E-J)), dimerization of βarr2 and biased signaling through the B1R are promoted by B1R-coupled, endogenously derived NO (Kuhr et al., 2010).
S-nitrosylation by n/iNOS of βarr1 and βarr2 Regulates Ligand/Receptor-independent Function
Signaling by βarrs is mediated in significant part through cytoplasmic scaffolding by βarrs of transcriptional co-factors and regulators (Ma and Pei, 2007). Among the best characterized interactions of βarrs is that of βarr2 with the proto-oncogene MDM2 (Boularan et al., 2007); MDM2 binds the tumor-suppressor transcription factor p53 to inhibit its activity (Kruse and Gu, 2009). We found that co-expression of either nNOS or iNOS substantially increased the interaction of βarr2 with MDM2 (Figures 5A and 5B), dependent upon Cys253 (Figures 5C and 5D), as assessed by co-immunoprecipitation (which did not require GPCR stimulation). The extent of interaction between MDM2 and WT and C253S βarr2 did not differ in the absence of n/iNOS (Figure 5D). In addition, confocal immunofluorescence microscopy revealed that, whereas MDM2 was localized to both cytoplasm and nucleus in control cells, nuclear MDM2 was greatly diminished by co-expression of nNOS/iNOS and WT βarr2 (Figure 5E). By contrast, co-expression of nNOS/iNOS and βarr2-C253S did not alter MDM2 distribution (Figure 5E). Co-immunoprecipitation of p53 and MDM2 was greatly diminished in cells expressing n/iNOS and WT, but not C253S βarr2 (Figures 5F and 5G) (without altering the predominantly nuclear localization of p53; Figure S6E). These findings are consistent with previous reports that MDM2 is more efficiently partitioned from nucleus to cytoplasm by a mutant form of βarr2 that cannot interact with GPCRs (Song et al., 2006) and that oligomerization of βarr2 is required for titration of MDM2 out of the nucleus (Boularan et al., 2007). Thus, S-nitrosylation-mediated cytosolic sequestration of MDM2 may represent an example of allosteric regulation through which βarrs can operate independently of particular GPCRs.
Figure 5. S-nitrosylation of Cys253 within βarr2 Enhances Ligand-independent Interaction of βarr2 and MDM2 and Suppresses Binding of MDM2 and p53.
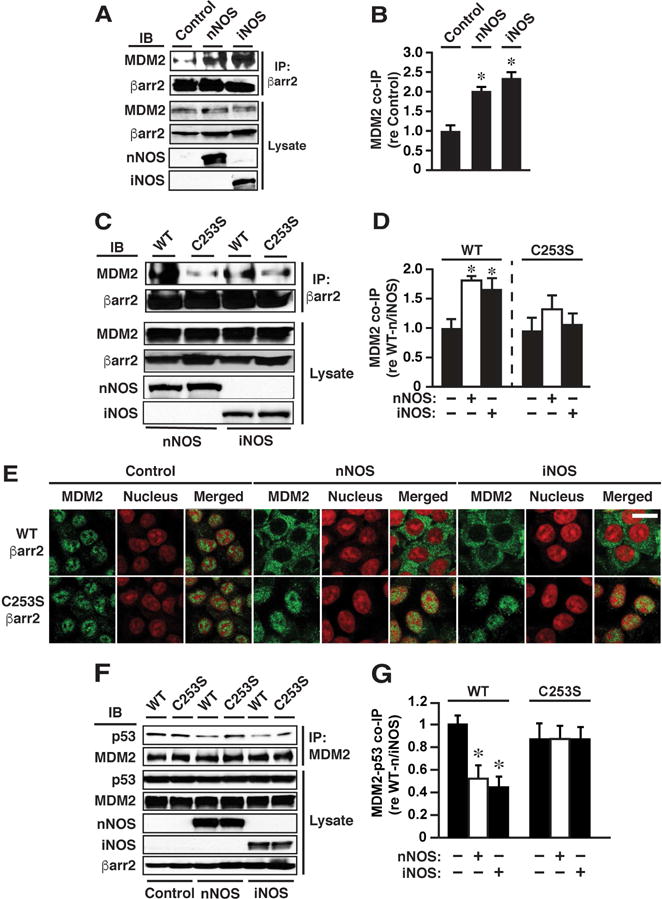
(A and B) Co-immunoprecipitation of βarr2 and MDM2 is enhanced by n/iNOS expression. Control=without n/iNOS (empty vector). In (B), n=3; *p<0.05 versus Control. (C and D) n/iNOS-dependent enhancement of βarr2-MDM2 co-immunoprecipitation is dependent upon Cys253 within βarr2. Note that co-immunoprecipitation of MDM2 with WT versus C253S βarr2 did not differ significantly in the absence of n/iNOS. In D, n=3; *p<0.05 versus WT or C253S in the absence of n/iNOS.
(E) Confocal immunofluorescence microscopy reveals that the predominantly nuclear localization of MDM2 is abrogated in cells expressing n/iNOS and WT βarr2 but is unaltered in cells expressing n/iNOS and C253S βarr2. Each condition is represented by three images: MDM2 (green), nuclear propidium iodide staining (red) and the merged image. Scale bar (lower right)=20μm.
(F and G) Expression of n/iNOS and WT but not C253S βarr2 greatly diminishes co-immunoprecipitation of p53 and MDM2. Note that co-immunoprecipitation of MDM2 and p53 did not differ significantly in cells expressing WT versus C253S βarr2 in the absence of n/iNOS. In G, n=3; * p<0.05 versus WT without n/iNOS (ANOVA).
Up-regulation of iNOS and/or nNOS and Enhanced S-nitrosylation and Dimerization of βarr2 in a Mouse Model of Heart Failure, in Failing Human Hearts and in Aging Mice
Altered signaling by GPCRs, particularly βARs, and by p53 are hallmarks of heart failure. To examine the possibility that altered S-nitrosylation of βarrs might represent a previously unappreciated aspect of the pathophysiology of heart failure, we assessed changes in βarr S-nitrosylation (and corresponding dimerization) in both a mouse model of heart failure (induced by transverse aortic constriction; TAC) (Rockman et al., 1991) and in failing human hearts. In the myocardium of mice subjected to TAC versus sham-operated mice (3 wk survival; Figure 6A), both nNOS and iNOS (but not eNOS) were up-regulated as assessed by Western blotting (Figure 6B), and both S-nitrosylation (Figures 6C and 6D) and dimerization (Figures 6E and 6F) of both βarr2 and βarr1 were significantly enhanced. We carried out a similar analysis on samples of human myocardium obtained from non-failing (control) hearts not accepted for transplant (n=12) and hearts from transplant patients with either ischemic (n=12) or non-ischemic (n=12) heart failure (Duke Human Heart Repository; see Experimental Procedures for patient demographics). In both ischemic and non-ischemic failing hearts, iNOS (but not nNOS or eNOS) was up-regulated (Figure 6G) and both S-nitrosylation (Figures 6H and 6I) and dimerization (Figures 6J and 6K) of both βarr2 and βarr1 were substantially enhanced versus control hearts; notably, the magnitude of enhancement was similar in ischemic and non-ischemic heart failure. Remarkably, about 70-75% of βarrs were dimerized in failing human hearts. Differences in the degree of dimerization in mouse and human hearts may reflect different etiologies and/or duration of pathology as well as differences in tissue preparation. These findings indicate that selective suppression of βarr-versus G protein-mediated signaling by GPCRs that is mediated by S-nitrosylation of βarr may be a common feature of heart failure.
Figure 6. Enhanced S-nitrosylation of βarr1/2 by n/iNOS in Mouse and Human Heart Failure and in Aging Mice.
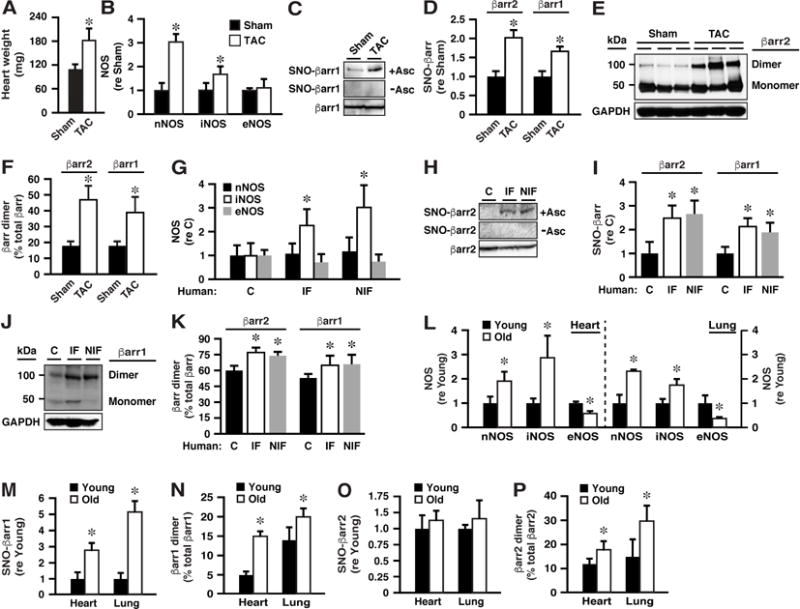
(A) Increased heart weight in mice subjected to TAC versus sham-operated mice (3 wk survival) verifies cardiac hypertrophy.
(B) Myocardial nNOS and iNOS are significantly up-regulated in TAC mice, whereas eNOS is unchanged.
(C and D) S-nitrosylation of both βarr1 and βarr2 is enhanced in the myocardium of TAC mice. (E and F) Dimerization of both βarr1 and βarr2 is enhanced in the myocardium of TAC mice. In A, B, D and F, n=8-10; *p<0.05 versus Sham.
(G-K) In human myocardium from non-failing (C; control) hearts versus hearts with ischemic heart failure (IF) or non-ischemic heart failure (NIF): (G) iNOS is up-regulated; (H and I) S-nitrosylation of βarr1 and of βarr2 is enhanced; (J and K) dimerization of βarr1 and of βarr2 is enhanced; n=8-12. *p<0.05 versus C.
(L-P) In heart and lung of old versus young mice: (L) nNOS and iNOS are induced and eNOS expression declines; (M and N) S-nitrosylation and dimerization of βarr1 are enhanced; (O and P) Dimerization but not S-nitrosylation (as detected by SNO-RAC) of βarr2 is enhanced. In L-P, n=3-5; *p<0.05 versus Young.
Altered expression of NOSs accompanies aging in numerous tissues where it has been associated with a broad range of pathophysiologies including heart failure (Cau et al., 2012). Our findings raise the possibility that altered S-nitrosylation of βarr might contribute to age-related alterations in GPCR function including those associated with cardiovascular decline. In old (50-53 wks of age) versus young (14-16 wks of age) mice, we found that, in both heart and lung, expression of both nNOS and iNOS was increased (whereas expression of eNOS was decreased) (Figure 6L), and that S-nitrosylation (Figure 6M) and dimerization (Figure 6N) of βarr1 were enhanced, consistent with S-nitrosylation of Cys251 by n/iNOS (eNOS does not S-nitrosylate βarr1). S-nitrosylation of βarr2 did not differ significantly in young and old mice (Figure 6O), consistent with down-regulation of eNOS in association with up-regulation of n/iNOS (the SNO-RAC method would not distinguish between S-nitrosylation of Cys410 by eNOS and of Cys253 by n/iNOS). However, dimerization of βarr2 in heart (and lung) was significantly enhanced in old versus young mice (Figure 6P), consistent with enhanced S-nitrosylation of Cys253.
TAC-induced Cardiac Decompensation is Enhanced in Mice Lacking S-nitrosylation by n/iNOS of βarr2
To evaluate the patho/physiological roles of βarr S-nitrosylation by n/iNOS, we generated a mouse strain in which Cys253 within βarr2 was replaced with Ser (βarr2-C253S; InGenious Targeting Laboratory, Ronkonkoma, NY) (Figures S7A and S7B). Levels of expression of WT and C253S βarr2 were indistinguishable in heart samples (Figure S7C). Blood pressure and heart rate were unchanged versus wild type under basal conditions, as further detailed below.
We subjected WT and βarr2-C253S mice to TAC, with assessment of cardiac function by echocardiography (as in Figures 7C-F and Figures S7H-K). Prior to TAC, no differences were observed between WT and βarr2-C253S mice in functional measures (Figures 7C-E and S7H-K; Sham=sham operated). Two weeks following TAC, S-nitrosylation (Figure 7A; Figures S7D and S7E) and dimerization (Figure 7B; Figures S7F and S7G) of βarr2 were significantly diminished by C253S mutation, without effects on βarr1, and βarr2-C253S mice were significantly more compromised than WT mice, including lower ejection fraction, fractional shortening and cardiac output (Figures 7C–7E; see also Figures 7L and 7M) as well as greater ventricular dilation (Figures 7F and S7H-K); significant differences between WT and βarr2-C253S mice on all measures were enhanced at 4 weeks following TAC (Figures 7C-F and S7H-K). Thus, S-nitrosylation by n/iNOS of Cys253 within βarr2 in vivo, which would affect transduction through multiple GPCRs, including in particular the βARs that control cardiac output, is cardio-protective in the setting of pressure overload-induced heart failure, and strikingly, this effect was demonstrated in the presence of WT βarr1.
Figure 7. S-nitrosylation of βarr2 by n/iNOS Is Cardio-protective in the Setting of Heart Failure.
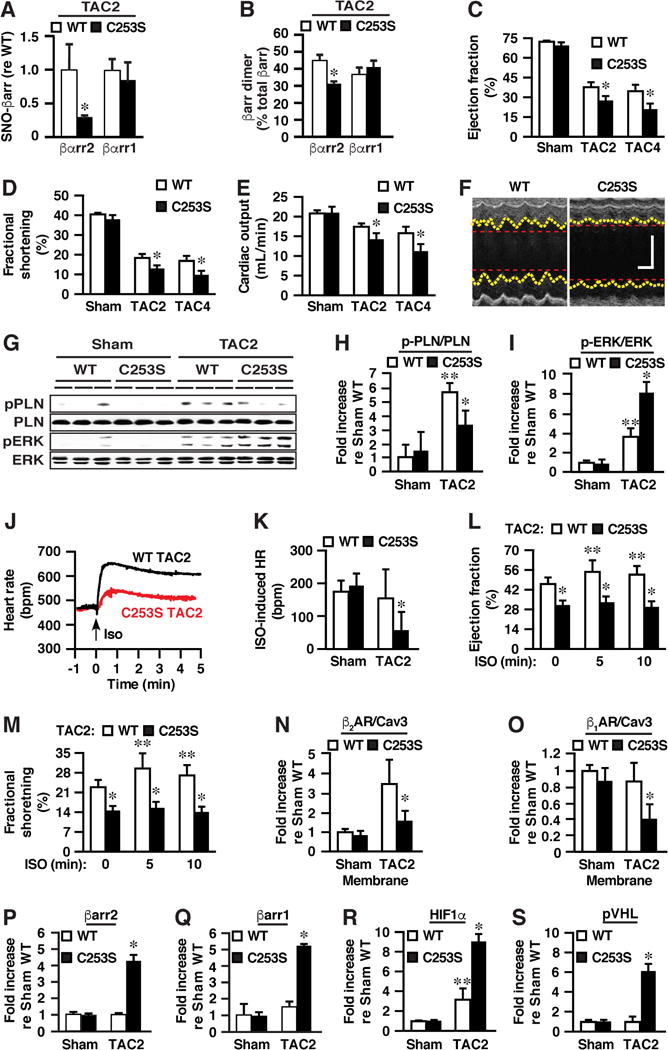
(A and B) After two weeks of TAC (TAC2), (A) S-nitrosylation and (B) dimerization of βarr2 are significantly diminished in βarr2-C253S versus WT (WT) mice, without effects on βarr1 (see Figures S7D and S7E, and Figures S7F and S7G for raw data). (C-F) As assessed by echocardiography, impairment of heart function in TAC2 and TAC4 mice is significantly exacerbated in βarr2-C253S versus WT mice, with respect to diminished (C) left ventricular (LV) ejection fraction, (D) LV fractional shortening and (E) calculated cardiac output, as well as (F) greater LV dilation (representative echocardiograms from TAC4 mice; see Figures S7H and S7I for quantification of end-systolic and end diastolic diameter and Figures S7J and S7K for end-systolic and end-diastolic volume). In F, the vertical scale bar represents 2 mm and the horizontal scale bar represents 100 msec. See also Figures 7L and 7M below. (G and H) TAC-induced enhancement of phospholamban phosphorylation representing cAMP production (pPLN, normalized with respect to total phospholamban) is significantly diminished whereas (G and I) enhancement of ERK phosphorylation (pERK, normalized with respect to total ERK) is significantly enhanced in βarr2-C253S versus WT mice. (J) Representative electrocardiographic tracings illustrating greatly suppressed chronotropic response to ISO in βarr2-C253S versus WT following two weeks of TAC. (K) Quantification of electrocardiographic data. (L and M) Echocardiography reveals that enhancement of contractility (LV ejection fraction (L) and fractional shortening (M)) induced by ISO is eliminated in TAC2 βarr2-C253S mice. (N) TAC results in up-regulation of the β2AR in myocardial membranes from WT mice that is greatly diminished in βarr2-C253S mice. (O) TAC results in down-regulation of the β1AR in βarr2-C253S mice but not in WT mice. See Figure S7N for raw data. (P and Q) The abundance of myocardial (P) βarr2 and (Q) βarr1 is enhanced by TAC in βarr2-C253S but not in WT mice. See Figure S7O for raw data. (R) Up-regulation of HIF-1α by TAC is greatly enhanced in βarr2-C253S versus WT mice (see Figure S7O for raw data). (S) TAC induces expression of pVHL in βarr2-C253S mice. See Figure S7O for raw data. In A and B, n=3-4; in C-E, n=11; in H and I, n=4; in K, n=4-6; in L and M, n=4-5; in N and O, n=3-6; in P-S, n=4. Data are mean +SEM. *p<0.05 versus WT and **p<0.05 versus sham.
TAC-induced signaling bias, adrenergic dysfunction, receptor down-regulation and ligand independence regulated by S-nitrosylation by n/iNOS of βarr2
We sought to determine whether signaling bias created by S-nitrosylation of Cys253 within βarr2 by n/iNOS, observed in vitro, would be evident in the context of heart failure in vivo. We found that TAC, which is characterized by adrenergic overstimulation (Liao et al., 2012), resulted in enhanced cAMP production as evidenced by both phospholamban phosphorylation (Figures 7G and 7H) and cAMP ELISA (Figure S7L), but that this enhancement was significantly diminished in βarr2-C253S versus WT mice (Figures 7G and 7H; Figure S7L). Further, ERK phosphorylation was also enhanced by TAC but the enhancement was significantly greater in βarr2-C253S versus WT mice (Figures 7G and 7I). Thus, TAC results in G protein-biased signaling mediated by S-nitrosylation of βarr2 in wild type mice and in βarr-biased signaling in βarr2-C253S mutant (versus wild type) mice, consistent with enhanced membrane recruitment of βarr2, ERK activity and densensitization of the βAR in the absence of the suppressive influence of S-nitrosylation (Figure 3).
βARs are the principal effectors of cardioregulation through chronotropic and ionotropic enhancement that is mediated by cAMP. We therefore examined β-adrenergic functionality in WT versus βarr2-C253S mice. ISO (10 μg/kg; via inferior vena cava) resulted in a rapid and sustained increase in heart rate in both WT and βarr2-C253S mice (Figure S7M), providing a physiological read-out of G protein/cAMP signaling. However, remarkably, whereas the chronotropic effect of ISO was not significantly altered by TAC in WT mice, it was largely eliminated in βarr2-C253S mice (Figures 7J and 7K). Furthermore, in a second set of experiments (TAC, 2 weeks), the positive inotropic effect of ISO (10 μg/kg; via jugular vein) on LV ejection fraction and fractional shortening observed in WT mice was abrogated in C253S mice (Figures 7L and 7M). Thus, S-nitrosylation of C253 within βarr2 is required to maintain β-adrenergic responsiveness (though G proteins) in experimental heart failure.
In addition to biasing signaling away from transduction via cAMP, we reasoned that enhanced βarr function in βarr2-C253S mice might contribute to cardiopathology by enhancing down-regulation of βARs (a sine qua non of heart failure). It has been reported that total myocardial βAR levels were enhanced in a rat model of TAC (Limas, 1979), likely reflecting the up-regulation of the β2AR (Xie et al., 2009). We found in WT mice that the abundance of β2ARs was significantly enhanced (versus sham-operated) and that this enhancement was largely eliminated in βarr2-C253S mice (Figures 7N and S7N). The abundance of β1ARs was unaltered by TAC in WT mice, but was greatly decreased by TAC in βarr2-C253S mice (Figures 7O and S7N). Therefore, under the adrenergic stress that characterizes heart failure (Triposkiadis et al., 2009) particularly that induced by TAC (Liao et al., 2012), βAR abundance is greatly diminished by the absence of βarr2 S-nitrosylation, consistent with the role of βarr2 S-nitrosylation in preventing βAR desensitization/internalization and down-regulation observed in vitro (Figures 3 and S3). Indeed, down-regulation and dysfunction (diminished chronotropy and inotropy) is not observed in our heart failure model unless S-nitrosylation of βarr is compromised. These results raise the idea that S-nitrosylation level may be a previously unrecognized determinant of classical down-regulation and desensitization (loss of adrenergic responsivity) in human heart failure. Moreover, we found that TAC resulted in substantial (~5-fold) increases in the abundance of both βarr2 and βarr1 in βarr2-C253S but not in WT mice (Figures 7P and 7Q; Figure S7O), emphasizing that S-nitrosylation of βarr2 specifically represents a central mechanism to suppress βarr function in heart failure, inasmuch as up-regulated Cys253Ser βarr2 would not be subject to control by S-nitrosylation. In addition, qPCR revealed that up-regulation of the βarrs was not a result of enhanced transcription (Figures S7P and S7Q), pointing to the likelihood of post-transcriptional regulation by altered degradation. Thus, S-nitrosylation of βarr2 is a critical mechanism regulating βAR function and cardiac performance in the context of experimental heart failure.
We then sought evidence for alterations in receptor-independent βarr activity in heart failure. In the TAC model, pressure overload induces expression of the transcription factor hypoxia-inducible factor 1 (HIF-1) (Semenza, 2014), which promotes adaptive cardiac angiogenesis; subsequent induction of p53 suppresses HIF-1 and drives heart failure (Sano et al., 2007). Our finding that S-nitrosylation of βarr2 by n/iNOS, which is increased by TAC (Figure 6D), suppresses nuclear interaction between MDM2 and p53 (Figures 5F and 5G), predicts that βarr2 S-nitrosylation might regulate HIF-1 activity in heart failure. Indeed, we found that HIF-1α expression was greatly enhanced by TAC in βarr2-C253S versus WT mice (Figures 7R and S7O), as was expression of the von Hippel-Lindau protein (pVHL) (Figures 7S and S7O), which is transcriptionally upregulated by HIF-1 (Karhausen et al., 2005). Thus, our findings provide new insight into regulation by βarrs of the HIF-p53 axis including a central role for S-nitrosylation.
Discussion
Our findings provide several major advances. First, we establish a general mechanism for biasing GPCR-mediated signals. The ability of synthetic ligands to induce biased signaling through GPCRs points to the possibility that endogenous mechanisms might operate to bias signaling, which would represent a novel, fundamental aspect of GPCR biology, but general mechanisms for biasing GPCRs had not heretofore been established. Our results establish that S-nitrosylation of βarr1/2 by n/iNOS provides such a mechanism. Second, as a consequence of the wide-ranging impact of βarrs across most GPCRs, our findings support the proposition that regulation by S-nitrosylation of GPCR-mediated signal transduction may represent a broadly operating mechanism through which NO exerts its ubiquitous cellular influence. Third, we also demonstrate that βarrs can be regulated allosterically by NO and thereby function independently of ligand-receptor interactions. The idea that nature may exploit allostery in βarrs (rather than an orthosteric mechanism in GPCRs generally) to regulate bias, and that βarrs can, through posttranslational modification, be disconnected from classical activities and endowed with new function, provides novel insight into GPCR-related machinery. In addition, our results may provide novel insight into the molecular pathophysiology of heart failure, where S-nitrosylation-regulated signaling bias may play a previously unappreciated role in sine qua non features of the disease, including receptor down-regulation and adrenergic dysfunction, that influence the severity of heart failure and the response to commonly employed therapeutic agents.
Previous analyses have revealed that regulation by S-nitrosylation of GPCR-mediated signaling is evidently multiplexed with respect to signaling elements affected, which include, in addition to βarrs, GRKs, dynamin, and GPCRs (Aronstam et al., 1995; Haldar and Stamler, 2013; Leclerc et al., 2006; Nozik-Grayck et al., 2006; Ozawa et al., 2008; Wang et al., 2006; Whalen et al., 2007). Our new findings indicate that multiplexed regulation reflects as well the enzymatic source of NO: all three isoforms of NOS bind both βarr1 and βarr2, but the consequences for GPCR signaling are different, reflecting differential S-nitrosylation of βarr1/2 by n/iNOS versus eNOS. Because S-nitrosylation by n/iNOS of either βarr1 or βarr2 suppresses recruitment to multiple GPCRs, S-nitrosylation of βarrs may represent a prevalent mechanism through which signaling, desensitization and internalization of GPCRs are regulated. The expression of dedicated NO synthesizing enzymes and of multiple GPCRs characterizes most, if not all, metazoan cells and tissues. Thus our results, in combination with prior demonstrations of the role of S-nitrosylation in regulating GPCR transduction and fate, support the proposition that regulation of GPCR function by S-nitrosylation may represent a principal mechanism through which NO exerts its ubiquitous influence in cells.
The ligand-induced interaction between βarrs and GPCRs is best characterized in the case of the β2AR, (Gurevich and Gurevich, 2004; Shukla et al., 2013). Recent structural analysis has shown that a short loop of residues (242-246) within the C-terminal domain (of βarr1) is in a position to interact with the ligand-activated β2AR (Shukla et al., 2014). As shown in Figure S2G, this motif is conserved across metazoan phylogeny where βarrs have been identified. Furthermore, the region of the β2AR that can interact with the C-terminal loop of bound βarr1 (Shukla et al., 2014) includes the highly conserved DRY motif important for agonist-induced recruitment of βarrs to the β2AR (Valentin-Hansen et al., 2012) as well as to dopaminergic GPCRs (Kim and Caron, 2008). Thus, perhaps S-nitrosylation of Cys253/251 within βarrs negatively regulates directly the binding of βarrs to the β2AR, and a similar mechanism may operate to regulate βarr binding to other GPCRs including AT1aR and the B1R (although the operation of this mechanism across a broader range of GPCRs remains to be tested). In addition, residues 242-246 lie within the dimerization domain of βarr1/2, and our finding that S-nitrosylation of Cys253/251 is associated with homo-dimerization of βarrs suggests that dimerization may contribute to the suppressive effect of S-nitrosylation on recruitment of βarrs to GPCRs by masking the interaction domain. Sequestration of βarrs in the cytosol by enhanced interaction with cytosolic partners including Mdm2 would also contribute to suppressed agonist-induced membrane recruitment.
Although homo/heterodimerization of βarrs in vitro and in situ is now well documented (Boularan et al., 2007; Milano et al., 2006; Storez et al., 2005), the regulation and physiological significance of βarr dimerization have remained in question. We show here that homodimerization is enhanced by S-nitrosylation of βarr1/2 and represents a previously unappreciated signature of S-nitrosylation under conditions in which n/iNOS are upregulated, including inflammation, aging and heart failure. Our finding that S-nitrosylation of βarr2 enhances its affinity for Mdm2 and results in the titration of the transcriptional regulator Mdm2 from nucleus to cytosol is consistent with previous findings that it is the dimerized form of βarr2 that interacts with Mdm2 (Boularan et al., 2007). The profound subcellular redistribution of MDM2 induced by S-nitrosylation of βarrs may signify NO-dependent control of MDM2-dependent transcription and of the βarr interactome more broadly, comprising hundreds of proteins that subserve transduction of diverse GPCR signals (Xiao et al., 2007).
Our results suggest that the balance between G protein-vis-à-vis βarr-mediated transduction, reflecting the status of βarr S-nitrosylation, serves as a previously unrecognized determinant of adrenergic responsivity in the heart, and of cardiac function generally (Figure S7R). It is important to note that the molecular bases of compromised adrenergic responsivity and βAR down-regulation that are sine qua nons of human heart failure are in fact not fully understood, and in our TAC model, down-regulation and compromised responsivity are not observed unless S-nitrosylation of βarr is impaired. Thus, our findings suggest new mechanistic understanding of the basis of altered adrenergic function in heart failure, and emphasize that key characteristics of the disease may be accompanied by biased transduction. Indeed, our finding of enhanced S-nitrosylation of βarrs in both ischemic and non-ischemic human heart failure suggests that G protein bias may be a common feature of heart failure.
In addition, our results point to the possibility that heart failure may represent different clinical conditions depending on NO bioavailability, which will be reflected in G protein versus βarr bias (Figure S7R). Differences in n/iNOS expression (notably beneficial in this model) or degree of adrenergic stress could then determine the basis of adrenergic dysfunction (resulting from excessive adrenergic stimulation and/or impaired S-nitrosylation) and thus the efficacy of therapeutic agents that target GPCRs (e.g. beta blockers) or induce S-nitrosylation (nitrates). Further, one might anticipate limited efficacy in heart failure of βarr-biased ligands (e.g. carvedilol) in settings of enhanced βarr S-nitrosylation (and thus suppressed βarr function). Altogether, our findings in cellular systems, tissues, experimental heart failure and human heart failure suggest that NO-based regulation of βarrs and thereby GPCR function may be an unanticipated determinant of numerous clinical conditions in which GPCR dysfunction is implicated.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Jonathan Stamler at jonathan.stamler@case.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines employed and culture conditions
HEK293 (female in origin) and RAW264.7 cells (male in origin) were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) plus penicillin/streptomycin. HEK-nNOS cells were maintained in the presence of neomycin. RAW264.7 cells were treated with LPS (0.1 μg/ml) and IFγ (100 U/ml) overnight to induce iNOS. HEK293-iNOS cells were maintained in the presence of blasticidin and zeocin and iNOS expression was induced with doxycycline. W9 cells (derived from HEK cells) were maintained in the presence of neomycin. HEK293-AT1aR cells were maintained in the presence of zeocin. All cells were grown at 37 deg C under 5 % CO2.
βarr2-C253S mice
Male mice were employed in all experiments. All experimental procedures employing mice were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University School of Medicine and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Committee on Care and Use of Laboratory Animals (1996) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No. (NIH) 85-23).
βarr2-C253S mice were generated on a C57BL/6J background by InGenious Targeting Laboratory (Ronkonkoma, NY) and verified by genotyping. βarr2 expression levels were indistinguishable in the hearts of WT (C57BL/6J; Jackson Laboratory, Bar Harbor, ME) and βarr2-C253S mice (Figure S7C). WT and βarr2-C253S mice were subjected to TAC at 9 weeks of age.
Analysis of old versus young mice
The heart and lungs were obtained from C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) at 14-16 weeks of age (“young”) or 50-53 weeks of age (“old”).
Human Heart Samples
The collection and use of tissues for this study was approved by the Duke University Health System Institutional Review Board.
Patient demographics were as follows:
| Non-failing | Non-ischemic failing | Ischemic failing | |
|---|---|---|---|
| Age, mean (SD) | 52 (14) | 55 (6) | 62 (6) |
| Males/females | 8/4 | 9/3 | 12/0 |
METHOD DETAILS
Murine TAC model
WT and βarr2-C253S mice were subjected to TAC at 9 weeks of age, carried out as described (Rockman et al., 1991) with minor modifications. In brief, mice were anesthetized by IP injection of ketamine (100 mg/Kg body weight) and xylazine (10 mg/Kg body weight). The aortic arch was exposed and a ligature of 7-0 silk suture was placed around the arch between the right innominate artery and left carotid artery. The ligature was tightened around the arch and a segment of 27G needle apposed to the arch in the case of samples prepared for biochemical analysis and of 28G needle in the case of mice employed for electrocardiographic analysis, and the needle was removed immediately. The resulting reduction in the cross-sectional area of the arch was ∼90% and ∼92% with 27G and 28G needles respectively. The procedure followed for sham-operated control subjects was identical, except that no suture was placed.
For biochemical analysis, mice were sacrificed 3 wk post-TAC and the heart was removed and weighed before storage in liquid nitrogen until processed. Lysates were prepared from finely chopped individual hearts with a Polytron homogenizer and, following determination of protein concentration, processed for SNO-RAC, Western blotting for NOSs and assessment of βarr dimerization. For assessment of βarr dimerization, chopped tissue was incubated with 4% paraformaldehyde for 15 min prior to homogenization. Samples from individual mice were assessed for NOS expression and βarr dimerization, and samples were pooled from 2 individual mice for SNO-RAC.
For echocardiography, mice were examined at two or four weeks post-TAC. Mice were anesthetized with isoflurane, and transthoracic echocardiography was performed using a Vevo 770 High-Resolution Imaging System equipped with an RMV-707B 30-MHz probe (VisualSonics). Standard M-mode sampling was used through the left ventricular short axis at the midpapillary level. Ejection fraction, fractional shortening, and other parameters were determined using the system’s software.
cAMP and pERK measurement in the mouse heart
cAMP was assayed both by ELISA (cAMP complete ELISA kit, ENZO Life Sciences, according to the manufacturer’s instructions) and by Western blotting for PKA-phosphorylated phospholamban (anti-phospho-Ser16, Badrilla, Leeds, UK), a well-established indirect method (Mangmool et al., 2010). The results of blotting for phospho-phospholamban were normalized with respect to total phospholamban (anti-phospholamban, Badrilla). pERK was assessed by Western blotting as described above. To prepare samples, the heart was flash frozen in situ, extracted and separated into three segments. One segment was ground to a fine powder with a mortar and pestle under liquid nitrogen, homogenized in 0.1 M HCl and centrifuged. The supernatant was used for cAMP detection. The second segment was homogenized in lysis buffer for Western blotting.
Realtime qPCR for βarr1 and βarr2 expression
Total RNA was extracted from the third segment of heart tissue by homogenization in TRIzol reagent (Fisher Scientific), according to the manufacturer’s instructions. cDNAs were synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosciences). The relative abundance of β1AR and β2AR mRNAs was quantified with the TaqMan Gene Expression Assay (Mouse βarr1: Mm00617540, Mouse βarr2: Mm00520666, Mouse GAPDH: Mm99999915). GAPDH expression was used for normalization.
Myocardial membrane isolation to assess β1AR and β2AR abundance
Hearts were frozen in situ as above and a membrane fraction was isolated as previously described (Zhou et al., 1998), with modifications. Hearts were homogenized in hypotonic lysis buffer and the homogenate was centrifuged at 2,000 G for 10 min. The supernatant was collected and centrifuged at 9,000 G for 20 min, and the resultant supernatant was centrifuged at 180,000 G for 90 min. Essentially all β1AR and β2AR were recovered in this final pellet. The pellet was reconstituted with RIPA buffer containing protease inhibitor (Roche) then running buffer for Western blotting (anti-β1AR (Santa Cruz), anti-β2AR (as above) and anti-Caveolin3 (BD Biosciences)).
Assessment of ISO-induced changes in heart rate and contractility
Heart rate was monitored by electrocardiography as described previously (Zhang et al., 2015) in mice anesthetized with isoflurane. The electrocardiogram (lead II) was continually recorded with a data acquisition system (PowerLab 8/30, AD instruments) and analyzed with LabChart 7 Pro software (version 7.3.7, AD instruments). Measurements were obtained continuously before and after bolus injection of ISO (10 μg/Kg, 30 μl) via an inferior vena cava or jugular catheter.
Analysis of old versus young mice
The heart and lungs were obtained from C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) at 14-16 weeks of age (“young”) or 50-53 weeks of age (“old”). Tissue processing and analysis were as for the analysis of TAC model mice. Samples from individual mice were assessed for NOS expression, βarr dimerization and SNO-RAC.
Human heart samples
Samples were acquired from the anterolateral left ventricular free wall of explanted ischemic failing (IF) or non-ischemic failing (NIF) hearts, or from non-failing (Control, C) hearts not utilized for transplant. Samples were processed and analyzed as for mouse tissues essentially as above. For IF hearts, the area of infarct was identified at the time of tissue procurement and only sites remote from the infarct with minimal scar were sampled. IF and NIF hearts underwent cold ischemic times of about 10-15 min and C hearts underwent cold ischemic times of about 10 min-2 hr before samples were procured and immediately flash frozen in liquid nitrogen before storage at -80 deg C. Samples from individual patients were assessed for NOS expression and βarr dimerization, and samples were pooled from 2-3 individual patients for SNO-RAC.
Receptor ligands and stimulation
ISO, angiotensin II and DAKD were obtained from Sigma-Aldrich. ISO was applied at 10 μM for 5-10 min unless otherwise indicated. Angiotensin II was applied at 0.1 μM for 0.5-5 min. DAKD was applied at 1 μM for 2 min.
Biotin-switch and SNO-RAC assays of protein S-nitrosylation
The biotin-switch method (employed in a limited number of early experiments) and the SNO-RAC method were employed essentially as described (Forrester et al., 2009; Ozawa et al., 2008). In brief, samples were incubated in HENS buffer (100 mM HEPES, 1 mM EDTA, 0.1 mM neocuproine, pH7.7) containing SDS (1%) and the thiol-blocking agent methyl methanethiosuflonate (0.1 %) for 20 min at 50 deg C. Samples were then collected by acetone precipitation, washed with 70 % acetone and re-suspended in HENS buffer containing sodium ascorbate (50 mM) before incubation with streptavidin-agarose (Pierce) or thiopropyl Sepharose in the dark for 4 hr. Washed beads were eluted and samples analyzed by Western blotting.
Immunoprecipitation, Western blotting and immunostaining
For immunoprecipitation, cells were washed X2 with chilled PBS and lysed with lysis buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% NP40, 10% glycerol and protease inhibitor cocktail (Roche)). After 30 min incubation on ice followed by sonication, samples were centrifuged at 20,000 g for 5 min. The supernatant was collected and protein concentration was determined. Immunoprecipitation from samples containing equal amounts of protein was with primary Ab followed by agarose-coupled protein G or with monoclonal M2 anti-FLAG-affinity agarose, prior to analysis by Western blotting. Blotted membranes were incubated for 1 hr at RT or overnight at 4 deg C with primary antibodies, and washed with PBS containing 0.05% Tween-20 before incubation with HRP-conjugated secondary antibody (anti-mouse or anti-rabbit IgG (Promega)) for 1 hr at RT followed by chemiluminescent detection (ECL (GE Healthcare)). Densitometric analysis was performed using Image J software. Densitometric values were normalized with respect to total antigen content. For immunostaining, cells on glass-bottom dish were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 2 min. The samples were blocked in 5% skim milk, followed by incubation with primary antibodies overnight at 4 deg C. The corresponding secondary antibodies were labeled with AlexaFluor 488 or 546 (Molecular Probes, Eugene, OR, USA). Nuclear staining was with propidium iodide.
Co-immunoprecipitation and detection of βarr dimerization
In general, effective demonstration of dimerization of βarr2 or βarr1, and of co-immunoprecipitation of βarr2 and the β2AR, required chemical cross-linking (although the results shown in Figures 2A–2D and quantified in Figure 2F were obtained without cross-linking). The hetero-bifunctional cross-linker DSP (Pierce, Rockford, IL, USA) was employed with intact cells, added after stimulation (ISO) but before lysis. In the mouse TAC model and in human heart samples, the cross-linker paraformaldehyde was employed, added before lysis. Co-immunoprecipitation of βarr1/2 with eNOS/iNOS/nNOS, and of MDM2 with βarr2 or p53, did not require cross-linking. Densitometric values were normalized with respect to total antigen content in the case of co-immunoprecipitations and to β–actin or glyceraldehyde 6-phosphate dehydrogenase in analysis of dimerization.
Cell-surface labeling to assess ligand-induced internalization of the β2AR
W9 cells transfected with WT or C253S βarr2 and nNOS or iNOS were stimulated with ISO (10 M; 10 min), then biotinylated with Sulfo-NHS-SS-Biotin (0.25 mg/ml; Pierce) in Hank's balanced salt solution (HBSS; Gibco) for 30 min at 4 deg C following the manufacturer’s instructions. Following cell-surface biotinylaton and washing X3 with 100 mM glycine in HBSS, cells were lysed and protein content was determined. Equal quantities of protein were incubated with streptavidin-agarose (Pierce) for 2 hr at 4 deg C. Washed beads were eluted with 2xSDS loading buffer containing 2% SDS and 5% 2-mercaptoethanol at 42 deg C for 10 min. Samples were analyzed by Western blotting for the β2AR (Santa Cruz).
Flow cytometry to assess ligand-induced internalization of the β2AR
Flow cytometry was performed as described previously (Whalen et al., 2007) with modifications. W9 cells were transfected with WT or Cys253Ser βarr2 and nNOS or iNOS using Lipofectamine2000 (Invitrogen). After 4 hr of starvation, cells were stimulated with ISO (10 M; 10 min), washed with PBS and collected by centrifugation. Re-suspended-cells were fixed with 1% paraformaldehyde for 15 min then incubated with primary antibody, M2 anti-FLAG, for 20 min at 4 deg C. Washed cells were then incubated with secondary antibody, phycoerythrin-conjugated goat anti-mouse IgG (BioLegend), for 20 min at 4 deg C. After washing, stained cells were analyzed by FACS (LSRII, BD Bioscience). Cells incubated with isotype-matched non-fluorescent secondary antibody (BioLegend) served as controls.
Confocal microscopy for βarr2 recruitment to membrane receptors
HEK293 cells stably expressing Flag-β2AR or HA-AT1aR and cultured on 35 mm glass bottom dishes were transfected with plasmids encoding HA-tagged or Flag-tagged βarr2 (WT or C253S) employing Lipofectamine2000 (Invitrogen). After 24 h, cells were stimulation with agonist at 37 deg C, followed by fixation with 4% paraformaldehyde for 15 min and permeabilization with 0.2% Triton X-100 for 2 min. Samples were blocked in 5 % skim milk, followed by incubation with primary antibodies overnight at 4 deg C. The corresponding secondary antibodies were labeled with AlexaFluor 488 or 546 (Molecular Probes). The samples were imaged by confocal microscopy (Zeiss).
Assay of cAMP
W9 cells transfected with WT or C253S βarr2, nNOS or iNOS or empty vector were serum-starved for 4 hr then incubated with or without ISO (10 M) for 2 min. Washed cells were collected and cAMP was assayed with a Parameter cAMP assay kit (R&D Systems) according to the manufacturer’s instructions.
Assessment of iNOS-induced biased signaling through the B1R
BAEC (Cell Applications; gender of origin unknown) were cultured in DMEM containing 10% FBS plus penicillin/streptomycin and treated with LPS (20 μg/ml) and IFγ (1000 U/ml) for 2 days with or without 1400W (10 μM) to induce iNOS and the B1R. Cells were then transferred to serum-free DMEM for 2 hr prior to stimulation with DAKD (1 μM). B1R and β-actin expression were assessed by qPCR (Syber green; Applied Biosystems) with the primers: B1R F: 5′-ATTCCTGCTGCGCTCTGTCAA-3′; B1R R: 5′-AGAAAACGATCGCAGCCAAGG-3′; β-actin F: 5′-AGATCTGGCACCACACCTTCT-3′; β-actin R: 5′-TCATCTTCTCACGGTTGGCCT-3′. Western blotting for pERK and pPKD were carried out as above. To assess dimerization of βarr2, washed cells were treated with DSP prior to Western blotting, as above.
Plasmids, cysteine mutants and truncated βarr1/2
pcDNA3-βarr1-Flag, pcDNA3-βarr2-Flag, pcDNA3-eNOS, pcDNA3-nNOS, pcDNA3-iNOS and cysteine mutants and truncated forms of βarr1/2 were employed in a previous study (Ozawa et al., 2008). pcDNA3-βarr2 C253S-HA was generated from pcDNA3-βarr2 HA by site-directed mutagenesis using the QuickChange XL site-directed kit (Stratagene, La Jolla, CA). Transfection was performed with Lipofectamine2000 (Invitrogen, Grand Island, NY).
Antibodies employed in immunoprecipitation, Western blotting and immunostaining
Antibodies employed in immunoprecipitation, Western blotting and immunostaining included: monoclonal M2 anti-FLAG (Sigma-Aldrich Aldrich), anti-HA (Covance), anti-β2AR (Santa Cruz), anti-nNOS (rabbit polyclonal (Santa Cruz) or mouse monoclonal anti-nNOS (BD Bioscience)), anti-iNOS (rabbit polyclonal (Santa Cruz) or mouse monoclonal (BD Bioscience)), anti-eNOS (rabbit polyclonal (Santa Cruz) or mouse monoclonal (BD Bioscience)), anti-ERK and anti-phospho ERK (phospho-Thr202/Tyr204) (Cell Signaling), anti-PKD and anti-phospho-PDK (phospho-Ser916) (Cell Signaling), anti-MDM2 (Santa Cruz), anti-p53 (Santa Cruz), anti-clathrin (heavy chain) (BD Transduction), anti-AP2 (BD Transduction), anti-β-actin (Sigma-Aldrich) and anti-GAPDH (Millipore), anti-HIF-1α (R&D Systems), anti-pVHL (BD Bioscience). Anti-βarr (A1CT and A2CT) rabbit polyclonal antibodies were provided by R.J. Lefkowitz, and additional antibodies against βarr1/2 were obtained from Santa Cruz, Cell Signaling and BD Transduction. Validation of the specificity of βarr1/2 antibodies in the setting of Western blotting is presented in Figures S1D and S1E.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values are expressed as mean ± SEM and n represents experimental replications. In each histogram showing normalized experimental values, the denominator was the mean of control replicates. Student’s t-test was employed for paired observations and one-way ANOVA in conjunction with the Tukey-Kramer test was employed for multiple observations. Values of p<0.05 were considered statistically significant. Figure legends provide n’s and p-values.
DATA AND SOFTWARE AVAILABILITY
Data analysis was carried out with Image J (NIH) and Microsoft Excel.
Supplementary Material
Highlights.
S-nitrosylation of β-arrestins (βarrs) by n/iNOS suppresses canonical βarr function.
S-nitrosylation provides a general mechanism to bias GPCR signaling via G proteins.
S-nitrosylation of βarrs also enables ligand-independent βarr function.
S-nitrosylation of βarr2 critically supports adrenergic function in failing hearts.
Acknowledgments
This work was supported by Grants P01 HL075443 and R35 HL135789 from the National Institutes of Health. HH was supported by an Overseas Research Fellowship from the Uehara Memorial Foundation. We thank Howard Rockman, Duke University Medical Center, for valuable advice and input; Robert J. Lefkowitz, Duke University, for providing advice and multiple cell lines and antibodies; Michael J. Watson, Duke University, for facilitating acquisition of human heart samples; Masakazu Ishikawa, Joao Pedro Lopes and Michael Sramkoski, Case Western Reserve University, for assistance with FACS analysis; Zhaoxia Qian, Case Western Reserve University, for assistance with mouse tissue samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.H. designed and performed experiments, analyzed results and wrote the manuscript; D.T.H. designed experiments, analyzed results and wrote the manuscript; R.Z., K.S., H.G. and B.L.T. performed experiments; D.E.B. and C.A.M. provided critical materials; M.K.J. and W.J.K. assessed results; J.S.S. designed experiments and wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ahn S, Nelson CD, Garrison TR, Miller WE, Lefkowitz RJ. Desensitization, internalization, and signaling functions of β-arrestins demonstrated by RNA interference. Proc Natl Acad Sci USA. 2003;100:1740–1744. doi: 10.1073/pnas.262789099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, Milligan G. Targeted elimination of G proteins and arrestins defines their specific cotributions to both intensity and duration of G protein-coupled receptor signaling. J Biol Chem. 2016;291:27147–27159. doi: 10.1074/jbc.M116.754887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronstam RS, Martin DC, Dennison RL, Cooley HG. S-nitrosylation of m2 muscarinic receptor thiols disrupts receptor-G-protein coupling. Ann NY Acad Sci. 1995;10:215–217. doi: 10.1111/j.1749-6632.1995.tb17477.x. [DOI] [PubMed] [Google Scholar]
- Boularan C, Scott MG, Bourougaa K, Bellal M, Esteve E, Thuret A, Benmerah A, Tramier M, Coppey-Moisan M, Labbe-Jullie C, et al. β-arrestin 2 oligomerization controls the Mdm2-dependent inhibition of p53. Proc Natl Acad Sci USA. 2007;104:18061–18066. doi: 10.1073/pnas.0705550104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cau SB, Carneiro FS, Tostes RC. Differential modulation of nitric oxide synthases in aging: therapeutic opportunities. Front Physiol. 2012;3:218. doi: 10.3389/fphys.2012.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Haldar SM, Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest. 2013;123:101–110. doi: 10.1172/JCI62854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- Karhausen J, Kong T, Narravula S, Colgan SP. Induction of the von Hippel-Lindau tumor suppressor gene by late hypoxia limits HIF-1 expression. J Cell Biochem. 2005;95:1264–1275. doi: 10.1002/jcb.20489. [DOI] [PubMed] [Google Scholar]
- Kim KM, Caron MG. Complementary roles of the DRY motif and C-terminus tail of GPCRS for G protein coupling and β-arrestin interaction. Biochem Biophys Res Commun. 2008;366:42–47. doi: 10.1016/j.bbrc.2007.11.055. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci USA. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhr FK, Zhang Y, Brovkovych V, Skidgel RA. β-arrestin 2 is required for B1 receptor-dependent post-translational activation of inducible nitric oxide synthase. FASEB J. 2010;24:2475–2483. doi: 10.1096/fj.09-148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc PC, Lanctot PM, Auger-Messier M, Escher E, Leduc R, Guillemette G. S-nitrosylatin of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br J Pharmacol. 2006;148:306–313. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Liao Y, Bin J, Asakura M, Xuan W, Chen B, Huang Q, Xu D, Ledent C, Takashima S, Kitakaze M. Deficiency of type 1 cannabinoid receptors worsens acute heart failure induced by pressure overload in mice. Eur Heart J. 2012;33:3124–3133. doi: 10.1093/eurheartj/ehr246. [DOI] [PubMed] [Google Scholar]
- Limas CJ. Increased number of β-adrenergic receptors in the hypertrophied myocardium. Biochim Biophys Acta. 1979;588:174–178. doi: 10.1016/0304-4165(79)90382-9. [DOI] [PubMed] [Google Scholar]
- Ma L, Pei G. β-arrestin signaling and regulation of transcription. J Cell Sci. 2007;120:213–218. doi: 10.1242/jcs.03338. [DOI] [PubMed] [Google Scholar]
- Mangmool S, Shukla AK, Rockman HA. β-arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J Cell Biol. 2010;189:573–587. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C. Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem. 2006;281:9812–9823. doi: 10.1074/jbc.M512703200. [DOI] [PubMed] [Google Scholar]
- Nozik-Grayck E, Whalen EJ, Stamler JS, McMahon TJ, Chitano P, Piantadosi CA. S-nitrosoglutathione inhibits alpha1-adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am J Physiol. 2006;290:L136–143. doi: 10.1152/ajplung.00230.2005. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al. p-53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and MDM2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storez H, Scott MG, Issafras H, Burtey A, Benmerah A, Muntaner O, Piolot T, Tramier M, Coppey-Moisan M, Bouvier M, et al. Homo- and hetero-oligomerization of β-arrestins in living cells. J Biol Chem. 2005;280:40210–40215. doi: 10.1074/jbc.M508001200. [DOI] [PubMed] [Google Scholar]
- Tan M, Xu X, Ohba M, Cui MZ. Angiotensin II-induced protein kinase D activation is regulated by protein kinase Cδ and mediated via the angiotensin II type 1 receptor in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:2271–2276. doi: 10.1161/01.ATV.0000148449.92035.3a. [DOI] [PubMed] [Google Scholar]
- Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–1762. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen L, Groenen M, Nygaard R, Frimurer TM, Holliday ND, Schwartz TW. The arginine of the DRY motif in transmembrane segment III functions as a balancing micro-switch in the activation of the β2-adrenergic receptor. J Biol Chem. 2012;287:31973–31982. doi: 10.1074/jbc.M112.348565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. Endogenous allosteric modulators of G protein-coupled receptors. J Pharmacol Exp Ther. 2015;353:246–260. doi: 10.1124/jpet.114.221606. [DOI] [PubMed] [Google Scholar]
- Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci USA. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, et al. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci USA. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS. Oxygen-regulated β2-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2:ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Hess DT, Qian Z, Hausladen A, Fonseca F, Chaube R, Reynolds JD, Stamler JS. Hemoglobin βCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc Natl Acad Sci USA. 2015;112:6425–6430. doi: 10.1073/pnas.1502285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sevilla L, Vallega G, Chen P, Palacin M, Zorzano A, Pilch PF, Kandror KV. Insulin-dependent protein trafficking in skeletal muscle cells. Am J Physiol. 1998;275:E187–196. doi: 10.1152/ajpendo.1998.275.2.E187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.